
Abstract
Bilirubin (BR), a metabolite with increased concentrations in plasma during viral hepatitis, has been recognized as a potential immune-modulator. We recently reported that conjugated BR (CB) augments regulatory T cell (Treg) suppressor activity during acute hepatitis A virus (HAV) infection. However, the mechanisms related to the effects of CB on Treg function in the course of hepatotropic viral diseases have not been elucidated. T cell immunoglobulin domain and mucin domain 3 (TIM-3), via its interactions with galectin-9 (GAL-9), is a receptor associated with enhanced Treg function. Thus, TIM-3 expression may be related to the crosstalk between CB and Tregs during HAV infection. Herein, in vitro treatment with high concentrations of CB upregulated TIM-3 expression on Tregs from healthy donors. CB treatment in vitro did not induce de novo Treg generation, and in vitro stimulation with TGF-β, which shows increased secretion during HAV infection, resulted in a trend toward increased TIM-3 expression on Tregs and CD4+ T lymphocytes (TLs) from healthy donors. Interestingly, an upregulation of TIM-3 expression on CD4+CD25+ T cells and an increase in the proportion of CD4+ TLs expressing GAL-9 were found in HAV-infected patients with abnormal CB values relative to healthy controls. In addition, a statistically significantly reduction in IL-17F production was observed after treatment of CD4+ TLs from healthy donors with high doses of CB in vitro. In summary, our results suggest that CB might regulate Treg activity via a TIM-3-mediated mechanism, ultimately leading to an anti-inflammatory hepatoprotective effect.
Introduction
T
Infections with hepatotropic viruses, including hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis E virus (HEV), result in elevated serum aminotransferase activity and the deregulation of internalization and excretion of BR by hepatocytes, which results in increased CB values (>0.3 mg/dL). These changes in normal BR concentration are related to liver disease (16).
In addition, BR has recently been recognized as an immune-modulatory metabolite able to modulate CD4+ T lymphocyte (TL) function by promoting the induction of de novo regulatory T cells (Tregs) after the administration of BR to transplant recipients in murine models (15,26,36,39), and this correlates with an increase in the proportion of Tregs in cirrhotic patients with hyperbilirubinemia (13). However, the mechanisms underlying the effect of CB on Treg function in the course of hepatotropic viral diseases have not been elucidated.
Each year, HAV infects ∼1.5 million people worldwide, and the pediatric population is at a greater risk of contracting the infection (4). Hepatitis A is typically cleared after acute infection without liver damage in immunocompetent individuals (23).
The exact mechanisms that contribute to the resolution of HAV infection during the acute phase have not been determined, but it is accepted that the final elimination of the virus is mediated by a specific and effective host immune response. In this regard, the humoral immune response to HAV is characterized by the specific production of IgM and IgG antibodies that target viral proteins. IgM antibodies are typically short-lived during HAV infection, and their maximum levels coincide with a marked elevation in serum aminotransferase activity, viremia, and maximum serum levels of BR, with CB values above normal (35). The adaptive immune response relies on CD4+ TLs to control HAV infection (42). Moreover, an effective resolution may also be mediated by host metabolic components.
We have recently reported that in the context of HAV infection, high CB levels (>2 mg/dL) result in a lower degree of phosphorylation of intracellular proteins, such as CD3 epsilon and Syk in CD4+ TLs. Furthermore, our data reveal that CB levels ≥2 mg/dL increase the suppressive capacity of Tregs, thereby contributing to the nonoptimal functional status of CD4+ TLs during HAV infection (5) through CB-mediated modulation of STAT-1 and STAT-5 function that in turn determines specific cytokine profiles (1,5,6). These observations strongly suggest that during HAV infection, BR can regulate the inflammatory immune response via Treg control. Consistent with this hypothesis, decreased Treg frequency is associated with liver injury in acute hepatitis A (3).
Because of the high lipophilic chemical nature of BR, its function has been associated with membrane receptors (33), resulting in a powerful suppressive function on T cells and inhibiting costimulatory molecules such as CD28, B7-1, and B7-2 (17). During HAV infection, increased levels of CB result in T cell immunoglobulin domain and mucin domain 1 (TIM-1) upregulation on Tregs (5), and a direct interaction between HAV and TIM-1 may partially explain the changes in Treg function (19).
In addition, high expression of PD-1 and CTLA-4 has been reported during acute HAV infection, which suggests that inhibitory molecules may suppress cytotoxic T cells and thus prevent the destruction of virus-infected hepatocytes (2). In this regard, the T cell immunoglobulin domain and mucin domain 3 (TIM-3) inhibitory receptor, via its interactions with galectin-9 (GAL-9), has been associated with enhanced Treg function (7). Moreover, TIM-3 negatively regulates T cells, resulting in exhausted T cells and subsequent cell death of TIM-3+ T cells (10,28) suggesting a double-edged effect of TIM-3 in cellular immune responses. However, the effect of CB on the expression of TIM-3 on Tregs during acute HAV infection has not been determined to date.
Materials and Methods
Study population
A total of 71 pediatric patients (1–15 years old) were recruited from May 2015 to February 2016 and included in this study. Hepatitis was defined as hepatomegaly, fever (>38°C), and/or jaundice with elevated serum aspartate aminotransferase (AST) (>38 IU/L) and alanine aminotransferase (ALT) (>35 IU/L) values, as previously described (6). A total of 40 healthy pediatric donors (1–15-year-old children with normal hepatic enzymatic activity in the absence of HAV serological markers) who had been admitted to the Unidad de Vacunacion of the Hospital Civil Fray Antonio Alcalde (HCFAA) were included in this study. CB, alkaline phosphatase (ALP), and total protein (TP) values were measured in patients and controls.
Demographic and clinical features were recorded from all participants using a structured questionnaire. Patients and healthy donors who were undergoing treatment with hepatotoxic, immunosuppressive or anti-inflammatory drugs, those with acute or chronic HEV, HBV, or HCV infections, and those diagnosed with autoimmune hepatitis were excluded from the study. HAV vaccination is not mandatory in Mexico, and none of the participants in the study had been vaccinated against HAV. All the participants had been vaccinated against HBV (a mandatory vaccine for children in Mexico).
