
Abstract
Ebolavirus (EBOV) is the etiology of Ebola hemorrhagic fever (EHF). A major EHF outbreak in 2014–2015 in West Africa claimed >11,000 lives. A licensed vaccine is not available for EHF, although several vaccines have undergone clinical trials. We developed a human adenovirus (Ad) serotype 5-based candidate EHF vaccine based on controlled expression of the EBOV (Makona strain) glycoprotein (GP) as the immunogen. Two clones, AdGP72 and AdGP75, and a control Ad515 vector, were generated and tested for protein expression in vitro and immunogenicity in mice. Eight groups of mice were immunized with three doses of buffer, Ad515, AdGP72, and AdGP75, by two different dose regimens. Three different antigens (AdGP75-infected Vero E6 cell extract and two baculovirus expressed EBOV GP antigens, namely, GP alone or GP with EBOV VP40) were used to evaluate the immune response. Expression studies indicated that full-length GP was cleaved into its component subunits when expressed in mammalian cells through the Ad vectors. Moreover, in coimmunoprecipitation studies, EBOV GP was found to be associated with VP40 when expressed in baculoviruses. The candidate vaccines were immunogenic in mice, as evaluated by enzyme-linked immunosorbent assay using mammalian- or baculovirus-derived antigens. Further characterization and development of the candidate vaccines are warranted.
Introduction
E
The EBOV has a nonsegmented negative-sense linear single-stranded RNA genome of 18–19 kb, encoding genes in the order 3′-NP-VP35-VP40-GP-VP30-VP24-L-5′. The virion contains seven structural proteins, namely, nucleoprotein (NP), viral protein 35 (VP35), VP40, glycoprotein (GP), VP30, VP24, and the polymerase (L). Two nonstructural proteins, the secretory GP (sGP) and the small soluble GP (ssGP), are also expressed in infected cells. The NP forms a complex with genomic RNA and protects it from nucleases. VP35 acts as a cofactor of viral RNA polymerase complex (41), and is an indispensable component for virus assembly (42). The matrix protein VP40 can oligomerize, bud independently, and form virus-like particles (43), and it forms the essential link between the ribonucleoprotein and the viral envelope during virion morphogenesis. VP30 is involved in transcription (5). Both VP35 and the secondary matrix protein VP24 act as interferon antagonists, dampening the host cell's antiviral pathways, thus facilitating virus replication (76). The L protein is an RNA-dependent RNA polymerase and functions in transcription and genome replication (62).
Three different forms of GP are expressed in infected cells through the GP open reading frame. The majority of GP mRNAs transcribed from the first reading frame lead to the synthesis of sGP, whereas the expression levels of the membrane-anchored surface GP and ssGP are a consequence of transcriptional stuttering by viral polymerase (39,60). The homodimeric sGP (63) inhibits neutrophil activation (73) and binds to neutralizing antibodies, potentially aiding in virus propagation in vivo (23). The function of ssGP is not clear (39). Another form of GP, the shed or truncated GP, is formed due to post-translational processing of the full-length GP (14), and is thought to activate noninfected immune cells and affect vascular endothelial function (15).
The membrane-anchored GP is initially expressed as a ∼110 kDa endoglycosidase H (Endo-H)-sensitive precursor GP (preGPer), which, upon entry into the Golgi apparatus, is converted into a ∼160 kDa Endo-H-resistant preGP. The preGP is cleaved in the trans-Golgi network, by the host protease furin, into GP1 (∼130–140 kDa) and GP2 (∼24–26 kDa) subunits (52,61). The GP2 is anchored to the membrane, whereas GP1 is associated with GP2 by a single disulfide bond between Cys53 of GP1 and Cys609 of GP2. This mature heterodimeric GP forms a trimeric complex of ∼450 kDa on the surface of infected cells and on the virion envelope (27), and is responsible for the attachment, fusion, and entry of the virus particle into the host cell.
The EBOV can spread through direct contact with infected body fluids. The virus enters the host through cuts or broken skin or upon exposure with mucous membranes. Dendritic cells and cells of monocyte–macrophage lineage are the primary sites of initial virus infection and replication (18). Virus infection disrupts host innate and adaptive immune cell function, leading to a dysregulated inflammatory response, which further exacerbates disease pathogenesis (65). Blood and the lymphatic system facilitate dissemination of the virus to various parts of the body, enabling infection of multiple cell types (35). The incubation period may range from 2 to 21 days (71). The initial symptoms include high fever, myalgia, malaise, and nausea. As the disease progresses, vomiting, watery diarrhea, and impaired liver and kidney functions are observed. Terminal symptoms are characterized by respiratory and gastrointestinal distress, neurological complications, and increased vascular permeability. Death usually occurs because of hypovolemic shock resulting from the severe loss of body fluids and multiorgan failure (24).
Since the first outbreak of EHF in 1976 (67), the disease has been confined to Central Africa with a few hundred deaths. The 2014–2015 outbreak, caused by ZEBOV, was the most complicated, occurring for the first time in western Africa, with >20,000 confirmed human cases and >11,000 deaths (69), and also landing outside Africa for the first time. Lack of awareness among people about EHF and its spread and the unavailability of a licensed vaccine were the two major reasons for the massive spread of EHF in 2014–2015. In terms of vaccine development, initial attempts utilized inactivated EBOV, but it was partially efficacious in guinea pigs; however, a killed vaccine is difficult to manufacture considering the potential risk of handling live virus (32). In addition, the protective efficacy of EBOV proteins VP24, VP30, VP35, and VP40 was found to be not satisfactory (69) as assessed in animal models. In contrast, GP has been shown to be a target of protective immune responses (72), and has, therefore, been the focus of efforts to develop vaccines against EHF.
