
Abstract
A cross-sectional study on hepatitis B patients in Indonesia showed association of pre-S2 start codon mutation (M120 V) with cirrhosis and hepatocellular carcinoma (HCC), which was dissimilar from studies from other populations where pre-S2 deletion mutation was more prevalent. Different mutation patterns were attributed to different hepatitis B virus (HBV) subgenotypes in each population study. HBV surface proteins are reported to induce the activation of NF-κB, a transcriptional factor known to play an important role in the development of liver disease. This study aimed to see the effects of HBs variants in HBV subgenotype B3 on the expression and activation of NF-κB as one of the mechanisms in inducing advanced liver disease. HBV subgenotypes B3, each carrying wild-type (wt) HBs, M120 V, and pre-S2 deletion mutation were isolated from three HCC patients. HBs genes were amplified and cloned into pcDNA3.1 and were transfected using Lipofectamine into a Huh7 cell line. NF-κB activation was measured through IκB-α expression, which is regulated by NF-κB. RNA expressions for HBs, IκB-α, and NF-κB subunit (p50) were evaluated using real-time PCR. M120 V mutant had a significantly higher mRNA level compared with wt and pre-S2 deletion mutant; however, there were no significant differences in HBs protein expressions. The transcription level of p50 was higher in M120 V mutation compared with HBs wild-type and pre-S2 deletion mutant. NF-κB activation was higher in HBs wild-type compared with the two mutant variants. Pre-S2 mutations had no effect on the increment of NF-κB activation. However, M120 V mutation may utilize a different pathway in liver disease progression that involves high expression of NF-κB subunit, p50.
Introduction
H
Viral factors associated with severe liver disease in HBV-chronic patients are seroconversion status, viral titer, genotype, and HBV variant (5). Double mutation at HBV precore region (T1762/A1764) and mutation at pre-S regions have been known to play a role in liver disease progressions (18,43). A previous cross-sectional study on hepatitis B patients in Indonesia showed association of pre-S2 start codon mutation with cirrhosis and HCC (41,42). This result was dissimilar to other population studies, where pre-S2 deletion mutation was more prevalent (11,13). The different mutation patterns were attributed to the different HBV subgenotypes in each population study. HBV subgenotype B3 is the major and endemic strain found in Indonesia (26,40).
HBV surface (HBs) proteins are composed of three proteins, large (L), middle (M), and small (S) HBs. The three proteins are expressed from one open reading frame. S protein is the smallest of the three proteins with 226 amino acids (aa) (SHBs). The two other proteins consist of SHBs protein with additional 55 aa (MHBs) and 163 aa (LHBs) at the N-terminal, respectively (35). Certain types of mutations at the HBs gene have been reported to be associated with the severity of liver disease. Three of the most common HBs mutations are deletion mutation at pre-S1 and pre-S2 gene and amino acid changes at pre-S2 start codon (13,18). Some studies have reported the effect of pre-S deletion and its role in progressive liver disease, but not the pre-S2 start codon mutation specifically. Surface proteins from HBV pre-S1 and pre-S2 deletion mutant were found in liver tissue with ground glass hepatocyte characteristics, which is the histological hallmark for HBV infection (45). The deletion mutants, in particular the pre-S2 deletion mutant, were reported to induce oxidative stress and DNA damage due to HBs mutant accumulation in endoplasmic reticulum (ER) of hepatocyte that could activate signal transduction by ER stress (9,31). Pre-S2 deletion mutant has been shown to play a role in upregulating expression of aberrant cyclin A and centrosome duplication, which activates cell proliferation and leads to HCC development (44,46). Pre-S2 start codon mutation in HBV genes caused the loss of MHBs protein but not LHBs and SHBs proteins. HBV with its inability of producing MHBs has been associated with fulminant hepatitis, although studies confirming its role are still lacking (31,32).
NF-κB is a transcriptional factor known to play a major role in many aspects of chronic liver disease such as cirrhosis and HCC. NF-κB is activated in almost every chronic liver disease, including alcoholic liver disease, nonalcoholic fatty liver disease, viral hepatitis, and biliary liver disease (20). There are five members of the mammalian NF-κB subunits, including p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2). NF-κB may present in the form of heterodimer or homodimer bound with its inhibitor (IκB-α) when a cell is in an unstimulated condition (7). NF-κB activation occurs via canonical and noncanonical pathways. The canonical pathway is induced by TNF-α, IL-1, or lipopolysaccharide that activates IKK (IκB kinase). IκB-α phosphorylation by IKKβ causes NF-κB release, which enables it to translocate to the nucleus and induce DNA transcription. In the noncanonical pathway, the TNF cytokine family activates IKKα via NF-κB-inducing kinase (NIK). IKKα selectively phosphorylates p100 and leads to proteasomal processing of p100 to p52. Activated p52 dimers are then translocated to the nucleus and targeted specific NF-κB elements in the DNA. Termination of NF-κB is not fully understood yet. One of the known mechanisms is through the synthesis of IκB-α, which is regulated by NF-κB activation. Newly synthesized IκB-α can enter the nucleus, where it binds with NF-κB and the complex relocates to the cytosol.
