
Abstract
Influenza virus infections can be complicated by bacterial superinfections, which are medically relevant because of a complex interaction between the host, the virus, and the bacteria. Studies to date have implicated several influenza virus genes, varied host immune responses, and bacterial virulence factors, however, the host–pathogen interactions that predict survival versus lethal outcomes remain undefined. Previous work by our group showed that certain influenza viruses could yield a survival phenotype (A/swine/Texas/4199-2/98-H3N2, TX98), whereas others were associated with a lethal phenotype (A/Puerto Rico/8/34-H1N1, PR8). Based on this observation, we developed the hypothesis that individual influenza virus genes could contribute to a superinfection, and that the host response after influenza virus infection could influence superinfection severity. The present study analyzes individual influenza virus gene contributions to superinfection severity using reassortant viruses created using TX98 and PR8 viral genes. Host and pathogen interactions, relevant to survival and lethal phenotypes, were studied with a focus on pathogen clearance, host cellular infiltrates, and cytokine levels after infection. Specifically, we found that the hemagglutinin gene expressed by an influenza virus can contribute to the severity of a secondary bacterial infection, likely through modulation of host proinflammatory responses. Altogether, these results advance our understanding of molecular mechanisms underlying influenza virus–bacteria superinfections and identify viral and corresponding host factors that may contribute to morbidity and mortality.
Introduction
S
Streptococcus pyogenes or group A streptococcus (GAS) is a β hemolytic, Gram-positive bacterium that can be carried asymptomatically by humans on the skin and in the respiratory system (69). Interestingly, GAS and influenza share a common seasonality, and work with an influenza virus: GAS model of superinfection has shown an association with necrotizing fasciitis (49,51). Our group has been working with an influenza virus–GAS model of superinfection in mice (12) to evaluate the contribution of influenza viruses and bacteria toward the severity of secondary bacterial infections (22,81). Our previous comparison of influenza viruses and bacterial species showed that the virus strain used for primary inoculation can have a profound effect on susceptibility to secondary infection with GAS, Streptococcus pneumoniae, and Staphylococcus aureus (81). Most notably, data from this study showed that the primary swine influenza virus isolate, A/swine/Texas/4199-2/98-H3N2 (TX98), does not significantly predispose an infected host toward death after inoculation with GAS, S. pneumoniae, and S. aureus, whereas the model laboratory influenza virus isolate, A/Puerto Rico/8/34-H1N1 (PR8), induces a lethal synergism (81). Based on this observation, we developed the hypothesis that specific influenza virus genes and host immune responses work together to influence the severity of a secondary bacterial infection. In this report, we show that the TX98 influenza virus is cleared more rapidly than PR8 from the lungs of infected mice, and that this rapid clearance limits bacterial burden both locally in lungs and systemically in spleens. Our work also shows that cellular recruitment profiles and cytokine responses differ between the two viruses in a manner that implicates host responses against the invading virus as a contributor to secondary infection severity. Work with reassortant influenza viruses that we created using reverse genetics shows that the hemagglutinin (HA) gene expressed by the virus correlates with susceptibility to secondary bacterial infections. We will discuss our findings in the context of HA protein contributions that can direct lung inflammatory responses to influence the severity of a secondary bacterial infection.
Materials and Methods
Mice
Adult (6- to 8-week-old) female BALB/cJ mice were obtained from Harlan Laboratories (Indianapolis, IN) and housed in groups of four, with 24-h access to food and water.
Ethics statement
All animal experiments were performed following the guidelines established and approved by the Animal Care and Use Committee at the University of South Dakota (Vermillion, SD) under protocol numbers 44-02-09-12E, 99-12-11-14D, and 21-12-14-17E.
Influenza virus–GAS superinfection model
Mice were inoculated intranasally (i.n.) with 100 μL of a sublethal dose (0.1 LD50) of PR8, TX98, or the indicated single gene segment (7:1) reassortant viruses (Table 1). On day 7 after virus inoculation, the MGAS315 strain of S. pyogenes (GAS; American Type Culture Collection [ATCC], Manassas, VA) was delivered at 0.1 LD50 (106 CFU) in a 100 μL volume, i.n., as described previously (12). Weight loss (morbidity) and survival (mortality) were monitored daily after infection, and mice were humanely euthanized if their body weight dropped below 30% of their initial body weight.
Values are reported as log10 TCID50 per milliliter.