Several technological platforms have been exploited for the development of candidate vaccines for EHF. The major antigen delivery platforms that underwent clinical trials recently were the recombinant chimpanzee adenovirus (ChAd)3 (16,57), the recombinant vesicular stomatitis virus (rVSV) (20,21), and the recombinant human adenovirus (Ad)5 (29,77,78) systems. However, all these systems are based on the constitutive expression of GP. At least with the Ad vectors, high level expression of heterologous viral glycoproteins has been shown to be detrimental to the production of the Ad vector or to the consequent expression levels of recombinant proteins (38,74). In addition, EBOV GP is known to be toxic when constitutively expressed in mammalian cells (7). We, therefore, generated Ad vectors through a regulated gene expression system wherein the expression of EBOV GP could be downmodulated during the generation of Ad vectors, similar to the earlier described instance of rabies virus glycoprotein expression (38). The Ad vectors encoding the EBOV GP gene were evaluated in vitro for protein expression and cleavage, and in vivo for immunogenicity in mice.
Materials and Methods
Cells and gene
The 293 and 293IQ cells were obtained from Microbix Biosystems, Inc. (Canada). The Vero E6 cells (ATCC C1008) were a kind gift from Prof. David C. Johnson, Oregon Health & Science University. The Sf21 cells, plasmids pFastBac HTA and pFastBac 1, and Escherichia coli strain DH10Bac were a kind gift from Prof. M.S. Shaila, Department of Microbiology and Cell Biology, Indian Institute of Science (Bangalore, India).
The 293 and 293IQ cells were maintained in Eagle's minimum essential medium (Gibco), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), and antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin). The Vero E6 cells were maintained in Dulbecco's modified Eagle's medium (Gibco) with 10% FBS and antibiotics. The 293, 293IQ, and Vero E6 cells were maintained at 37°C with 5% CO2. The Sf21 cells were maintained at 26–27°C in TC100 medium (Gibco) supplemented with 10% FBS and antibiotics.
GP synthetic constructs
The full-length EBOV GP gene was obtained as a human codon-optimized synthetic construct (GenScript), based on the sequence of ZEBOV Makona strain (GenBank Accession No. KM034550.1). Two unique restriction sites, XhoI at position 214 and BamHI at position 1958, were introduced in the full-length GP gene without altering the amino acid sequence, so as to allow inserting the intervening region after the construction of tagged ends of GP, as described hereunder.
The tagged ends of GP were generated through synthetic constructs that contained the 5′ and 3′ fragments, corresponding to nucleotide positions up to 214 and beyond 1958 (thus lacking the region between XhoI and BamHI sites of the full-length GP), along with HA and FLAG tags, respectively. Nucleotides encoding the anti-influenza virus hemagglutinin epitope tag (HA-tag) were introduced between sequences encoding the signal sequence and the mature GP. Similarly, nucleotides encoding FLAG-tag were introduced at the 3′ end before the stop codon. The XhoI and BamHI fragment of full-length GP was then introduced into this synthetic construct to obtain GP with N-terminal HA-tag and C-terminal FLAG-tag.
Antibodies
The monoclonal anti-EBOV GP antibody, 15H10 (75), was obtained from BEI Resources. The mouse anti-HA and anti-FLAG antibodies were obtained from Sigma-Aldrich. The mouse monoclonal antiadenovirus 5 hexon antibody was obtained from Thermo Fisher Scientific, Inc.
Recombinant adenovirus generation
The AdMax™ Hi-IQ system (Microbix) was used for the generation of replication-defective (RD) Ad5. The EBOV GP was inserted into the Ad shuttle plasmid pDC515(io) to generate pDC515(io)EBOVGP72 and pDC515(io)EBOVGP75 clones, and verified by restriction digestion and DNA sequencing. The RD-Ad5 was generated by cotransfecting pDC515(io), pDC515(io)EBOVGP72 or pDC515(io)EBOVGP75, and Ad genomic plasmid (pBHGfrtΔE1,3FLP) into 293IQ cells as described previously (58). The three recombinant RD adenoviruses, Ad515 (without EBOV GP) and two RD-Ad5 clones with GP (AdGP72 and AdGP75), were generated. Stocks of these Ad viruses were prepared and 50% tissue culture infective dose (TCID50) was determined in 293IQ cells by applying the Reed and Muench method (48).
Baculovirus generation
The pcDNA3.1 vector encoding β-lactamase fused to ZEBOV VP40 (pcDNA3.1-BlaMVP40) (34) was obtained from BEI Resources. The recombinant baculoviruses were generated by following a procedure similar to the Bac-to-Bac® system (Thermo Fisher Scientific, Inc.), following cloning of the synthetic EBOV GP or a 5′ HA-tagged VP40 (obtained by polymerase chain reaction from pcDNA3.1-BlaMVP40) into the donor plasmids pFastBacHTA and pFastBac1, respectively. The baculoviruses were titrated on Sf21 cells and the TCID50 was calculated by the Reed and Muench method (48).