After the children's parents provided informed consent, blood samples were obtained by venipuncture. The Ethics Committees of the HCFAA and the Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, approved this study (IRB: HCG/CI-883). The study was conducted at the Centro de Referencia de Hepatitis Virales del Occidente de Mexico and the Unidad de Inmunovirologia in the Servicio de Biologia Molecular, Hospital Civil de Guadalajara Fray Antonio Alcalde (HCFAA) in Guadalajara, Jalisco, Mexico.
Serological tests
To detect acute hepatitis A infection, serum samples from patients diagnosed with hepatitis were screened for the presence of anti-HAV IgM and for the absence of anti-HAV IgG.
The presence of anti-HAV IgM and the absence of anti-HAV IgG, the surface antigen of HBV (HBsAg), and anti-HCV antibodies were determined using a third-generation microparticle immunoenzymatic assay (Ax-SYM HAVAB-M 2 0, AxSYM HBsAg [V2], and AxSYM HCV 3 0; Abbott Laboratories, Chicago, IL) with an Ax-SYM analyzer (Abbott Laboratories). The absence of total anti-hepatitis B core antigen anti-HBc (total IgM and IgG) and anti-HEV antibodies was measured using immunoenzymatic assays (Monolisa Anti-HBc PLUS, Bio-Rad Laboratories, Chicago, IL, and MP Diagnostics, Geneva, Switzerland and MyBioSource, San Diego, CA, respectively). Final readouts were obtained with a WHYM201 Microplate reader (Poweam Medical Co., Ltd., Nanjing, China).
Serum levels of TP, ALT, AST, ALP, and CB were measured using routine clinical laboratory procedures by using an Ortho Clinical Vitros 250 Chemistry System (GMI, Ramsey, MN).
In vitro analysis
Cell purification
Ficoll-Paque PLUS (Healthcare, Uppsala, Sweden) gradient centrifugation was used to isolate peripheral blood mononuclear cells (PBMCs) from six anticoagulated blood samples from healthy pediatric donors. In brief, EDTA-anticoagulant-treated blood was diluted with an equal volume of a balanced salt solution (phosphate-buffered saline [PBS] 1 × ), layered carefully over Ficoll-Paque PLUS (without intermixing), and centrifuged at 400 g for 30 min. The buffy coat of each sample was washed three times with PBS 1 × (300 g; 10 min; room temperature) and resuspended in auto MACS Running Buffer (Miltenyi Biotec, Bergisch Gladbach, Germany).
CD4+ TLs were purified by negative magnetic selection with CD8, CD19, CD123, and CD127 antibodies, followed by Treg (CD4+CD25+CD127low) positive selection with anti-CD25 Micro Beads (Miltenyi Biotec). Before experimentation, CD4+ TLs and Tregs were arrested for 2 h in RPMI 1640 (HyClone, Logan, UT) supplemented with 2% (v/v) fetal bovine serum (FBS) with 2 mM L-glutamine, 50 μg/mL penicillin, 50 μg/mL streptomycin, and 50 μM β-mercaptoethanol (Sigma, St. Louis, MO).
Frequencies of CD4+ TLs expressing Foxp3, CD4+ TLs, and Tregs expressing TIM-3
Purified CD4+ TLs (1 × 106) and CD4+CD25+CD127low Tregs (0.5 × 106) from six healthy pediatric donors were incubated with different protocols. Cells were stimulated with anti-CD3 (1 μg/mL) and anti-CD28 (1 μg/mL) antibodies in the absence or presence of various doses of CB (0.3, 2, 4, or 15 mg/dL) or Transforming growth factor (TGF)-β1 (35, 350, or 3,500 pg/mL) in a total volume of 1 mL of RPMI 1640 (HyClone) (10% FBS) for 72 h at 37°C and 5% CO2.
Unstimulated cells were included as a negative control, and TPA 12-O-tetradecanoylphorbol-13-acetate (50 ng/mL) and ionomycin (1 μg/mL) were used as positive controls (data not shown). Cells were recovered and incubated with 2.0 μL of TIM-3-PE mAb for 20 min at 2–8°C under dark conditions for surface staining; for intracellular staining, the cells were subsequently fixed/permeabilized and were incubated with 2.0 μL Foxp3-Alexa Fluor 488 mAb for 30 min at 2–8°C under dark conditions. Finally, the percentages of CD4+ TLs expressing Foxp3, CD4+ TLs expressing TIM-3, and Tregs (Foxp3+) expressing TIM-3 were determined by flow cytometry. The percentage of positive cells was obtained from the acquisition of 10,000 events and analyzed using a GUAVA EASYCYTE 6 with INCYTE 2.0 software (Merck-Millipore). Independent experiments were performed in duplicate.
Cytokine determination assay
IL-17, IL-21, IL-10, and TGF-β
Purified CD4+ TLs (1 × 106) from six healthy pediatric donors were incubated according to different protocols. Cells were stimulated with anti-CD3 (1 μg/mL) and anti-CD28 (1 μg/mL) antibodies in the absence or presence of various doses of CB (0.3, 2, 4, or 15 mg/dL) in 1 mL RPMI 1640 medium supplemented with 10% FBS and 5% CO2 at 37°C for 72 h. Unstimulated cells were included as a negative control, and TPA 12-O-tetradecanoylphorbol-13-acetate (50 ng/mL) and ionomycin (1 μg/mL) were used as positive controls (data not shown). After the incubation, the cell supernatant (100 μL) was recovered and clarified by high-speed centrifugation.
We used the 3-Plex IL-17F, IL-21, and IL-10 and the Single-Plex TGF-β1 Magnetic bead MAGPIX Kits. Cytokine analysis was performed using a MAGPIX system powered by xMAP Luminex Technology with xPONENT® software from EMD (Merck-Millipore). The assay was performed according to the supplier's instructions. In brief, following plate prewetting, IL-17F, IL-21, IL-10, and TGF-β cytokine beads were combined and then added to the plate. The plate was washed twice, and cell supernatants (25 μL) from distinct stimulation conditions were diluted 1: 2 with the assay buffer and added to the plate. The plate was shaken for 30 s at 300 g and subsequently incubated overnight on a plate shaker at 300 g at room temperature. The plate was washed twice, 25 μL of detection antibody was added per well, and the plate was then incubated for 1 h on a plate shaker. Subsequently, 50 μL of a streptavidin-PE conjugate was added per well and incubated for 30 min at room temperature. Finally, the plate was washed three times, 150 μL of sheath fluid was added to each well, and then 50 precombined beads for each cytokine from the plate were read using the MAGPIX machine (Merck-Millipore). Independent experiments were performed in duplicate.