Previous studies showed the role of hepatitis B proteins particularly HBx and HBs with NF-κB activation. HBx was reported to activate different members of NF-κB that play a role in apoptosis and in antiapoptotic pathways (37,38). Furthermore, HBx was shown to bind with voltage-dependent anion channel, which alters mitochondrial potential and subsequently generates reactive oxygen species (ROS), leading to the activation of STAT-3 and NF-κB (47). Truncated MHBs and LHBs proteins were demonstrated to play roles in NF-κB activation through PKC-dependent activation of the c-Raf-1/MEK signal transduction cascade and could act as oncogene as shown in Huh7 cells and transgenic mice (17,23). This other function of viral surface proteins was due to the interaction of pre-S2 domain of truncated MHBs and LHBs that remains on the cytosolic side of the ER after translation with PKC, which induced NF-κB activation (8,17,23). Our study in Indonesian HBV patients has shown the high prevalence of pre-S2 start codon mutation in patients with advanced liver disease. This mutation resulted in the loss of MHBs and resulting LHBs and SHBs as surface proteins. A study on the effect of this mutation and its role in liver disease is still lacking. Previous studies have demonstrated surface protein association with NF-κB activation in liver pathogenesis. Therefore, this study was designed to evaluate the effects of pre-S2 variants of HBV subgenotype B3, as the endemic strain in Indonesia, on the expression and activation of NF-κB in the Huh7 cell line.
Materials and Methods
Samples
Full HBs genes were amplified from archived HBV subgenotype B3 samples isolated from HCC patients, which have been reported in the previous study (41,42). Three HBs genes each harboring wild type (JQ429073), pre-S2 start codon mutation M120 V (JQ429023), and pre-S2 deletion mutation with deletion at nt 133–141 (JQ428949) were used in this study (Fig. 1A, B). HBV sequence subgenotype B3 from GenBank NCBI M54923 (
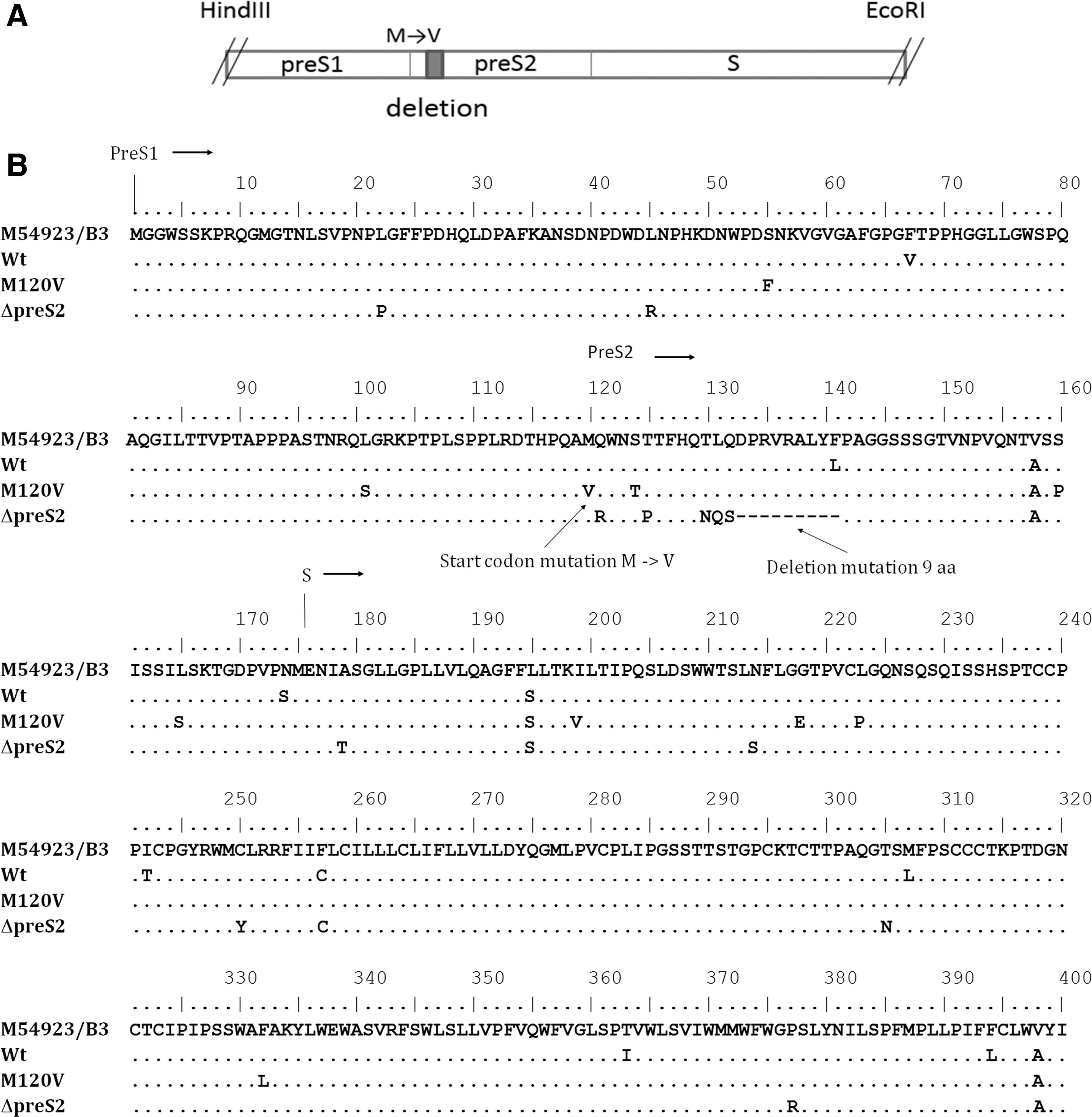
Illustration of digested HBs region (HindIII-EcoRI) showing positions of pre-S2 start codon mutation (M→V) and deletion mutation (shaded box) subcloned to expression plasmid, pcDNA3.1
Plasmids
Plasmids for HBs protein expressions were constructed by amplifying the full HBs gene with nested PCR and subcloned to pcDNA3.1. First round of nested PCR was done by using primer sets nt2343–2368 5′-CGAATTCGATGCCCCTATCTTATCAACACTTCC-3′ and nt1595–1620 5′-GTCGTCGACCGGTGGTCTCCATGC TACGTGCAGA-3′ with PCR profile as follows: 94°C for 2 min; 94°C for 15 sec, 55°C for 30 sec, 68°C for 2 min for 35 cycles; and 68°C for 7 min. Second round was carried out using the first round product as template and primer sets 5′- GC
Cell culture and transient transfection