Analysis of GP expression
Adenovirus-mediated expression of EBOV GP in cells
Cells were infected with Ad vectors at a multiplicity of infection (MOI) of 10. For induction, the cells were treated with 10 mM isopropyl β-
Baculovirus-mediated expression of EBOV GP and HA-tagged VP40 in Sf21 cells
To assess baculovirus-mediated protein expression, Sf21 cells were infected at 4 MOI, harvested after 48 h, and lysed as described. Postnuclear supernatants were subjected to 10% SDS-PAGE and Western blotting using 15H10 or anti-HA antibody.
To evaluate the association between GP and VP40, extracts from 106 cells coinfected with baculoviruses encoding GP and HA-VP40 were incubated with either anti-GP (pooled mouse sera) or anti-HA antibody. The antigen–antibody complexes were captured with Protein A agarose beads, the complexes were released by boiling and subjected to 10% SDS-PAGE, followed by Western blotting for GP and HA-tagged proteins.
Transient expression of tagged-GP in 293 cells
The version of full-length GP, with N-terminal HA-tag and C-terminal FLAG-tag, was cloned into the mammalian expression vector pcDNA3.1 (pcDNA3.1-tagged GP). The pcDNA3.1 empty vector or pcDNA3.1-tagged GP construct was transfected into 293 cells using polyethyleneimine as described previously (58). In brief, 8 μg of plasmid DNA was mixed with 2 μg of polyehyleneimine and used for transfection of 0.7 × 106 cells. The transfected cells were harvested 48 h post-transfection, lysed, and processed as already mentioned for detection of HA-tagged or FLAG-tagged protein by Western blotting.
Purification, quantification, and electron microscopy of adenovirus
The Ad vectors were purified as described previously (6). In brief, the Q Sepharose®Fast Flow (Sigma-Aldrich) columns were equilibrated with 0.1 M NaCl, 50 mM Tris (pH 8.0), and 5% glycerol. The adenovirus preparations were loaded onto the column and eluted with increasing concentration (0.2–0.7 M) of NaCl in 50 mM Tris (pH 8.0) and 5% glycerol. The eluted fractions were subjected to optical density (OD) measurement at 280 nm, and to SDS-PAGE followed by Coomassie staining and Western blotting with mouse monoclonal antibody to detect adenovirus hexon.
The virus particles were enumerated as described previously (33), in comparison with Ad5 reference material (VR-1516; ATCC). In brief, the purified Ads were disassembled with 0.1% SDS (w/v) in Tris-EDTA (10 mM Tris-HCl [pH 8.0], 1 mM EDTA), and subjected to measurement of OD at 260 nm. Virus particles were extrapolated from the formula: Virus particles/mL = OD260 × dilution factor × 1.1 × 1012.
Virus particles were adsorbed on 400 mesh carbon-coated formvar-coated copper grids and negatively stained with sodium phosphotungstic acid. Imaging of virus particles was carried out under 120 KeV mode in a transmission electron microscope (Tecnai 12 Biotwin; FEi Co., the Netherlands), and digital images were acquired and processed using a side mounted 2k × 2k camera (SIS, Germany) and onboard software.
Mouse immunization
Mouse studies were carried out following approval of the Institutional Animal Ethics Committee. Balb/C mice (4–6 weeks old) were grouped randomly into eight, comprising three animals per group. Two immunization regimens with different routes were followed: (a) first dose intranasal (0 day), second dose subcutaneous (21st day), and third dose intranasal (59th day) and (b) first dose subcutaneous (0 day), second dose intranasal (21st day), and third dose intranasal (59th day). For intranasal inoculations, the respective immunogen in 10 μL volume was introduced drop by drop into each nostril with a micropipette. For subcutaneous inoculations, 100 μL of the immunogen suspension was injected with an insulin syringe. Each dose of the Ad515 or AdGP72 or AdGP75 contained 1 × 1011 particles. Blood samples were collected from the retro-orbital sinus on days 0, 14, 31, and 67.
Production of antigens
For mammalian cell-derived antigen, Vero E6 cells were infected with Ad vectors at 20 MOI and harvested 48 h postinfection. Clarified cells were resuspended in phosphate-buffered saline (PBS) containing protease inhibitor cocktail (Sigma-Aldrich). Cell extracts were prepared by three freeze–thaw cycles followed by clarification, and the supernatant was used as antigen for enzyme-linked immunosorbent assay (ELISA).
For baculovirus-derived antigen, Sf21 cells were infected with 4 MOI of baculovirus-GP, or 2 MOI each of baculovirus-GP and baculovirus-HA VP40. Cell extracts were prepared 72 h postinfection by three freeze–thaw cycles followed by clarification, and supernatants were overlaid on 20% sucrose cushion and concentrated by two cycles of ultracentrifugation at 1,00,000 g for 1 h each. The resultant pellet was resuspended in PBS containing protease inhibitor cocktail, and used as antigen for ELISA.
Enzyme-linked immunosorbent assay
Microwell plates (Thermo Fisher Scientific, Inc.) were coated with antigen in carbonate–bicarbonate buffer, pH 9.2. The wells were blocked with 3% skimmed milk powder in PBS. Pooled sera from each group were added in duplicate, washed, and reacted with antimouse IgG (Fab-specific)–peroxidase conjugate. The color was developed using o-phenylenediamine substrate and the OD was measured at 490 nm (iMark™ Microplate Reader; Bio-Rad). To determine the endpoint titers, serial twofold dilutions of the pooled preimmune and final bleed sera were used.
Statistical analysis
The results of the ELISA were subjected to one-way analysis of variance (ANOVA). The OD values of the first, second, or the final bleed were compared with the preimmune samples by Bonferroni's multiple comparison test (Prism 4, Version 4.00; GraphPad Software, Inc.).