Ex vivo analysis
Frequencies of CD4+CD25+ T cells expressing TIM-3 and CD4+ TLs expressing GAL-9
PBMCs from 18 HAV seropositive pediatric patients and 40 healthy pediatric donors were incubated with 2.0 μL of anti-CD4-Alexa Fluor 488 mAb, 2.0 μL of anti-CD25-PercPCy5.5 mAb, 2.0 μL of anti-TIM-3-PE mAb, and 2.0 μL of anti-GAL-9-PE mAb in dark conditions at 2–8°C for 30 min for surface staining. The cells were subsequently recovered by centrifugation at 300 g for 5 min, resuspended in assay buffer, and analyzed by flow cytometry. The percentages of positive cells were obtained from the acquisition of 10,000 events and analyzed using a GUAVA EASYCYTE 6 machine with INCYTE 2.0 software (Merck-Millipore).
Reagents
The following reagents were used: CB was from Merck-Millipore, Darmstadt, Germany. Recombinant human TGF-β1, anti-CD3 mAb, anti-CD28 mAb, anti-CD4-Alexa Fluor 488 mAb, anti-CD25-PercPCy5.5 mAb, anti-Foxp3-Alexa Fluor 488 mAb, Foxp3 Fixation/Permeabilization Buffer, anti-TIM-3-PE mAb, and anti-GAL-9-PE mAb were from BioLegend (San Diego, CA). The 3-Plex IL-17F, IL-21, and IL-10 and the Single-Plex TGF-β1 Magnetic bead MAGPIX Kits were from Merck-Millipore. Treg cell (CD4+CD25+CD127low) and CD4+ TL isolation kits were from Miltenyi Biotec.
Statistical analysis
Data are presented as the mean, standard deviation (SD) and mean SD. Statistical comparisons were performed by using GraphPad Prism software version 5.01 (GraphPad Software, Inc., San Diego, CA). Nonparametric Kruskal–Wallis and Mann–Whitney U tests for comparisons between groups were used to calculate the statistical significance of the assay results. A p value <0.05 was considered statistically significant. Post hoc methods were used to ensure that there were differences between the compared groups. To study associations between variables, Spearman correlation coefficients were calculated.
Results
TIM-3 expression on Tregs from healthy donors was augmented following CB treatment in vitro
TIM-3 expression on Tregs identifies a population of highly immunosuppressive cells able to modulate CD4+TL responses (7,8,29). We first determined whether CB could induce TIM-3 expression on T cells. CD4+ TLs and Tregs were purified from healthy donors stimulated with anti-CD3/anti-CD28 mAbs, treated with various doses of CB (0.3, 2, 4, or 15 mg/dL) and later cultured for 72 h; TIM-3 expression was evaluated by flow cytometry. TIM-3 expression was significantly augmented on Tregs treated with CB (4 and 15 mg/dL) compared with that on anti-CD3/anti-CD28-stimulated Tregs (Fig. 1A, B). No significant differences in TIM-3 expression on CD4+ TLs were found (data not shown). These results suggest that CB regulates TIM-3 expression on Tregs and might enhance its suppressive function.
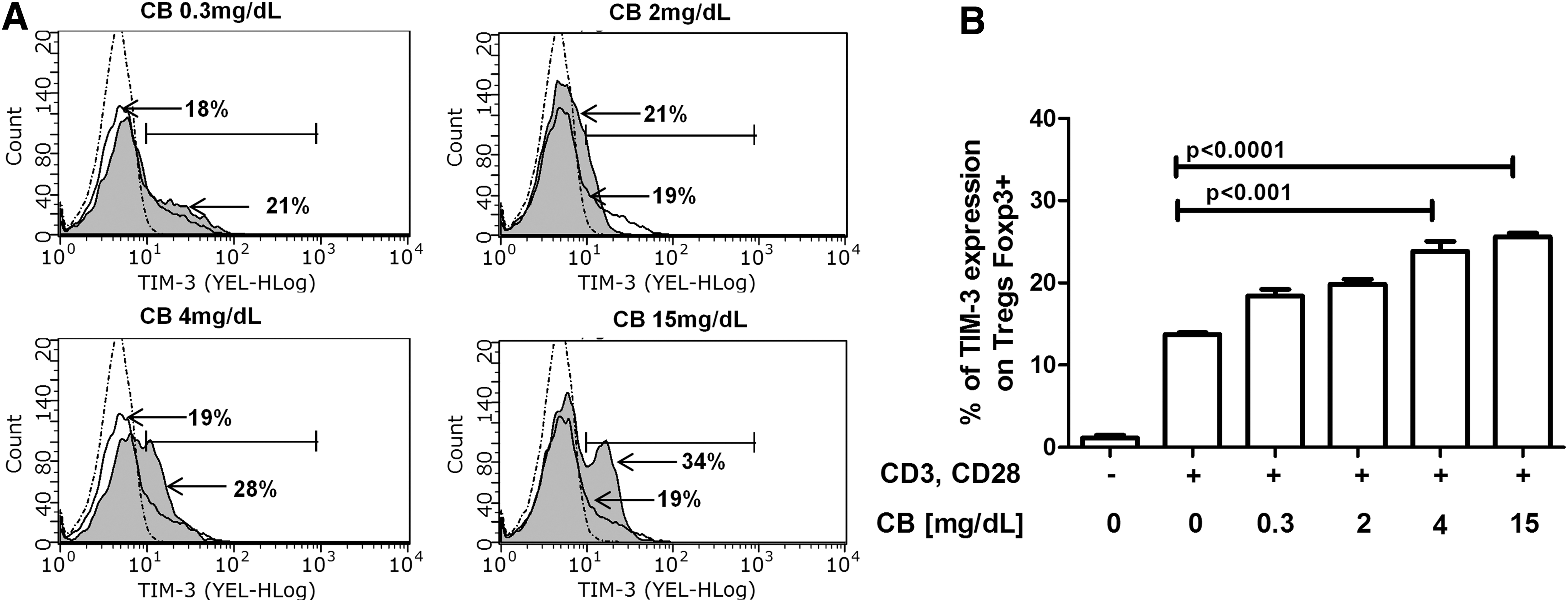
CB treatment in vitro induces augmented TIM-3 expression on Tregs from healthy donors. Purified Tregs from six healthy donors were incubated for 72 h at 37°C, 5% CO2 in RPMI 1640 medium supplemented with 10% FBS, and 1× antibiotics under the following conditions: Tregs without stimulation, Tregs stimulated with anti-CD3 and anti-CD28 (1 μg/mL), and Tregs stimulated with anti-CD3 and anti-CD28 (1 μg/mL) in the presence of different concentrations of CB (0.3, 2, 4, and 15 mg/dL).