Human hepatoma 7 (Huh7, RBRC-RCB1942) cell lines were maintained in MEM (Gibco), 10% FBS (Gibco), and 1% penicillin/streptomycin (Pen/Strep) 5000 U (Invitrogen). Huh7 cell lines (7 × 105 cells per well) were seeded into a six-well plate a day before transfection with 2 mL of medium (MEM, 10% FBS, 1% Pen/Strep 5000 U) incubated at 37°C, 5% CO2. The following day, plasmid DNA (1 μg per well) was transfected using Lipofectamine 2000 reagent (Invitrogen). Briefly, culture media were changed with 500 μL Opti-MEM (Gibco) and starved for 4 h at 37°C, 5% CO2. Plasmid DNA (pcDNA3.1, pcDNAs expressing wt HBs, M120 V HBs, and ΔpreS2 HBs, and pHBV1.3) (1 μg) was mixed with 200 μL Opti-MEM and incubated for 10 min at room temperature before subsequently being added with 5 μL of Lipofectamine in 200 μL Opti-MEM and further incubated for 20 min to allow the forming of DNA-Lipofectamine complexes. After 4 h of incubation, Opti-MEM culture media were changed with Opti-MEM containing DNA-Lipofectamine and incubated for another 4 h at 37°C, 5% CO2. The culture media were later changed with DMEM (Gibco), and transfected cell lines were maintained in 1.5 mL DMEM, 10% FBS, and 1% Pen/Strep 5000 U at 37°C, 5% CO2. Culture supernatants and cells were collected 24 and 48 h after transfection and used for further analysis with real-time PCR and enzyme-linked immunosorbent assay (ELISA). Transfection efficiency was determined by transfecting pMax-GFP (green fluorescence protein) (Lonza) to Huh7 cells. GFP expression was detected with flow cytometry, BD Accuri™ C6 Flow Cytometer (BD Biosciences) with software BD Accuri™ C6 (BD Biosciences). Transfection efficiency based on GFP expression was 81.27% ± 3.37% (data not shown).
NF-κB activation and real-time PCR assay
Total RNA was extracted from 1 × 106 Huh7 cells transfected with pcDNA plasmids expressing HBs and pHBV1.3 after 24 and 48 h of transfection using the High Pure RNA Isolation Kit (Roche) followed by cDNA synthesis using SuperScript® III First-Strand Synthesis Supermix (Invitrogen) according to the manufacturer's instructions. Real-time PCR synthesis was done using iQ SYBR Green Supermix (BioRad) and CFX96 Real-Time PCR (BioRad) machine for HBs, IκB-α, and p50. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was assayed as housekeeping gene. Reaction mix was made for a total volume of 50 μL, which comprised 25 μL IQ SYBR Green Supermix (2 × concentration); 0.5 μL of 10 μM forward primer; 0.5 μL of 10 μM reverse primer; cDNA (from 80 ng RNA); and dH2O up to 50 μL. PCR profile was as follows: 95°C, 5 min; 35 cycles (95°C, 30 sec; 61.8°C, 30 sec; 72°C, 30 sec). Annealing temperature was first optimized for all targets using melt curve analysis and gradient temperature at 45–95°C. For all target genes, optimized annealing temperature was achieved at 61.8°C (data not shown). Free nuclease dH2O was run every time as No Template Control. Length of product for each gene was as follows: GAPDH 250 bp, IκB-α 95 bp, NF-κB (p50) 180 bp, and HBs 250 bp. Relative gene expression was count manually using the Livak method 2−ΔΔCt (19). The primers used are listed in Table 1. Statistical analysis was done using unpaired t-test with software GraphPad Prism 6 and PASW Statistic 18. p < 0.05 was considered significant.