Values are reported as the log10 TCID50 that represented 0.1 LD50 in BALB/c mice.
Virus created with seven gene segments from TX98 and one gene segment from PR8.
LD50, 50% lethal dose; MDCK, Madin-Darby canine kidney; TCID50, 50% infectious dose in tissue culture.
Viral and bacterial titers in mouse lungs and spleens
Lungs were collected on days 0, 3, 7, 8, 9, 10, and 11 after inoculation with influenza, and both viral and bacterial titers were quantitated. For viral titers, confluent Madin-Darby canine kidney (MDCK) monolayers were inoculated with log10 serial dilutions of lung homogenates as described previously (26), and titers are reported with a range of detection from 103 to 108 TCID50/mL. For bacterial titers, log10 serial dilutions of lung homogenates were inoculated onto tryptic soy agar plates supplemented with 10% defibrinated sheep blood (Becton Dickinson, Sparks, MD) as described (12). Colony-forming units (CFUs) were determined after 24 h incubation at 37°C, 5% CO2 with a range of detection from 103 to 108 CFU/mL. To detect the systemic spread of bacteria from lungs, spleens were collected on days 0, 3, 7, 8, 9, 10, and 11 after inoculation with influenza virus. Titers of bacteria in spleen homogenates were quantitated as described for lung bacterial titers.
Host cell populations
Host cell populations were evaluated as previously described (11,18). Specifically, at the times indicated after infection lungs were perfused with phosphate-buffered saline (PBS), excised, finely minced, and digested enzymatically for 60 min at 37°C in Roswell Park Memorial Institute media containing 0.02 mg/mL collagenase (Worthington, Lakewood, NJ), 10 μg/mL DNase (Sigma, St. Louis, MO), and 5% fetal calf serum (Atlanta Biologicals, Lawrenceville, GA). After being filtered through a 100 μm mesh, and centrifuged at 1,000 rpm for 10 min, suspensions were depleted of erythrocytes by osmotic shock using red blood cell lysis buffer (10 mM KHCO3, 150 mM NH4Cl, 0.1 mM EDTA, pH 8.0). Cells were kept on ice, and all subsequent incubations were done at 4°C. To block Fc receptors, cells were incubated with anti-mouse CD16/CD32 antibody (clone 93; BioLegend, San Diego, CA). Cells were incubated with the following antibodies to detect cell surface markers: CD11c PE/Cy5 (clone N418; BioLegend), CD11b Alexa Fluor 488 (clone M1/70; BioLegend), Siglec-F PE (clone E50-2440; BD Biosciences, San Diego, CA), and GR-1 Alexa Fluor 647 (clone 1A8; BioLegend) in flow cytometry buffer (0.5% fetal bovine serum and 0.09% sodium azide in Dulbecco's PBS [DPBS]) for 30 min. Cells were washed with DPBS, and fixed with 4% paraformaldehyde in DPBS for 30 min. Enumerated cells were then washed and resuspended in DPBS and results were acquired using a C6 Flow Cytometer (Accuri Cytometers, Ltd., Ann Arbor, MI), with data analyzed using CFlow Plus software (Accuri Cytometers). Populations were defined as either alveolar macrophages (CD11chigh, Siglec-F+) or neutrophils [CD11bhigh, Ly-6G(Gr1high)].
Lung cytokines
Lung homogenates were centrifuged at 300 × g for 3 min and cytokines were detected using either the Th1/Th2/Th17 Cytometric Bead Array Kit (BD Biosciences) or ELISA (enzyme-linked immunosorbent assay) for IL-13 and IL-22 (eBioscience). Samples were analyzed using either a C6 Flow Cytometer with CFlow Plus software (Accuri Cytometers), or with a BioTek EL808 plate reader (BioTek Instruments, Inc., Winooski, VT). Levels are reported in pg/mL based on standards included with the kit, and all assays were performed following the manufacturer's instructions.