Results
Generation of RD-Ad5 and expression of EBOV GP
The GP gene was selected based on an analysis of the genome of EBOV strains from the 2014 outbreak (19). Three RD-Ad5 vectors, one negative control vector, Ad515, and two containing GP (Fig. 1), AdGP72 and AdGP75, were generated. Titers of Ad515, AdGP72, and AdGP75 ranged from 4 × 108 to 3 × 109 TCID50/mL. Negative stain transmission electron microscopy of the RD-Ad5 stocks revealed typical icosahedral symmetry (Fig. 2; ref. 25).
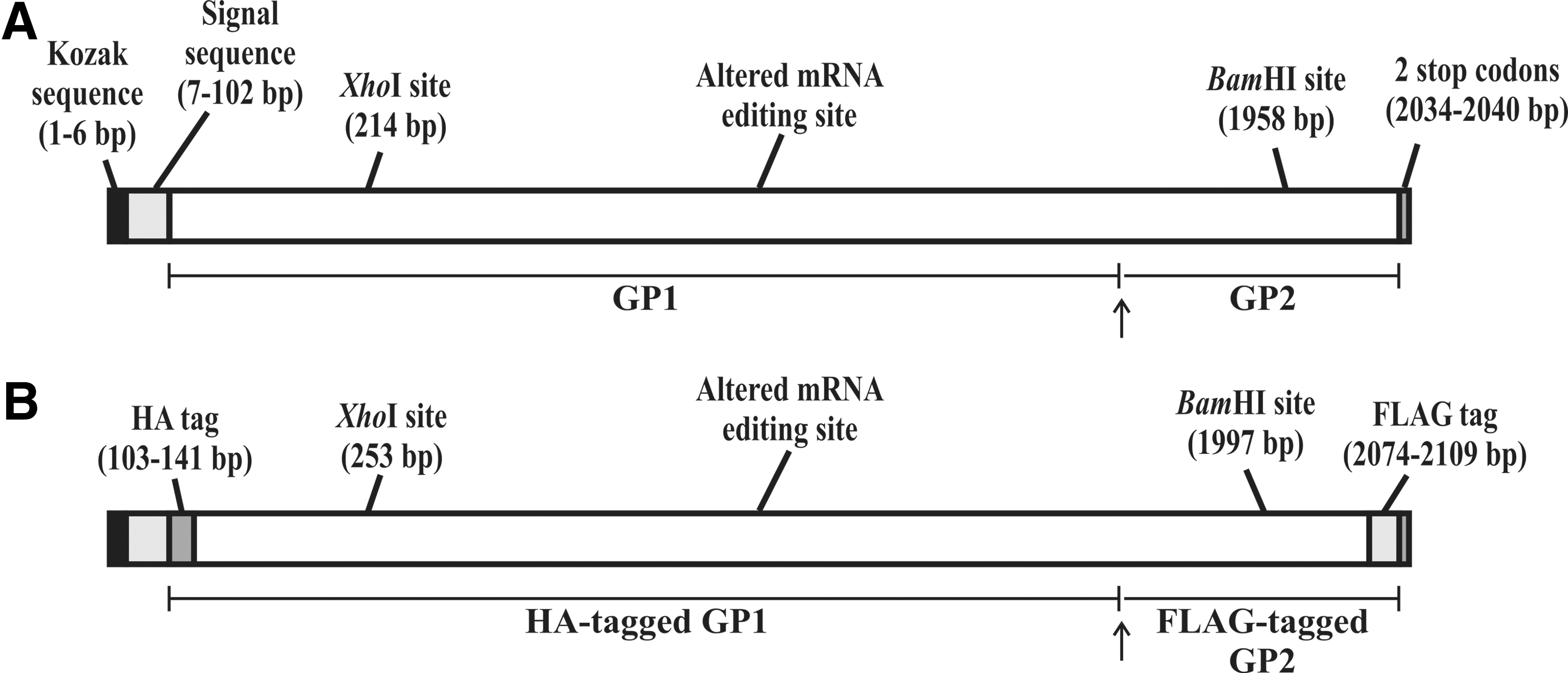
Schematic representation of the construct of Ebolavirus glycoprotein gene (not to scale). The construct contains the GP gene (2,040 bp) with a Kozak sequence (“GCCACC”) before the start codon (“ATG”). The open reading frame was altered by introducing “G” at the 882nd position, so as to change the reading frame to produce full-length GP
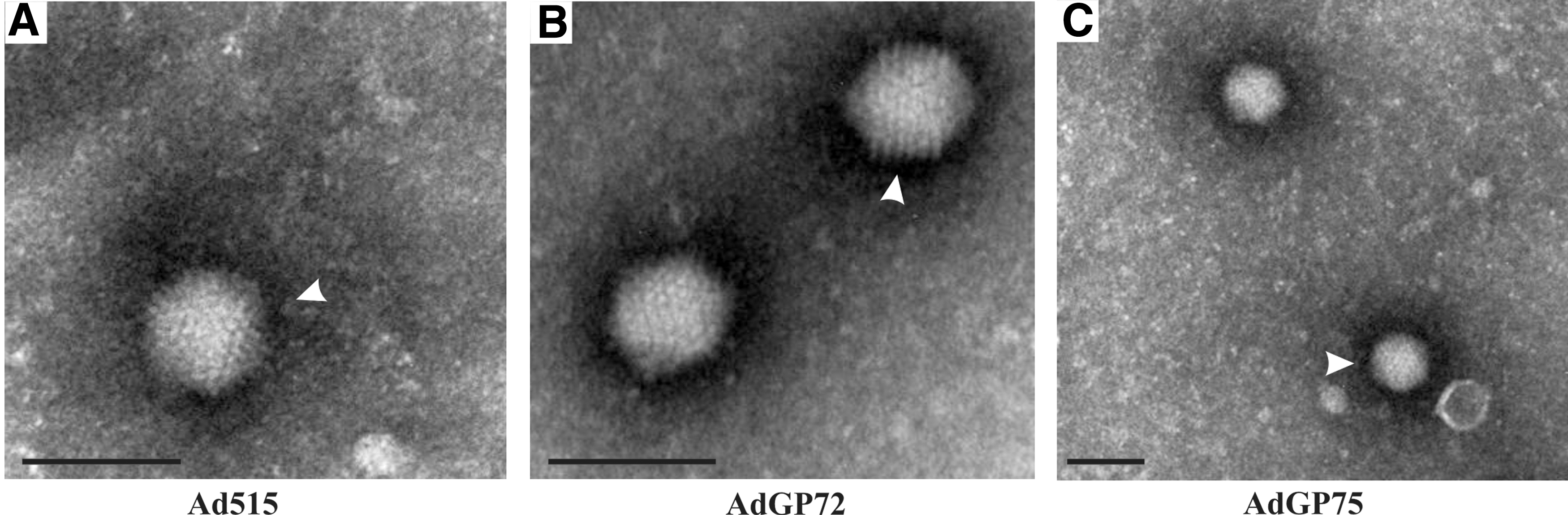
Morphology of the Ad vectors. Transmission electron micrograph images of
Cleavage of GP into the N-terminal GP1 and C-terminal GP2 was confirmed by using HA-tagged and FLAG-tagged GP, and detecting the proteins in plasmid-transfected cells by Western blot using anti-tag antibodies (Supplementary Fig. S1A; Supplementary Data are available online at
To assess the ability of the Ad vectors to express GP, infected Vero E6, 293, and 293IQ cell lysates were subjected to Western blotting with 15H10 antibody, which recognizes GP2 (74). The GP2 subunit was readily detected in all cell types when infected with AdGP72 or AdGP75 (Fig. 3B, lanes 3, 4, 7, 8, 11, 12, and 15, weak in 16; ref. 25). The higher molecular weight protein probably corresponds to the uncleaved GP, which contains both GP1 and GP2 (Fig. 3A, lanes 7 and 11, faint in 8 and 12; ref. 25). The GP expression in 293 cells was ∼12-fold higher than that in 293IQ cells, presumably because of its repression in the latter cells (Fig. 3B, lane 7 vs. 15). However, GP is expressed at a low level in 293IQ cells (Fig. 3B, lane 15; ref. 25), suggesting that repression was not absolute. In contrast, treatment of 293IQ cells with IPTG, which sequesters the lac repressor, reversed the suppression (Fig. 3B, lane 11 vs. 15). Although equal MOI of the AdGP72 and AdGP75 was used to infect the cells, GP expression by AdGP72 was ∼1.3-, 2.5-, and 4-fold higher than that of AdGP75 in Vero E6 (Fig. 3B, lane 3 vs. 4), 293 (Fig. 3B, lane 7 vs. 8), and 293IQ+IPTG (Fig. 3B, lane 11 vs. 12) cells, respectively. However, the difference in GP expression differed from experiment to experiment (data not shown). Similar amounts of GAPDH in Vero E6 and 293 cells reflect cell equivalents loaded in the lanes (Fig. 3C). Cell surface expression of GP was also confirmed by transfecting 293 cells with a plasmid encoding tagged GP construct (data not shown).
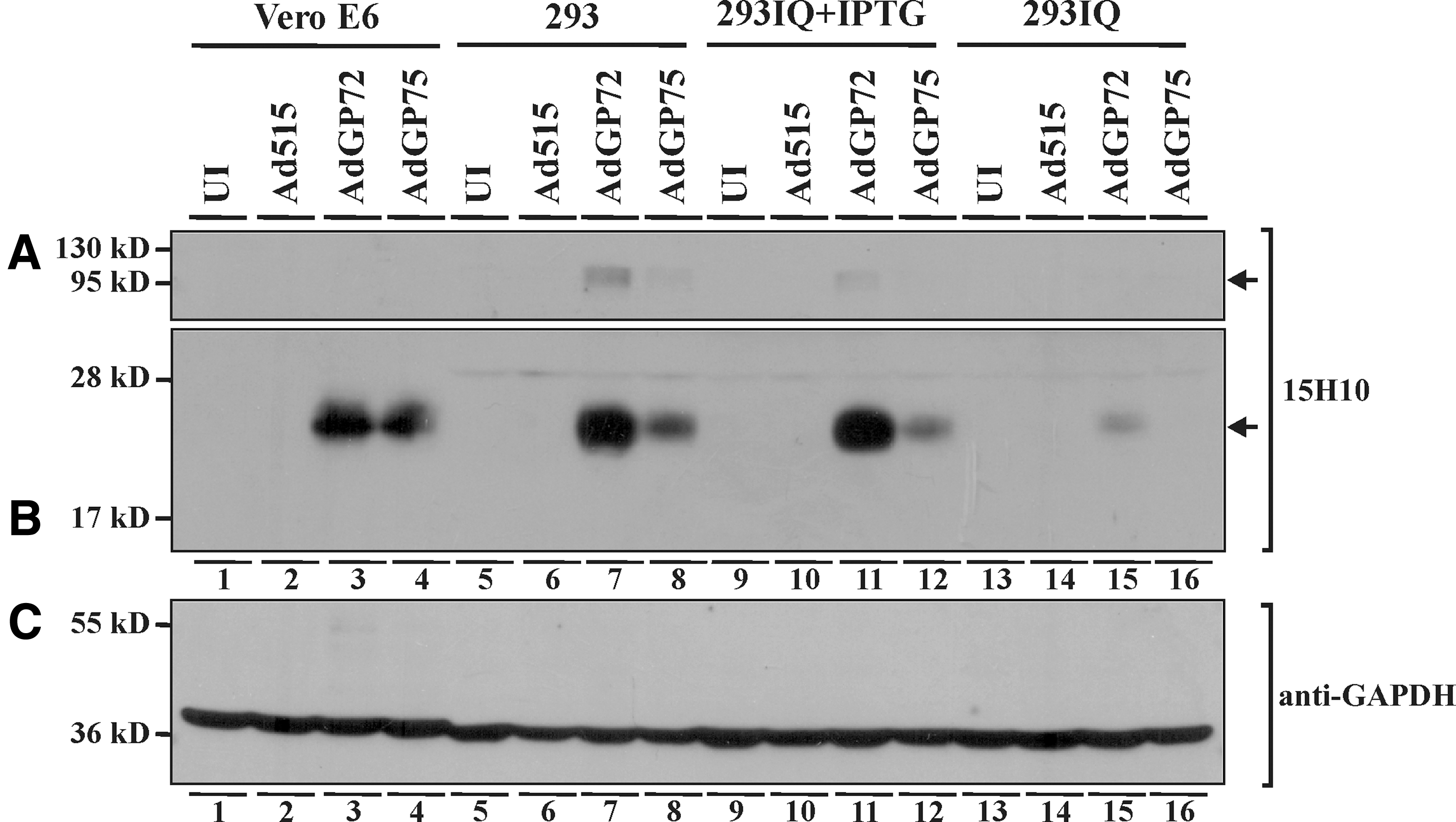
Glycoprotein expression by AdGP72 and AdGP75. Cells (Vero E6, 293, 293IQ, or 293IQ with 10 mM IPTG) were left uninfected, infected with Ad515, AdGP72, or AdGP75 and harvested 24 h postinfection. Lysates equivalent to 106 cells were subjected to 10% SDS-PAGE, and the proteins were transferred to PVDF membrane. The membrane was blocked with 5% SMP in PBS, and probed with 15H10
Expression of EBOV GP and VP40 through baculovirus
As the Ad viruses were produced using the human cell line 293 and the virus particles were not highly purified, it was possible that booster doses of the Ad virus preparation could induce antibodies to some mammalian proteins. To reduce this potential background reactivity anticipated in serological assays using antigen expressed in mammalian cells, we produced EBOV GP and VP40 through the baculovirus insect cell system. The expression of GP or HA-VP40 by baculoviruses was confirmed by Western blotting with 15H10 (Fig. 4A) or anti-HA (Fig. 4B) antibodies, respectively. Cleavage of GP into GP1 and GP2 was also confirmed by Western blotting (Supplementary Fig. S1B). The 15H10 antibody also detected a nonspecific protein between 35 and 40 kDa in uninfected cells (Supplementary Fig. S1B, lane 1). The difference in the molecular size of GP between the mammalian and insect cell systems is probably because of differential glycosylation (39,63).