HBs, hepatitis B virus surface protein; IκB-α, inhibitor NF-κB alpha; p50, subunit protein 50; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
ELISA for HBs protein
Total protein was extracted from Huh7 cells transfected with pcDNA, plasmid expressing HBs and pHBV1.3, which were collected at 24 and 48 h after transfection using radio immunoprecipitation assay (RIPA) buffer. Phosphatase inhibitor (Cell Signaling Technology) and protease inhibitor (Sigma Aldrich) were added at a concentration of 1:100 just before lysis. Total protein concentration was measured using Bradford assay (BioRad). About 100 μg of total protein was analyzed for HBsAg using ELISA HBsAg (Abnova). Total protein and 50 μL HRP-conjugated anti-HBs were added into each well coated with anti-HBs and incubated for 80 min at 37°C. Each well was washed with 350 μL washing buffer using Immunowash (BioRad). One hundred microliters of HRP substrate was added into each well and incubated in a dark room for 30 min at room temperature. Enzymatic reaction was stopped with the addition of 100 μL 2 N HCl. Absorbance was measured at 450 nm using SmartSpec Plus spectrophotometer (BioRad).
Results
HBs expression was affected by pre-S2 mutations
Full HBs genes were amplified from HBV subgenotype B3 isolated from three HCC patients that were archived samples from previous studies (41,42). Pre-S2 start codon mutation is a substitution mutation at amino acid position 120 where methionine was mutated into valine (M120 V) (Fig. 1B). M120 V was the most prevalent type of pre-S2 start codon mutation found in our previous study (42). Pre-S2 deletion mutant involved a deletion of nine amino acids at position 133–141 (Fig. 1B). Level of HBs mRNA expression was determined using RT-PCR with primers derived from the small HBs region resulting in a 250 bp cDNA product. There were significant differences of HBs mRNA expression between M120 V with wild type and Δpre-S2 (Fig. 2A). In cells expressing pHBV1.3, HBs mRNA was lower compared to cells expressing HBs genes. This might be due to the utilization of endogenous promoter (2) by pHBV1.3, whereas HBs plasmid expression utilized the CMV, a strong mammalian expression promoter.
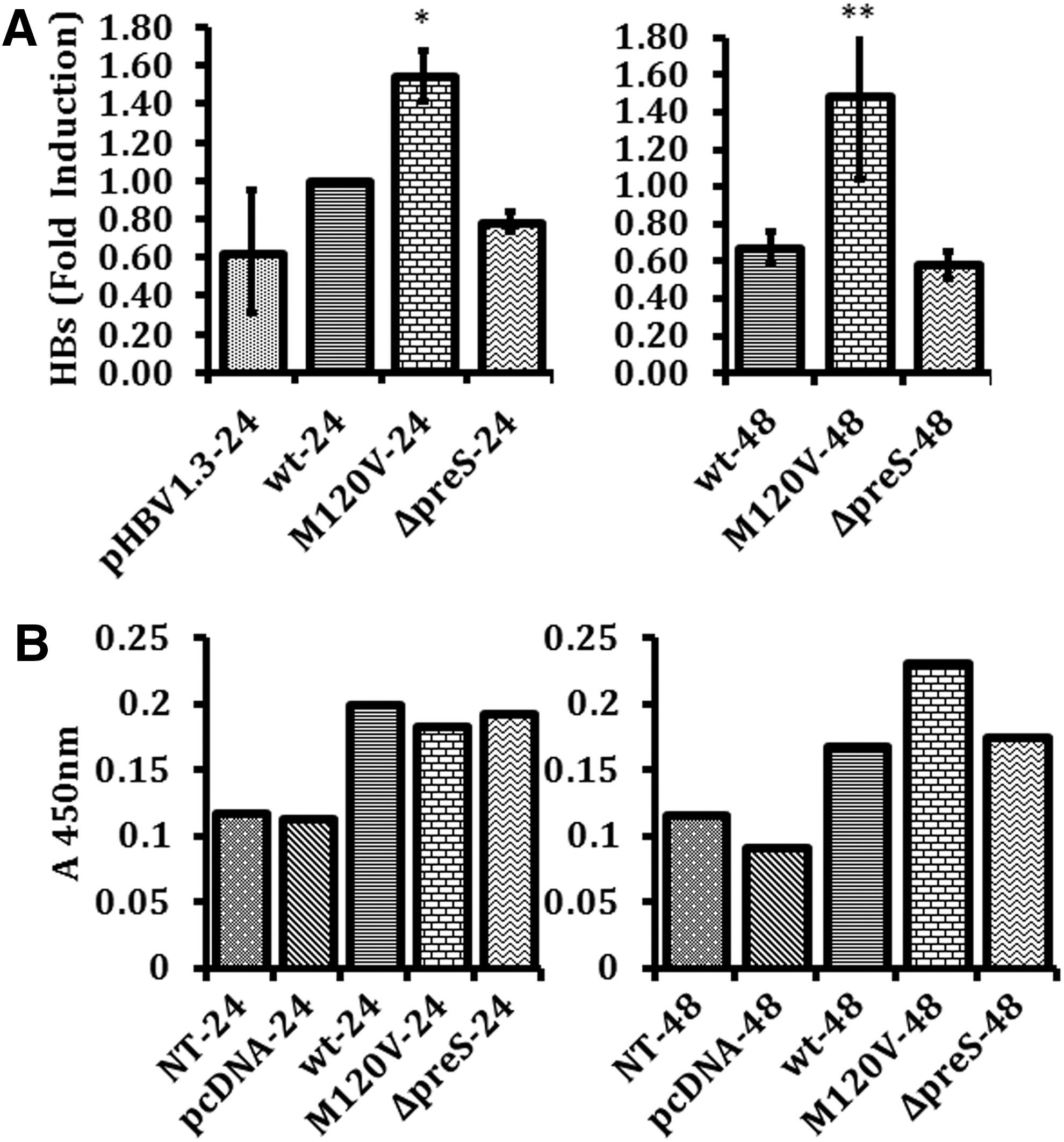
Expression level of HBs mRNA at 24 and 48 h. The calculation was made relative to wt at 24 h (mean ± SE)
Total protein of transfected Huh7 cells was subjected to ELISA for HBsAg. HBs protein expression was slightly higher in cells transfected with plasmid harboring M120 V mutation compared to wild type and Δpre-S2 (Fig. 2B). HBs protein was analyzed using a commercially available ELISA kit. This ELISA kit detected the HBsAg domain of HBs protein, which enabled it to detect all three types of HBs protein: LHBs, MHBs, and SHBs.