Treatment with CB in vitro did not induce Tregs de novo in healthy donors
CREB stabilizes Foxp3 expression to promote Tregs differentiation (14,27). We have previously shown an increased phosphorylation of CREB in CD4+ TLs from HAV-infected patients with abnormal serum levels of CB (5). Thus, it is plausible that CB in the medium is crucial for regulating Treg population size and/or function.
To determinate the possible effects of CB in de novo Treg generation, CD4+ TLs from healthy donors were stimulated with anti-CD3/anti-CD28 mAbs and treated with various doses of CB (0.3, 2, 4, or 15 mg/dL) for 72 h. After treatment, the percentages of Foxp3-expressing cells in the CD4+ TL populations were subsequently evaluated by flow cytometry. As expected, anti-CD3/anti-CD28 treatment resulted in an increased expression of Foxp3 on CD4+ TLs (Fig. 2A, B). A wide variability in the Foxp3 expression on CD4+ TLs was observed after CB treatment, with a trend toward a CB dose-dependent reduction in Foxp3 expression. However, no significant differences were found in Foxp3 expression among the CB treatment groups (Fig. 2A, B). In addition, when cell supernatants were evaluated to determinate IL-10 and TGF-β secretion, no changes in the levels of these cytokines were found after CB treatment (Fig. 2C, D).
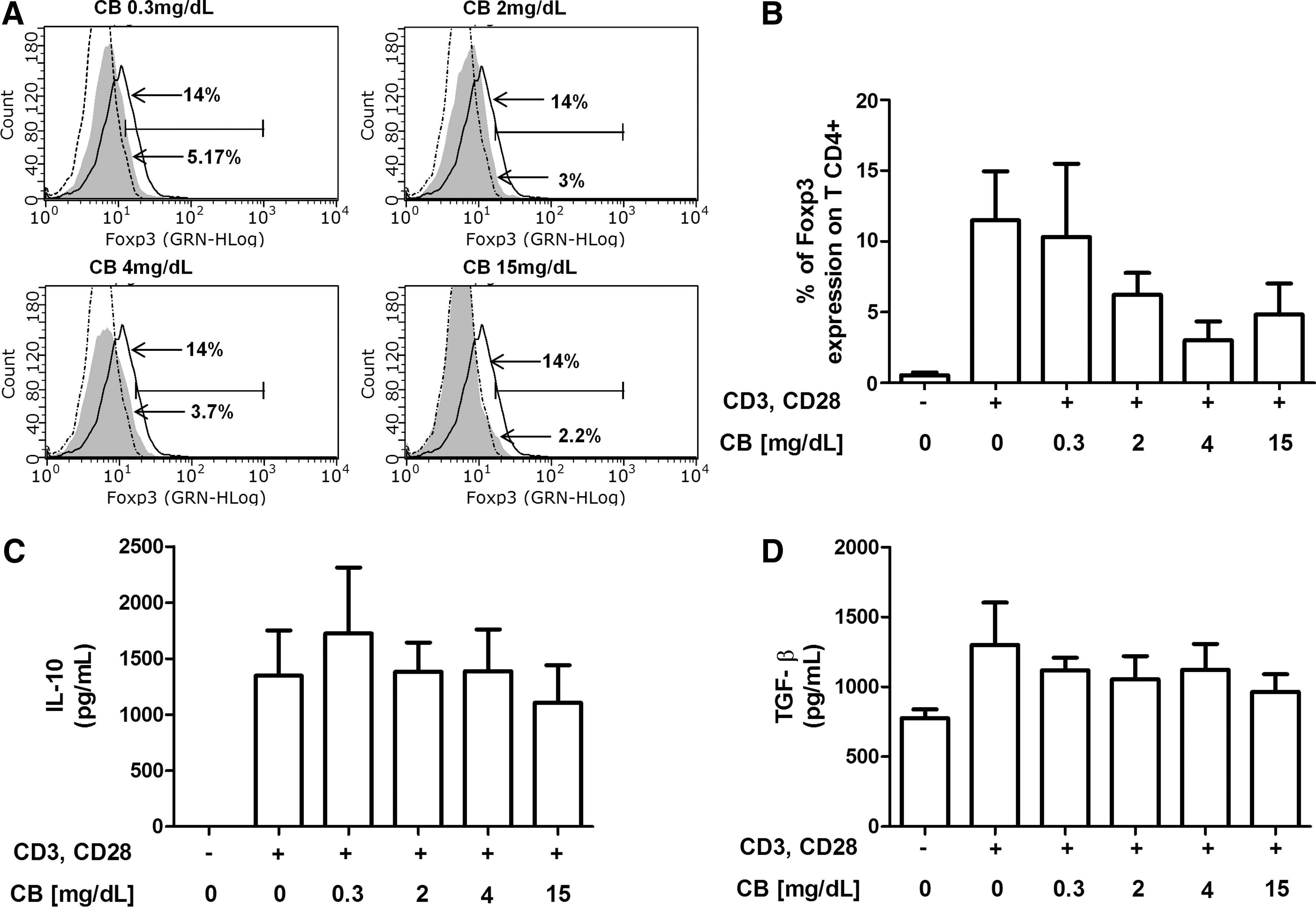
IL-10 and TGF-β secretion and Foxp3 expression in CD4+ TLs from healthy donors were not affected by CB treatment in vitro. Purified CD4+ TLs from six healthy donors were incubated for 72 h at 37°C, 5% CO2, in RPMI 1640 medium supplemented with 10% FBS and 1× antibiotics under the following conditions: CD4+ TLs without stimulation, CD4+ TLs stimulated with anti-CD3 and anti-CD28 (1 μg/mL), and CD4+ TLs stimulated with anti-CD3 and anti-CD28 (1 μg/mL) in the presence of different concentrations of CB (0.3, 2, 4, and 15 mg/dL).
Taken together, these data suggest that CB treatment in vitro does not induce de novo Treg generation or IL-10 or TGF-β secretion.
TGF-β treatment in vitro resulted in a trend toward increased TIM-3 expression in CD4+ TLs and Tregs from healthy donors
TGF-β plays a regulatory role during Treg differentiation, leading to Foxp3 expression and modulation of the function and induction of regulatory molecules on Tregs (34). We previously reported high TGF-β levels during HAV infection with both low and high CB values, a condition in which an augmentation in the percentage of CD4+CD25+ T cells has been observed (5,6).
To determine the possible effect of TGF-β on TIM-3 expression, CD4+ TLs and Tregs purified from healthy donors were stimulated with anti-CD3/anti-CD28 mAbs, treated with physiological doses of TGF-β (35, 350 or 3,500 pg/mL), and cultured for 72 h (6,18). Possible changes in TIM-3 expression were evaluated. No statistically significant differences were found in TIM-3 expression between CD4+ TLs and Tregs. However, a trend toward increased TIM-3 expression was observed in both populations, and TIM-3 expression on Tregs was higher than in CD4+ TLs (Fig. 3A–D). These results suggested that TIM-3 expression on Tregs may be modulated by TGF-β.
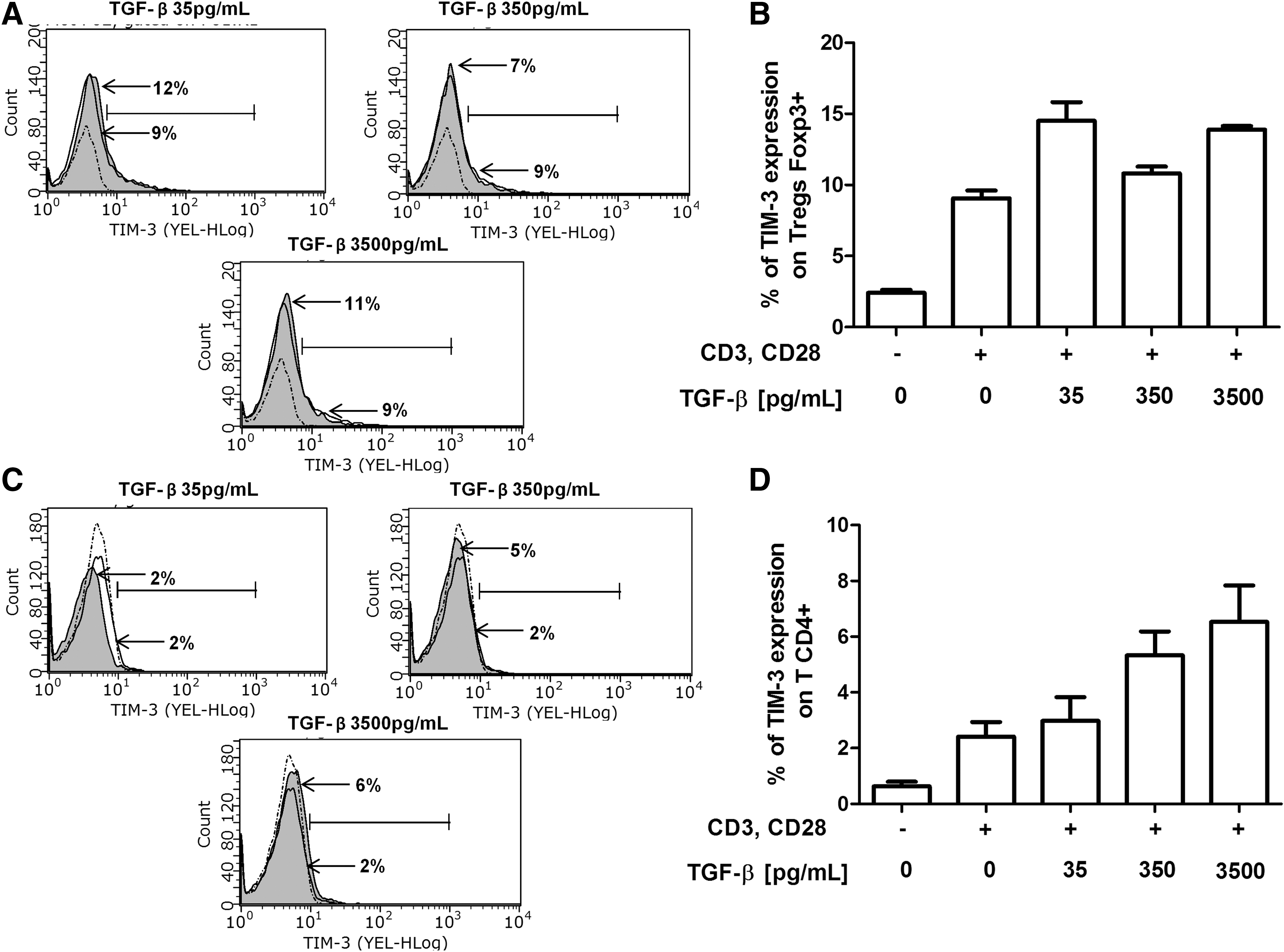
TGF-β treatment in vitro promoted a trend toward increased TIM-3 expression in both Tregs and CD4+ TLs from healthy donors. Purified CD4+ TLs and Tregs from six healthy donors were incubated for 72 h at 37°C, 5% CO2 in RPMI 1640 medium supplemented with 10% FBS, and 1× antibiotics under the following conditions: CD4+ TLs and Tregs without stimulation, cells stimulated with anti-CD3 and anti-CD28 (1 μg/mL) and cells stimulated with anti-CD3 and anti-CD28 (1 μg/mL) in the presence of different concentrations of TGF-β (35, 350 and 3,500 pg/mL).