Generation of reassortant viruses by reverse genetics
Stocks of pHW2000 plasmids containing individual gene segments, for either A/swine/TX/4199-2/98-H3N2 (TX98) or A/Puerto Rico/8/34-H1N1 (PR8), were kindly provided by Richard J. Webby (St. Jude Children's Research Hospital, Memphis, TN). Using reverse genetics as described previously (24), reassortants containing genes from PR8 and TX98 were generated in the combinations presented in Table 1. Briefly, cocultures of MDCK (ATCC) and 293T (ATCC) cells in Opti-MEM (Invitrogen, Grand Island, NY) were transfected with 1 μg of each of the eight plasmids indicated, using the transfection reagent TransIT (Mirus Bio, Madison, WI). After 6 h of incubation at 37°C, the transfection mixture was replaced with fresh Opti-MEM. TPCK-trypsin (Worthington, Lakewood, NJ) was added 24 h later, and culture supernatants were collected 72 h after plasmids were initially added. Virus stocks were propagated in the allantoic cavity of 10-day-old embryonated chicken eggs that were incubated for 3 days at 35°C, as described (26).
Virus characterization
Viral RNA was isolated (Qiagen, Germantown, MD), and genes were amplified by reverse transcription-polymerase chain reaction and sequenced (Eurofins, Louisville, KY) to confirm the desired reassortant genotype. After the genotype was confirmed, reassortant viruses were propagated in MDCK cells to determine the 50% infectious dose in tissue culture (TCID50), as described (12,26). The 50% lethal dose (LD50) was determined using mice, and 0.1 LD50 values are reported based on the TCID50 at the dilution used for inoculation. Table 1 presents the TCID50 and 0.1 LD50 values for TX98, PR8, and individual reassortant viruses. Calculations related to dose were made using the method of Reed and Muench (61).
Statistical analyses
Comparison of weight loss between groups of mice were made by calculating area under the curve. Survival between groups of mice was made using the log-rank chi-squared test on the Kaplan–Meier survival data. Data consisting of three or more groups were analyzed using a one-way ANOVA (analysis of variance) with post hoc Tukey's multiple comparison test. Values were accepted as significant if p < 0.05. Figures were created, and statistical analyses were performed, using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA).
Results
Weight loss, survival, and pathogen load during PR8:GAS and TX98:GAS superinfections