NF-κB subunit p50 expression by pre-S2 start codon mutant was higher than HBs wild type
NF-κB can be found in the form of a heterodimer or a homodimer. One of the protein subunits that play a major role in the NF-κB signaling pathway is p50, which could form a homodimer with another p50 or a heterodimer with p65. M120 V mutant showed a higher level of p50 mRNA compared with Δpre-S2 mutant and wild type after 24 and 48 h of transfection, respectively (Fig. 3).
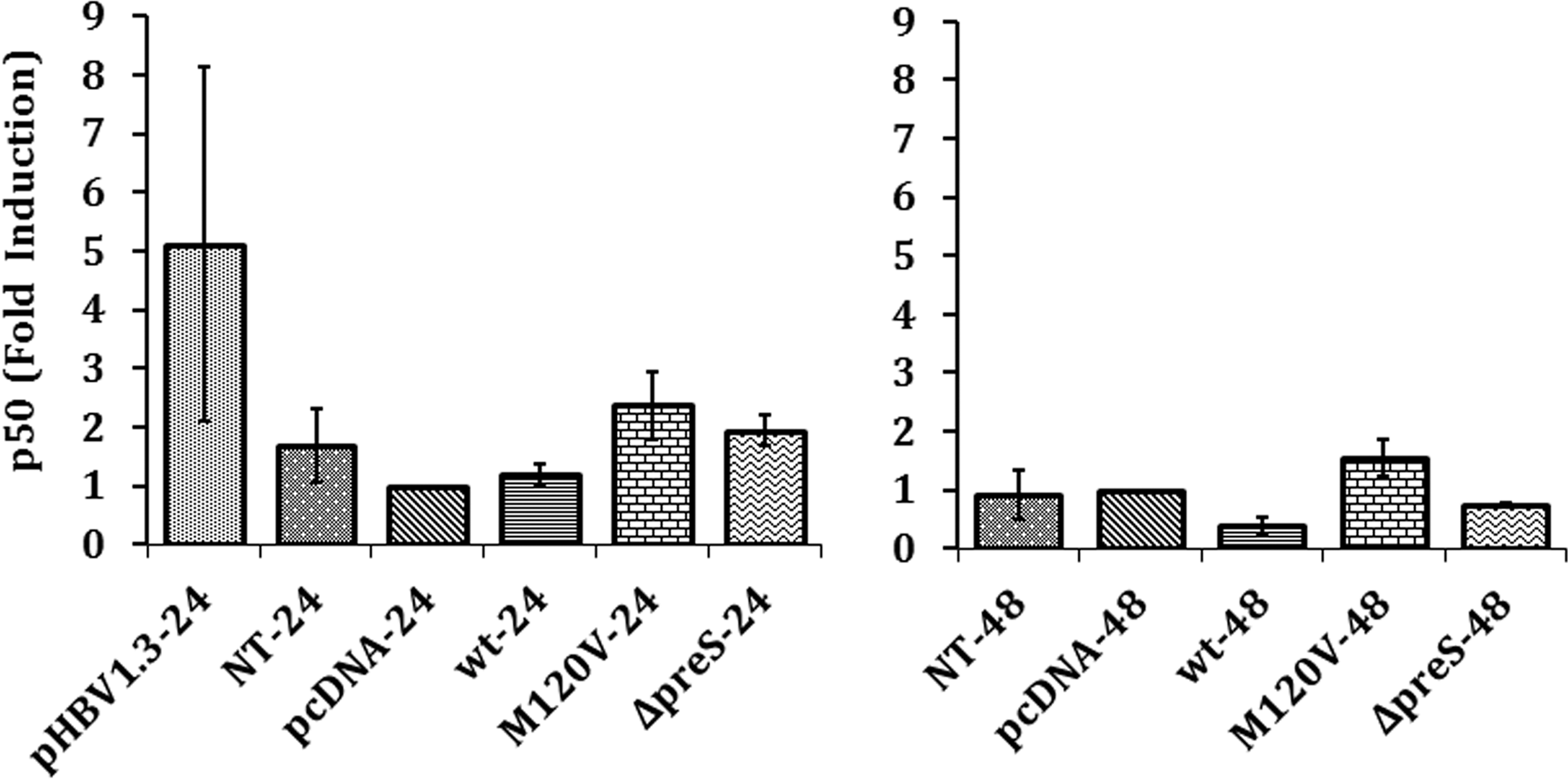
Expression level of p50 mRNA at 24 and 48 h. The calculation was made relative to negative control pcDNA at 24 h (mean ± SE). p < 0.05.