Following CB treatment in vitro, IL-17F secretion by CD4+ TLs from healthy donors was reduced
The adequate control of Tregs and Th17 phenotypes is required to maintain cellular homeostasis during infection (11,27,37). We have previously reported an increased secretion of IL-17F in the serum of acute HAV-infected patients with CB levels greater than 2 mg/dL (32).
To determine the role of CB in modulating the secretion of Th17 cytokines, CD4+ TLs from healthy donors were stimulated with anti-CD3/anti-CD28 mAbs and treated with various doses of CB (0.3, 2, 4, or 15 mg/dL) for 72 h. Following CB treatment, IL-17F and IL-21 secretion in the culture supernatant was determined by MAGPIX technology. A statistically significantly reduction was observed in IL-17F secretion from CD4+ TLs treated with 15 mg/dL compared with that from those treated with 2 mg/dL CB, and a trend toward reduced IL-21 secretion was also observed (Fig. 4A, B). These results support the anti-inflammatory properties of CB.
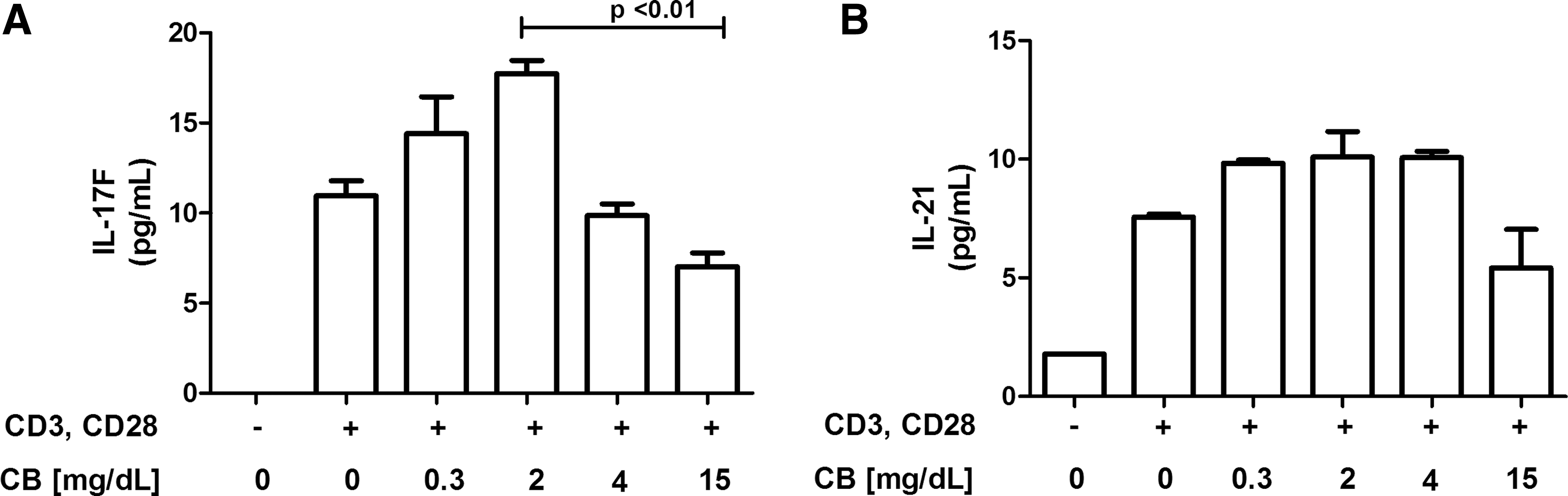
IL-17 secretion in CD4+ TLs from healthy donors was reduced by CB treatment in vitro. Purified CD4+ TLs from six healthy donors were incubated for 72 h at 37°C, 5% CO2 in RPMI 1640 medium supplemented with 10% FBS and 1× antibiotics under the following conditions: CD4+ TLs without stimulation, CD4+ TLs stimulated with anti-CD3 and anti-CD28 (1 μg/mL), and CD4+ TLs stimulated with anti-CD3 and anti-CD28 (1 μg/mL) in the presence of different concentrations of CB (0.3, 2, 4, and 15 mg/dL).
Acute HAV infection leads to an increase in the proportion of CD4+CD25+ T cells expressing TIM-3 and CD4+ TLs expressing GAL-9
Tregs play a major role in suppressing the activation of self-reactive T cells, and evidence from many human studies indicates that quantitative and qualitative defects in Treg populations are implicated in immune-mediated tissue injury (3). In addition, decreased Treg numbers result in severe liver injury during HAV infection (3). We previously reported an increase in the CD4+CD25+ T cell proportion in HAV-infected children and augmented Treg activity after CB treatment in vitro (5).
In this study, we evaluated TIM-3 expression on CD4+CD25+ T cells from HAV-infected children with abnormal liver injury markers (CB ≥0.3, ALT >35 IU/L, AST >38 IU/L, ALP >98 IU/L and, as expected for acute infections, normal TP values) and healthy individuals (Table 1). The percentage of TIM-3+ on CD4+CD25+ T cells was statistically higher in patients (1.52 ± 0.84) than in healthy pediatric donors (0.32 ± 0.32) with normal CB levels (<0.3 mg/dL) (Fig. 5A, B). We analyzed the possible correlation between TIM-3 expression on CD4+CD25+ T cells and liver injury markers in the HAV-infected children.