Previous work with PR8 and TX98 in influenza virus–bacteria superinfection models defined lethal (PR8) and survival (TX98) phenotypes associated with these viruses (81). In the present study, a sublethal dose of either PR8 or TX98 was delivered to mice on day 0, followed by a sublethal dose of GAS on day 7. Differences in weight loss patterns between the two viruses were observed with TX98-infected mice losing weight within 24 h after virus inoculation (Fig. 1A). This weight loss reached its nadir by day 2 or 3 after infection, with subsequent recovery for all infected animals between days 3 and 7. In contrast, PR8-induced weight loss was not consistently observed until day 6 after this sublethal infection. Following inoculation with GAS, both virus groups lost weight within the first 24 h, showing the effects of bacterial inoculation. As predicted (81), all mice in the PR8:GAS superinfection group died, whereas none of the mice in the TX98:GAS group succumbed to secondary bacterial infection (Fig. 1B).
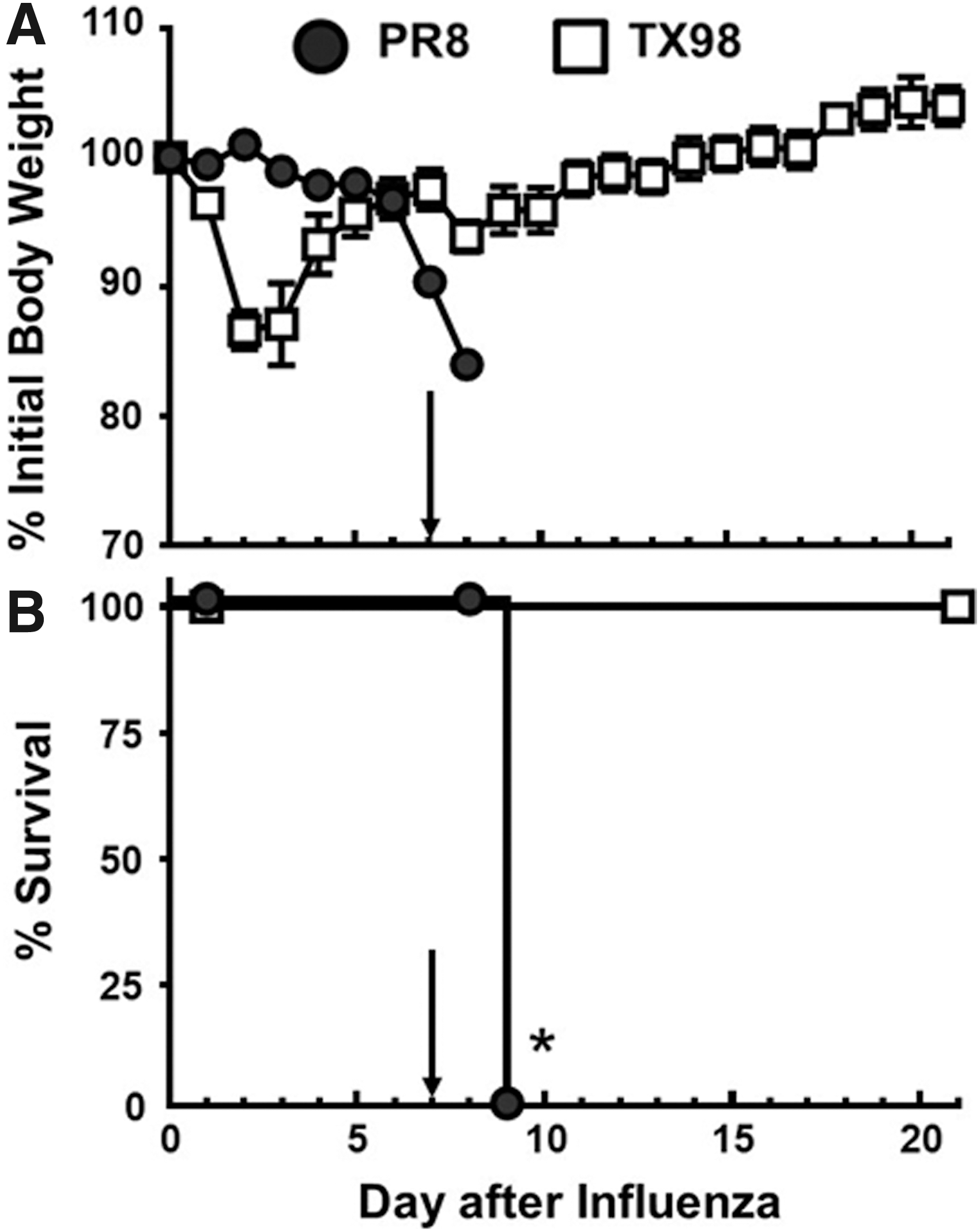
Morbidity and mortality for PR8:GAS and TX98:GAS superinfections in mice. Two groups of mice (n = 8 per group) were inoculated with either PR8 or TX98 on day 0, and subsequently inoculated i.n. with MGAS315 at day 7 (marked by arrow). Weights
We next determined pathogen titers in the lungs and spleens of mice during the course of an influenza virus–GAS superinfection (Fig. 2). Virus titers were equal at day 3 postinoculation for PR8 and TX98 (Fig. 2A), with titers for individual mice ranging between 105 and 107 TCID50/mL. Virus titers in the lungs of mice infected with TX98 were reduced by day 7 postinoculation, whereas PR8 persisted in lungs beyond day 7 after virus infection. In fact, virus could be detected in the lungs of PR8-infected mice 48 h after inoculation with GAS (day 9 postinfluenza). The presence of virus in the lungs of PR8-infected animals until at least day 7 correlated with levels of bacteria detected in the lungs (Fig. 2B) and spleens (Fig. 2C) after inoculation with bacteria, showing that PR8 can influence the persistence of bacteria locally and its spread systemically. These data suggest that survival after PR8- and TX98-induced secondary bacterial infection is associated with the kinetics of virus and bacteria clearance, an observation that prompted us to identify the contribution of host immunity to the differences observed.