Baculovirus expression of GP and HA-VP40. Sf21 cells were left uninfected or infected with 4 MOI of baculovirus encoding GP or HA-VP40, and clarified cell extracts (equivalent to 106 cells) were subjected to 10% SDS-PAGE and Western blotting with anti-GP
Furthermore, we wished to generate structures that would mimic EBOV, so that immunogenicity could be assessed against such higher order structures (HOSs). This could potentially also allow us to perform entry assays, which could possibly be used to carry out functional assay in terms of virus neutralization. The association between GP and HA-VP40 into a HOS was assessed by immunoprecipitation with anti-GP antibody (pooled mouse serum) and Western blotting with anti-HA antibody or vice versa in Sf21 cells coinfected with both the baculoviruses. Anti-HA antibody was able to precipitate HA-VP40 in complex with GP (Fig. 4C, top panel, lanes 5 and 6). Similarly, anti-GP serum was able to precipitate GP in complex with HA-VP40 (Fig. 4C, bottom panel, lanes 7 and 8). The presence of HA-VP40 and GP in the same complexes was confirmed by Western blotting for the same protein after the immunoprecipitation step (Fig. 4C, top panel, lanes 7 and 8 for GP, and bottom panel, lanes 5 and 6 for HA-VP40).
Immunogenicity of the RD-Ad5 vectors
Purified Ad viruses were used for immunization. Upon Q-Sepharose purification, the Ad viruses eluted at 0.4 M NaCl (Fig. 5; shown for AdGP75 only), and were confirmed to be Ad virus by Western blotting for the Ad hexon protein, in comparison with a reference virus (Fig. 5; shown for AdGP75 only). The particle count of the best fractions ranged from 1.64 to 1.8 × 1012 per milliliter.