NF-κB activation by HBs mutant
IκB-α mRNA expression was used as a parameter to investigate NF-κB activation as described by Bottero et al. (2). NF-κB induces transcription of the IκB gene. IκB acts as an inhibitor for NF-κB to remain in its inactive state. The negative feedback loop IκB-α is one of the mechanisms to terminate NF-κB activation. Eletrophoretic mobility shift assay (EMSA) is the method commonly used for detecting NF-κB activation. EMSA detects the presence of protein-DNA complex, particularly DNA that has the binding site for the transcription factor NF-κB. DNA that binds to the protein will move slower than the free DNA. The method used by Bottero et al. was compared with EMSA and Northern blot analysis, respectively. The level of IκB mRNA expression analyzed from different cellular models (Jurkat leukemic T cells, HeLa carcinoma cells, HDLM-2 Hodgkin lymphoma cells, and primary peripheral blood lymphocytes) correlated perfectly with the two other methods. Furthermore, IκB-α mRNA expression was regulated by NF-κB activation and was proven to have a direct correlation with the rate of transcription caused by NF-κB activation. Measurement of IκB-α mRNA expression, therefore, allows additional information that cannot be provided from other analyses such as EMSA that detects only the level of NF-κB binding to DNA.
Real-time analysis showed a lower level of IκB-α mRNA expression in Huh7 cells transfected with M120 V compared to wild type, which indicated lower level of NF-κB activation by pre-S2 start codon mutant (Fig. 4). Lower level of activation was also observed in Huh7 cells transfected with Δpre-S2 although statistically not significant (Fig. 4). In other words, pre-S2 mutants did not affect NF-κB activation. Higher level of NF-κB activation was also seen by cell line transfected with plasmid expressing full-genome HBV.
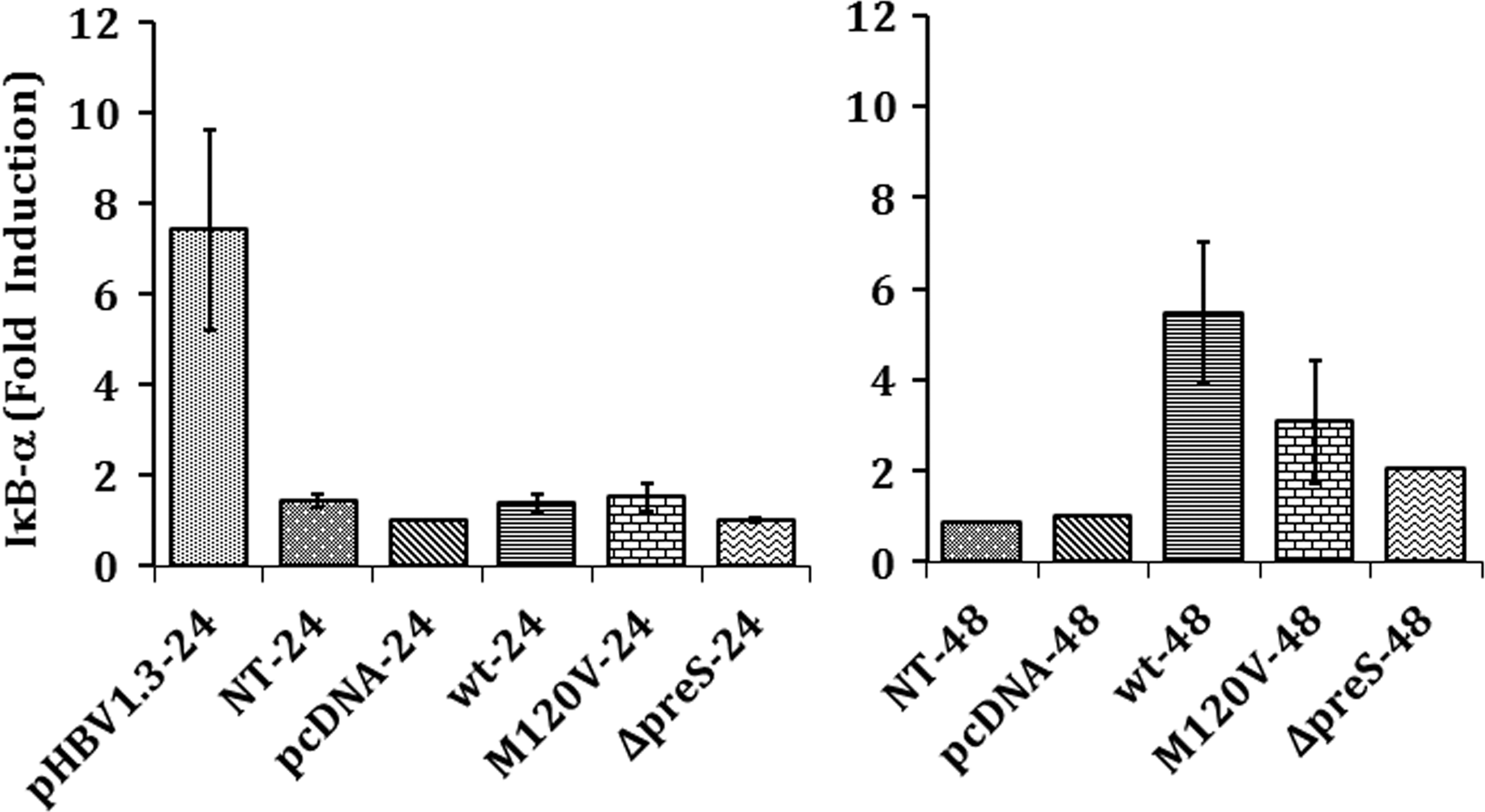
NF-κB activation based on IκB-α expression in Huh7 cells at 24 and 48 h. The calculation was made relative to negative control pcDNA at 24 h (mean ± SE). p < 0.05 was considered significant. HBs, hepatitis B virus surface protein; IκB-α, inhibitor NF-κB alpha; p50, subunit protein 50; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
HBV subgenotype B3 is the predominant subgenotype infecting hepatitis B patients in Indonesia. Different subgenotypes have different mutation patterns and different risks of developing HCC (4,21). In our previous studies, we found a different mutation pattern of HBV subgenotype B3 surface gene compared with other HBV subgenotypes in other populations. HBV polymerase lacks proofreading capacity, which causes different variations in the HBV genome. These variations can be lethal but can also be beneficial to the virus and may result in increased pathogenicity of the virus. The selection of these variations is affected by viral, host immune suppression and exogenous pressure, such as immunoprophylaxis and antiviral therapy (31). Our previous studies showed that commonly found mutations associated with advanced liver disease in Indonesian populations were mutations in HBV basal core promoter and the pre-S2 start codon mutation (3,17). Pre-S2 start codon mutation was the predominant mutation found in HBV subgenotype B3 surface gene. This mutation was associated with cirrhosis and HCC; in addition, it was an independent factor for advanced liver disease (41,42). Studies on pre-S mutant's role were mostly on pre-S deletion or pre-S deletion in combination with pre-S2 start codon mutant. This is the first study to observe the effect of pre-S2 start codon mutant alone.