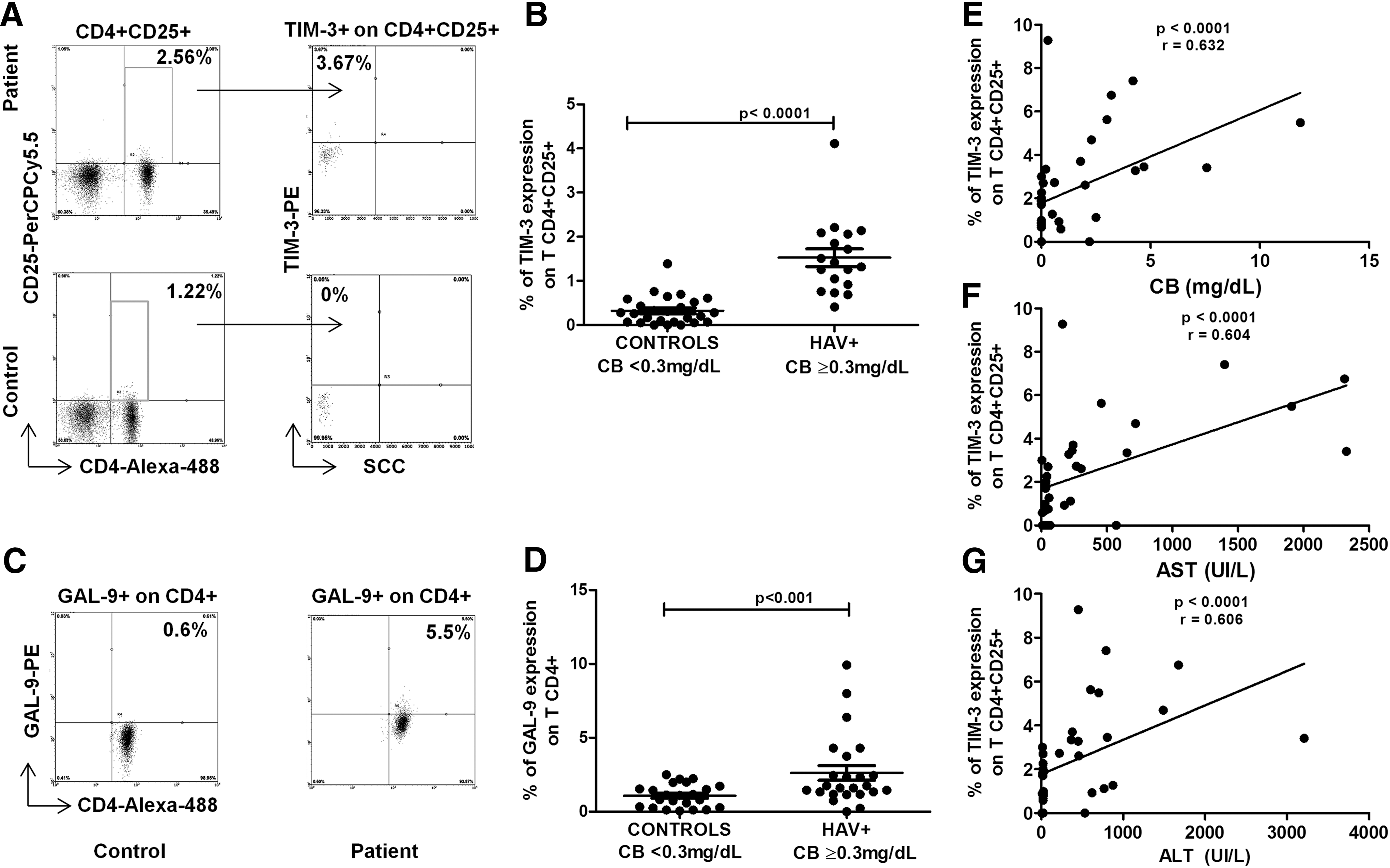
HAV infection leads to an increased frequency of TIM-3 expression on CD4+CD25+ T cells and CD4+ TLs expressing GAL-9. Purified peripheral blood mononuclear cells from healthy pediatric donors and HAV+ pediatric patients were stained with anti-CD4 Alexa Fluor 488, anti-CD-25 PerCP-Cy5.5, anti-GAL-9, and anti-TIM-3 PE antibodies and evaluated by flow cytometry.
Data are presented as the mean ± SD. The nonparametric Mann–Whitney U test for comparisons between groups was used to calculate statistical significance. p < 0.05 was considered statistically significant.
p < 0.0001.
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CB, conjugated bilirubin; HAV, hepatitis A virus; HAV+, HAV-infected children; IU/L, international units per liter; SD, standard deviation; TP, total protein.
Interestingly, TIM-3 expression significantly correlated with the alteration of liver injury markers (ALT, AST, and CB) (Fig. 5E–G). These findings suggest that upregulated TIM-3 expression on CD4+CD25+ T cells in the face of abnormal CB levels might have a role in balancing the immune response during HAV infection by modulating inflammation.
To understand how Tregs function to modulate the inflammatory process during HAV infection, we examined GAL-9 expression on CD4+ TLs. Statistically significant GAL-9 overexpression was observed on CD4+ TLs from HAV-infected children (2.62 ± 2.40) compared with that on those from healthy pediatric donors (1.06 ± 0.7) (Fig. 5C, D). These results suggest that TIM-3/GAL-9 ligand-receptor interactions may constitute a regulatory mechanism, by which effector CD4+ TLs are controlled during HAV infection via modulation of Treg function.
Discussion
Our results indicate that CB represents a molecule of immunological importance related to Treg activity that is also capable of modulating CD4+ TL activity during HAV infection. This capacity is related to the expression of inhibitory molecules, such as TIM-3, on Tregs.
TIM-3 expression on Tregs identifies a population of highly immunosuppressive cells capable of modulating pathogenic Th1 and Th17 responses (7,8,29). In our study, in vitro CB treatment of Tregs from healthy donors resulted in increased TIM-3 expression (Fig. 1), and the same effect was observed on CD4+CD25+ T cells from HAV-infected children with high CB levels, which correlated with liver injury markers (Fig. 5).
Interestingly, upregulated TIM-3 expression on Tregs has been reported in chronic HCV infection (21). Thus, the effects of this inhibitory receptor may be different according to the circumstances of infection, either acute or chronic. During acute HAV infection, TIM-3+ Tregs may suppress CD4+ TL function, thereby preventing exacerbated inflammation. This hypothesis is supported by the fact that a decreased frequency of Tregs is associated with liver injury in acute hepatitis A (3). In this regard, a high expression of inhibitory molecules, including CTLA-4 and PD-1, has been reported in acute HAV-infected patients, and a protective effect has been related to this phenomenon (2).