Q-Sepharose column purification of RD-Ad5. AdGP75 vector preparations were purified by anion exchange chromatography and the samples were subjected to 10% SDS-PAGE and Coomassie blue staining (top panel) or Western blotting with antiadenovirus hexon antibody (bottom panel). The degraded adenovirus hexon protein can be seen as multiple protein bands below the hexon protein (lanes 9, 10, and 11). The numbers indicate fractions collected at the respective elution conditions. Adenovirus reference material (VR-1516; ATCC) was used as positive control. Asterisk indicates the adenovirus hexon protein (∼116 kDa). RD, replication defective.
The Ad vectors were inoculated to mice through intranasal or subcutaneous routes (Fig. 6A; ref. 25). With mammalian cell (Vero E6)-derived GP, a clear response was observed after the second immunization with both AdGP72 (groups III and VII) and AdGP75 (groups IV and VIII) (Fig. 6B; ref. 25). There was no significant difference (p > 0.05) between the preimmune and the first bleed of groups III, IV, and VII, but in group VIII, the response to first immunization was significant (p < 0.001). In groups III, IV, VII, and VIII, the responses to second and third immunization were significant (p < 0.001) when compared with the respective preimmune time points. None of the pre- or postimmune sera reacted with antigen prepared from Vero E6 cells not infected with any Ad vector (Supplementary Fig. S2A) or infected with Ad515 (Supplementary Fig. S2B).
Immunogenicity ofAdGP.Eight groups ofmicewere immunized with three doses of PBS orAd515 orAdGP72 orAdGP75with the immunization and bleeding schedule as shownin

In case of baculovirus-derived GP (Fig. 6D), the immune response of first bleed of groups III, IV, VII, and VIII was below the cutoff value (0.248). The titers of the second and final bleed of the same groups were significant (p < 0.001) when compared with the respective preimmune sera. With HOS as antigen (Fig. 6F), the cutoff value was considerably high (0.316), suggesting background reactivity; consequently, the titers of first bleed of all groups were less than the cutoff. The titers of second and final bleeds of groups III, VII, and VIII and only the final bleed of group IV were statistically significant (p < 0.001).
The endpoint titration of the final bleed sera of groups III, IV, VII, and VIII indicates that the GP-specific antibody response was better in groups III and IV (but statistically not significant) than in groups VII and VIII (Fig. 6C, E, and G).
Discussion
Owing to the recent outbreak of EHF in West Africa, and the consequent international emergency, an urgent need was felt to develop vaccines for EHF. Three different platforms, the modified vaccinia virus Ankara (MVA), rVSV, and Ad vectors, have been used for this purpose. The MVA vector is replication incompetent, but has been demonstrated to undergo recombination with orthopoxvirus in vitro (45); however, it is considered safe as no adverse events have been recorded till date. The rVSV vaccine is replication competent, but human infection with VSV, as demonstrated by the elicitation of antibodies, appears to be inconsequential clinically (17,49,66). As far as Ad vectors are concerned, both Ad and ChAd have been used. These Ads are RD. In addition, Ads pose minimal, if any, risk as they are fairly innocuous, as evidenced by the use of live adenovirus (serotypes 4 and 7) vaccines in humans. This study was designed to develop an RD-Ad5-based EHF vaccine candidate based on GP of the 2014 outbreak virus, whereas the other vaccines (except the Chinese Ad5-based vaccine) are either based on the Mayinga strain of 1976 or on the Kikwit strain of 1995. The Chinese Ad5-based vaccine is based on the 2014 outbreak virus, but uses a system wherein GP is expressed constitutively (77,78). In fact, all Ad vectors that underwent clinical trials recently express GP constitutively. By contrast, in our system, the GP expression is suppressed during the generation of the Ad vector.
Ad vectors are one of the most common systems for high-level expression of heterologous antigens. Although this is advantageous from the vaccine point of view, it may negatively impact the production of high-expressing adenovirus clones (38). In this article, the Ad vectors were generated and produced in 293IQ cells, which constitutively express lac repressor to subdue the lac operator-controlled expression of the transgene. Endpoint titers of sera from mice immunized with our RD-Ad5-GP ranged from 1,600 to 3,200 or from 200 to 400 when mammalian cell- or baculovirus-derived antigens, respectively, were used. It was possible that the sera obtained by immunizing mice with 293 cell-produced Ad virus could cross-react with an antigen derived from another mammalian cell line, Vero. However, to our surprise, none of the sera reacted with extracts obtained from either Vero E6 cells or Vero E6 cells infected with Ad515.
As commercial GP antigens were unavailable during the course of this study, the EBOV GP was also expressed in Sf21 cells through recombinant baculovirus. Recombinant baculovirus expressing EBOV GP and HA-tagged VP40 was generated, and two ELISA antigens (GP and HOS) were generated by infecting Sf21 cells. Owing to the difference in the glycosylation between the insect and the mammalian cells (9,64), the GP antigen expressed through insect cells was smaller than the mammalian cell-expressed GP (Fig. 4C, lane 4 vs. lane 9).
Lower titers were obtained in ELISA with the baculovirus-derived antigen, possibly because of the presence of a heterogeneous mixture of VP40 associated with or without GP, GP alone studded on pleomorphic structures, baculoviruses, and various other compact cellular structures that could pass through the 20% sucrose cushion (43). Attempts to develop an entry assay using HOS containing HA-tagged VP40 failed despite several attempts using different methodologies. It is possible that both the low titers in ELISA and failure to demonstrate entry of the HOS into cells were because of the crude nature of the baculovirus-derived antigen preparation.