In this study, we cloned HBs genes of HBV subgenotype B3 into a mammalian cell expression plasmid. Our results show that pre-S2 start codon mutation has a higher level of mRNA compared with pre-S2 deletion mutant and wild-type HBs; however, there is only a slight increase in the HBs protein expression level compared to wild type (Fig. 2). Pre-S2 start codon mutation causes the loss of MHBs protein and the slightly higher HBs protein expression in Huh7 cells expressing M120 V mutant may comprise LHBs and SHBs proteins.
Unlike LHBs and SHBs, MHBs protein does not play a role in virion formation, secretion, and infectivity (31). Variations or the loss of MHBs would not affect HBV life cycle. However, epidemiology studies suggested association of MHBs variants or loss of MHBs with increased hepatitis B pathogenesis. Loss of MHBs also appeared to play a role in the cases of fulminant hepatitis. It is suggested that it causes hepatic failure due to intracellular retention of HB proteins (31).
The absence of MHBs causes an imbalanced proportion of LHBs and SHBs. A study reported on the regulatory function of MHBs on surface gene expression, in which the loss of MHBs increased LHBs production and decreased SHBs (12). A proper ratio of LHBs to SHBs is essential for correct assembly and virion release from the hepatocytes. Overproduction and accumulation of LHBs due to the incapacity of HBV in producing MHBs have been demonstrated in transgenic mouse models of fulminant hepatitis (1,32).
The LHBs protein has two different transmembrane topologies in the ER that serve two different functions (3,15,30,33). The first one is where the pre-S1/pre-S2 domain of LHBs faces the cytoplasmic region and serves as a docking site for virus' nucleocapsid. In the second, the pre-S1/pre-S2 region of LHBs faces the ER lumen and posttranslationally translocates across the ER membrane, which functions in virion assembly (3). Hildt et al. reported NF-κB activation induction by LHBs protein due to the binding of cytoplasmic pre-S2 domain with PKC and its subsequent activation (8).
Higher LHBs protein expression may result in higher NF-κB activation. However, our result showed less NF-κB activation in cells expressing M120 V mutant or pre-S2 deletion mutant compared to wild-type HBs. It is possible that LHBs expressed in cells with M120 V mutant may not be in the correct orientation to interact with PKC. The composition of produced LHBs, MHBs, and SHBs proteins by each HBs variant was also not determined in this study. The effect of M120 V mutation to the production of LHBs and SHBs proteins and their locations in the cells therefore requires further investigation.
Lower NF-κB activation compared to wild type was also shown in Huh7 cells expressing pre-S2 deletion mutant. Pre-S2 deletion mutant was reported to play a role in oxidative stress and DNA damage by the retention of misfolded HBs protein mutant in the ER as observed in Huh7 cells and transgenic mice expressing LHBS (9). NF-κB activation was caused by the orientation of its pre-S2 domain and not by the ER retention of LHBs resulting in the increased level of radicals as demonstrated by Hildt et al. (8). In their experiment, the addition of N-acetyl s-cysteine, an ROS inhibitor, in Huh7 cells expressing LHBs did not result in a significant influence on the transcriptional activator function. Hence, pre-S2 deletion, although inducing ER stress, may not be associated with the increase of NF-κB activation. However, oxidative stress and DNA damage were not analyzed in this study.