Although our previous data support the notion that CB promotes de novo Treg generation by modulating intracellular signals, specifically CREB phosphorylation, which is related to Foxp3 function (5), our in vitro data in this study showed that CB treatment of CD4+ TLs does not induce Foxp3 expression or the secretion of IL-10 and TGF-β (Fig. 2). In agreement with this observation, Liu et al. have reported that in vitro BR treatment of CD4+ TLs in an experimental autoimmune encephalomyelitis mouse model does not induce Treg expansion (17).
In contrast, the induction of de novo Tregs after the administration of BR to transplant recipients in murine models has been reported (15,26,36,39). The discrepancy in the results related to the role of BR in Treg differentiation may be due to the differences in methodologies and models used (mouse models of transplant and/or autoimmune disease vs. in vitro human CD4+ TL priming models) for each of the studies. Thus, it is possible that the effect of CB on Tregs differentiation is affected by the cellular microenvironment and specific receptors expressed on cells from different species.
We recently reported that during HAV infection, CB levels between 0.3 and 2 mg/dL result in TGF-β secretion and increased STAT-5 phosphorylation, whereas CB levels >2 mg/dL are correlated with a reduction in the proportion of PBMCs positive for STAT-5 phosphorylation and increased levels of IL-6, IL-1α MCP-2, and TNF-α (1,6).
STAT-5 is known to act as an important mediator of Foxp3 expression and Treg function, and the release of TGF-β into the serum during acute viral infection inhibits antigen-specific T cell activation and proliferation as a result of modulating Treg activity (31). Moreover, TGF-β antiviral activity has been described in HBV- and HCV-infection replication models (12,22). In this regard, variants in the TGF-β gene increase susceptibility to viral hepatitis A infection in Mexican Americans (41). These findings strongly suggest that TGF-β levels have an important role in viral clearance and cellular immune response modulation during viral hepatitis. In this study, TGF-β treatment in vitro of CD4+ TLs and Tregs resulted in a trend toward increased TIM-3 expression on CD4+ TLs and Tregs (Fig. 3).
The TGF-β-mediated upregulation of TIM-3 on CD4+ TLs relative to Tregs might have different effects on these cells, as suggested through studies in HCV infection where an overexpression of TIM-3 on CD4+ TLs has been correlated with an exhausted T cell phenotype (21), whereas the upregulation of TIM-3 on Tregs has been related to an increase in the suppressive function of this subpopulation (7).
To the best of our knowledge, this is the first report of upregulated TIM-3 expression on CD4+ TLs and Tregs as a result of TGF-β administration, and our results are consistent with the upregulation of this receptor in TGF-β-stimulated mast cells and macrophages (38,40). Large-scale studies are necessary to dissect the functional significance of various concentrations of TGF-β for TIM-3 expression on CD4+ TLs and Tregs in the context of HAV infection.
How CB may affect Treg function by modulating TIM-3 expression is still not clear. A specific interaction between BR and the aryl hydrocarbon receptor (AhR) has been proposed (30). AhR is expressed in a variety of tissues, including the liver, lungs, skin, gastrointestinal tract, and immune cells, and plays an important role in the control of the adaptive immune response.
Specifically, AhR has been suggested to have a key role in the development and function of Tregs through regulation of Foxp3 expression (24,25). Furthermore, AhR activation leads to a significant suppression of inflammation in zebrafish (24,25). Thus, it is plausible that during acute HAV infection, an interaction between CB and AhR expressed on Tregs occurs, resulting in the upregulation of TIM-3 in these cells and allowing for a highly immunosuppressive/anti-inflammatory Treg phenotype. This hypothesis is consistent with the anti-inflammatory properties proposed for CB (9,17) and with our data showing that in vitro CB treatment of CD4+ TLs resulted in reduced IL-17F secretion (Fig. 4). Moreover, this proposed mechanism is in agreement with our previous report indicating that CB treatment in vitro does not induce IL-17A secretion by CD4+ TLs (5).
The immune response against HAV relies on CD4+ TLs, in contrast to the immune response raised against most viral infections, in which CD8+ T cells play a predominant role (42). Thus, the effect of CB on IL-17 secretion by CD4+ TLs may be important in defining the role of this metabolite to control CD4+ TL function during infection. However, in the liver, distinct cells (Tc17, ILC3s) may be responsible for IL-17 production (11). Further studies are required to more fully dissect the source of IL-17 and characterize the potential direct link between CB/AhR and Treg activity in the context of HAV infection.
The potential protective effect of TIM-3 on Tregs in HAV infection, suggested by our data, is supported by the high GAL-9 expression on CD4+ TLs observed in HAV-infected patients (Fig. 5).
It is accepted that the TIM-3/GAL-9 pathway reduces CD4+ TL proliferation and induces CD4+ TL death, thereby decreasing the amount of T cell-mediated inflammatory IFN-γ production (10). In fact, blockade of TIM-3 expression during Th1 responses increases T cell proliferation and prevents the development of immunological tolerance (10,28). Gal-9 circulates in the serum; in the liver, Kupffer cells are an important source of Gal-9, which significantly increases in the serum of patients with chronic HCV, resulting in Treg expansion, compared with that in the serum of healthy controls (20). Thus, in the context of liver disease, the TIM-3/GAL-9 pathway represents an important checkpoint for modulation of CD4+ TL activity, and may therefore influence the response to intrahepatic pathogens.
In conclusion, our data support an anti-inflammatory and hepatoprotective role of BR during acute type A viral hepatitis through a mechanism dependent on the expression of TIM-3 on Tregs.
Footnotes
Acknowledgments
The authors thank Dr. Griselda Escobedo-Melendez for providing blood samples from patients during the setting up of initial experiments. The authors also thank Dr. Ana R. Rincon-Sanchez for her support. This work was funded by grants from the Consejo Nacional de Ciencia y Tecnología (CONACYT) # 239470 to N.A.F. K.F.C.-J. and J.L.T.-O. were supported by PhD scholarships from the CONACYT.
Author Disclosure Statement
No competing financial interests exist.