One of the problems of using Ad vectors is preexisting immunity (PEI). Adenoviruses are one of the major viral agents associated with common cold in humans, and the presence of neutralizing antibodies to Ad varies from one population to another (1,3,8,37,44,47). Thus, PEI to adenovirus may affect the outcome of an Ad-based vaccination. There are many approaches to bypass PEI, and these include (a) increasing the number of particles per dose (76), (b) switching adenovirus type to less prevalent human Ad26, Ad48 (30,31), Ad35 (59), Ad6 (10,12), or nonhuman ChAd3 (57), or (c) a prime-boost regimen with heterologous Ad (28) or ChAd (53). In the follow-up study of the phase I clinical trial of human Ad5-based EHF vaccine, it was observed that a homologous prime-boost regimen with 6 months interval elicited greater humoral response than cellular response (29). It has also been shown in mice (13), guinea pigs (50), and nonhuman primates (51) that intranasal inoculation can overcome systemic PEI to Ad. The immunization regimen in this study, where the first two doses were alternated between subcutaneous and intranasal routes and the final dose was delivered intranasally, was designed to increase the GP-specific immune response and negate the effect of PEI. Incidentally, the serum-neutralizing antibody titers to Ad virus were <1:200 (data not shown), even after three immunizations, possibly because of the immunization regimen we used. Moreover, there was minimum reactivity of the sera in ELISA to Ad515 virus-infected Vero E6 extract.
Both humoral and cellular immunity play an important role in protection of animals with lethal dose of EBOV in challenge models. However, the level of protection varies according to the animal species and the type of immunogen. Plasmid DNA encoding GP protected guinea pigs if the prechallenge ELISA IgG titers were 1:3,200 (56). In case of primates immunized with plasmid DNA expressing GP, 100% protection was obtained when the ELISA IgG titers were 1:3,700 (55). Cellular immunity, especially CD8 T cell response, has been a crucial component to protect primates immunized with RD-Ad expressing GP against lethal dose of EBOV (54), and antibodies and B cells have been shown to be necessary for protection when a rVSV was used (36). In mice, single-dose and two-dose regimen of RD-Ad constitutively expressing EBOV GP elicited ELISA IgG titers of 16,000 and 100,000, respectively. However, the correlates of protection in mice are not well defined and the use of mouse-adapted EBOV further complicates extrapolation of the results to other animals or humans. As pathophysiology of EHF in mice is different from that in humans, even well-defined correlates of protection in mice or guinea pigs could be misleading.
Human clinical trials based on RD-Ad vectors (whether Ad or ChAd) have clearly demonstrated that 1010 to 1011 Ad particles elicit serum antibody responses that can neutralize EBOV in vitro, with increasing doses eliciting higher responses (16,77). However, it is possible that a single immunization may not suffice for achieving robust levels of protective antibodies, which may be a prerequisite for prophylactic protection against HFs. In this context, a priming with RD-Ad followed by a booster with MVA vaccine has shown better results (16,57).
Although RD-Ad vectors are one of the well-studied expression vectors, assembly defective adenoviruses hold promise as the next-generation vectors. Limited reports on assembly defective adenoviruses are published till date. Notable among these are Ad vectors with 100K (22) and IIIa gene deletion (10,12). The adenovirus 100K gene is responsible for the trimerization and nuclear transport of the hexon, and hence, the 100K-deficient Ad vectors are assembly defective. Hodges et al. deleted the 100K gene on adenovirus ΔE1 genome (22), and further improvement can be expected with the 100K deletion on the wild-type Ad genome background. Recently, a single-cycle adenovirus (SC-Ad) with IIIa gene deletion, which can express the transgene >100 times more than that of RD-Ad, was reported (10,12). The SC-Ad was also more potent in that ∼33 times less of SC-Ad was required to elicit antibody responses similar to those elicited by RD-Ad expressing influenza virus antigen (11). Thus, besides providing dose-sparing effect, and the subsequent benefit in reduced cost of such a vaccine, it may also be possible to use a single dose of the SC-Ad to elicit robust and potentially long-lasting immunity.
In this study, we report the development of an RD-Ad5-based EHF vaccine candidate based on modulated expression of EBOV GP. The platform technology based on this and other Ad vectors can also be applied for the development of candidate vaccines for other human and animal diseases for which high containment facilities are required for inactivated or live attenuated vaccine production.
Footnotes
Acknowledgments
The authors thank BEI resources, United States, for the monoclonal antibody 15H10 and vector pcDNA3.1-BlaMVP40. The authors are grateful to Prof. David C. Johnson, Oregon Health & Science University, United States, for providing Vero E6 cells and Prof. M.S. Shaila, Department of Microbiology and Cell biology, Indian Institute of Science, Bangalore, India, for providing Sf21 cells, pFastBac HT A and pFastBac 1 vectors, and DH10Bac cells. This study was supported by Bharat Biotech International Ltd., Hyderabad, India.
Authors' Contributions
The work was conceptualized by K.M.E. and N.R.H., and the experiments were designed by D.K., S.G., M.U., P.P.R., A.B., and N.R.H. The experiments were carried out by D.K., S.G., M.U., K.N., and A.B., and the data were analyzed and interpreted by D.K., S.G., P.P.R., A.B., K.M.E., and N.R.H. The article was written by D.K. and N.R.H., and edited and revised by D.K., A.B., and N.R.H.
Author Disclosure Statement
Ella Foundation received financial support for other projects that were carried out for Bharat Biotech International Ltd., of which K.M.E. is the Chairman and Managing Director. No other conflicts of interest exist for any of the authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.