We later analyzed the expression of p50 as a major component of the NF-κB family that consists of the NF-κB dimer (16). Contrary to NF-κB activation result, the expression of p50 by pre-S2 mutants was higher than wild type in particular by M120 V mutant, although it was not significant. P50 lacks a transactivation domain and can only act as a transcription factor when it forms a dimer with other NF-κB family proteins that contain Rel homology domain. P65/p50 is the most common dimer of NF-κB. This heterodimer regulates the IκB-α transcription, which in this study was measured as the parameter for NF-κB activation (2). Transfection with full-length HBV (pHBV1.3) increased p50 mRNA expression and, concomitantly, we observed increased IκB-α mRNA expression indicating NF-κB activation. Transfection with plasmid containing HBs gene (wt, M120 V, pre-S2 deletion) resulted in p50 mRNA expression, which did not differ significantly with mock transfection. This might be caused by missing other cofactors such as HBx or other viral HBV proteins. NF-κB consists of p65 and p50 subunits. Their gene expressions are not regulated identically. Ideally to fully measure the effect of NF-κB activation, both p50 and p65 expressions need to be studied. Contrary to full-length HBV, we did not observe increased mRNA expression of p50 with the increased IκB-α mRNA expression using HBs gene construct. Meberg et al. (22) reported the inverse correlation between p50 mRNA expressions and IκB-α mRNA expression in hippocampal granule cells when NF-κB is activated. The inverse correlation could be potentially caused by the missing cofactors, or other mechanisms such as decrease of p50 mRNA stability. It is possible that the expression of p50 did not correlate proportionally with the activation of NF-κB, which should have been confirmed by measuring the expression of p65.
P50 can form a homodimer and is known to make a complex with Bcl-3 and plays a role in HCC pathogenesis (29). Moreover, Song et al. demonstrated that p50 has a regulatory function on protein modification independent of its transcriptional activity (36). In their report, p50 was shown to increase the stability of the GADD45α protein through suppressing its ubiquitination and proteasome-dependent degradation. This leads to the activation of the JNK pathway, which will lead to apoptosis. Although the mechanism of p50 in suppressing GADD45α ubiquitination remains unclear, they have shown that p50 can either activate or suppress apoptosis depending on the induction. Recently, the same group has shown other important roles of p50 in modulating p53 expression at the translational level (48). P53 is a tumor suppressor that plays an important role in cell cycle control, DNA repair, and apoptosis. It was shown that deletion of p50 resulted in the loss of p53 protein expression. In tumor-associated p53 mutations, the expression of p50 may enhance the severity of the disease. It has been apparent that mutant p53 protein not only results in the loss of its normal function but also may have developed oncogenic properties instead (25,34). Overexpression of the p50 subunit and the activation of its homodimer have been observed in several types of cancer, including cervical carcinoma, squamous cell carcinoma, nasopharyngeal carcinoma, nonsmall-cell lung carcinoma, lymphoma, and melanoma (14,39).
In this study, there were indications that HBs mutant M120 V can cause high expression of p50 but not NF-κB activation. It is possible that M120 V mutation may cause severe liver disease through different pathways involving p50 other than NF-κB activation. The presence of p50/p50 homodimer and Bcl-3 has been found with high prevalence in HCC, suggesting an important role in liver pathogenesis (28). It is tempting to speculate that, as pre-S2 start codon mutant of HBV subgenotype B3 was associated with advanced liver disease, p50/p50 homodimer might play an important role in liver pathogenesis in this mutant.
There were several limitations in this study. First, the pre-S2 mutations in this study were not generated mutations but were naturally occurring; hence, they carried other nucleotide variations (Fig. 1B). However, none of the other nucleotide variations has been reported in association with liver disease severity. Second, the protein expression analysis of the surface proteins did not differentiate between the LHBs, MHBs, and SHBs. The ELISA method used for protein analysis detected all the surface proteins. Therefore, the ratio between the three surface proteins was not determined in this study. Third, the p50 protein expression analysis to confirm the results from RNA expression analysis was not performed. Furthermore, expressions of other NF-κB subunits such as p65 could also be analyzed.
Conclusion
Pre-S2 mutations have lower NF-κB activation than wild type based on IκB-α expression analysis. This study shows that p50 mRNA expression in hepatoma cell lines is upregulated in cells with HBs M120 V mutant expression. High expression of p50 by HBs protein in association with HCC has never been reported. It can be postulated, therefore, that the pre-S2 start codon mutation of HBV subgenotype B3 plays a role in liver disease progression by higher expression of p50. However, due to the limitations of this study, further investigations are required to confirm the role of p50 in liver pathogenesis in association with pre-S2 start codon mutation of HBV subgenotype B3.
Footnotes
Acknowledgments
We thank Atul Philip Varghese, MSc, and Bruce G. Foster, MA, for their critical reading of the article and Teridah Ernala Ginting, DVM, PhD, for discussion. This study was supported by a grant from the Mochtar Riady Institute. Part of this study has been presented as a Poster Presentation at the 2015 European Society for Medical Oncology (ESMO) Asia Conference in Singapore.
Author Disclosure Statement
The authors declared no conflict of interest.