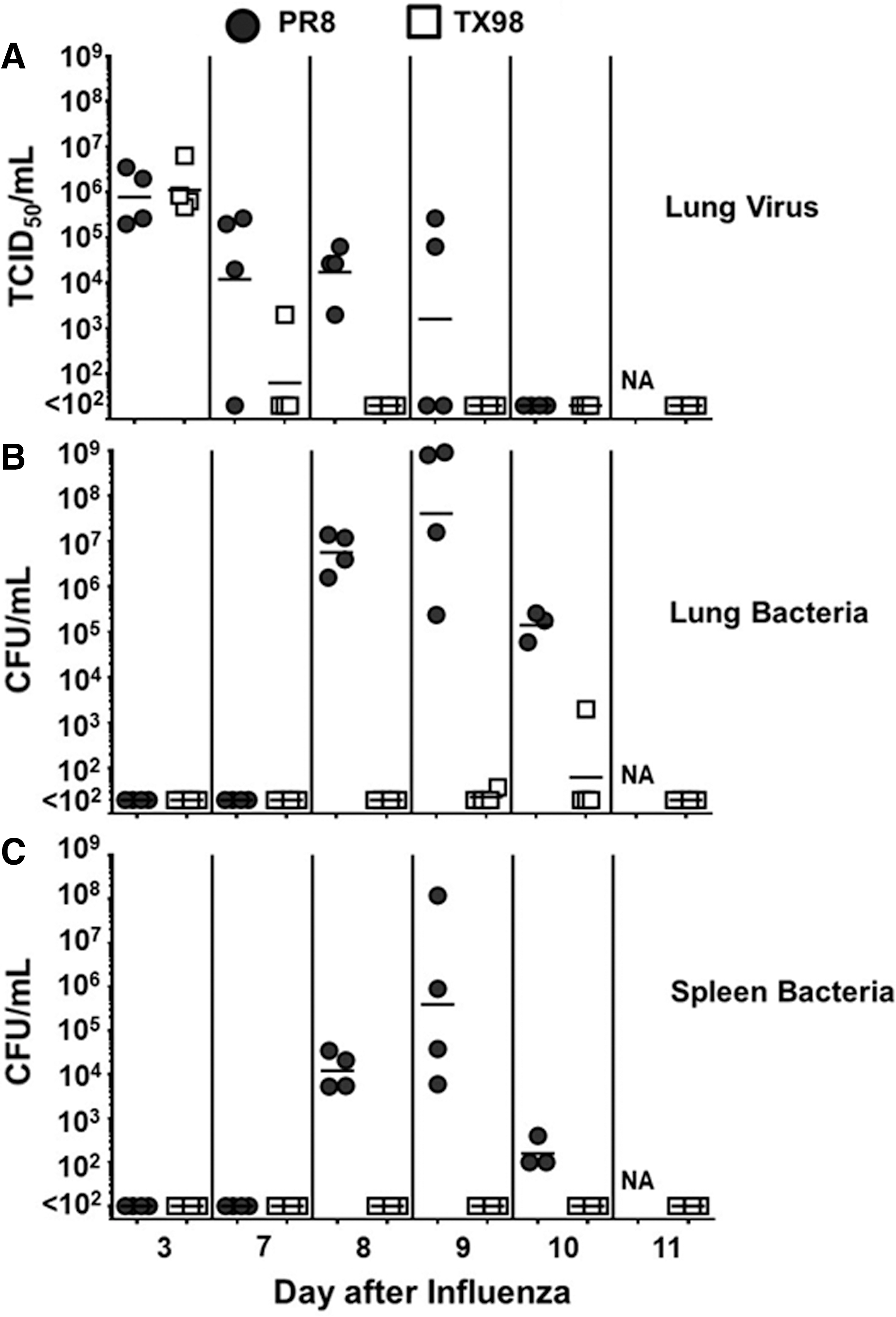
Pathogen titers in the lungs and spleen during a time course of PR8:GAS and TX98:GAS superinfection. Lung viral titers
Host cell responses after TX98 and PR8 infection and bacterial superinfection
Infection with TX98 was associated with an early influx of neutrophils in the lung, whereas the host response to PR8 was not associated with an increase in neutrophils during the first 24 h postinfection (Fig. 3A). As further evidence of this neutrophil influx, myeloperoxidase (MPO) levels in the lungs of mice were significantly increased (p = 0.0056) in TX98-infected mice (7.81 ± 0.39 pg/mL) compared with PR8-infected mice (5.00 ± 0.54 pg/mL). In response to both viruses, macrophage levels in the lungs of infected mice decreased compared with levels before infection, and their levels remained low through day 7 after influenza virus infection (Fig. 3B). The decrease in macrophages was significantly more pronounced in the lungs of TX98-infected mice during the first 3 days postinfluenza, but by day 5 both PR8 and TX98 had similarly reduced levels of macrophages in the lung. Ultimately, at the time of inoculation with bacteria (day 7 postinfluenza), the neutrophil and macrophage populations in the lungs of infected mice did not differ statistically from each other, yet both were significantly below the levels observed before infection.
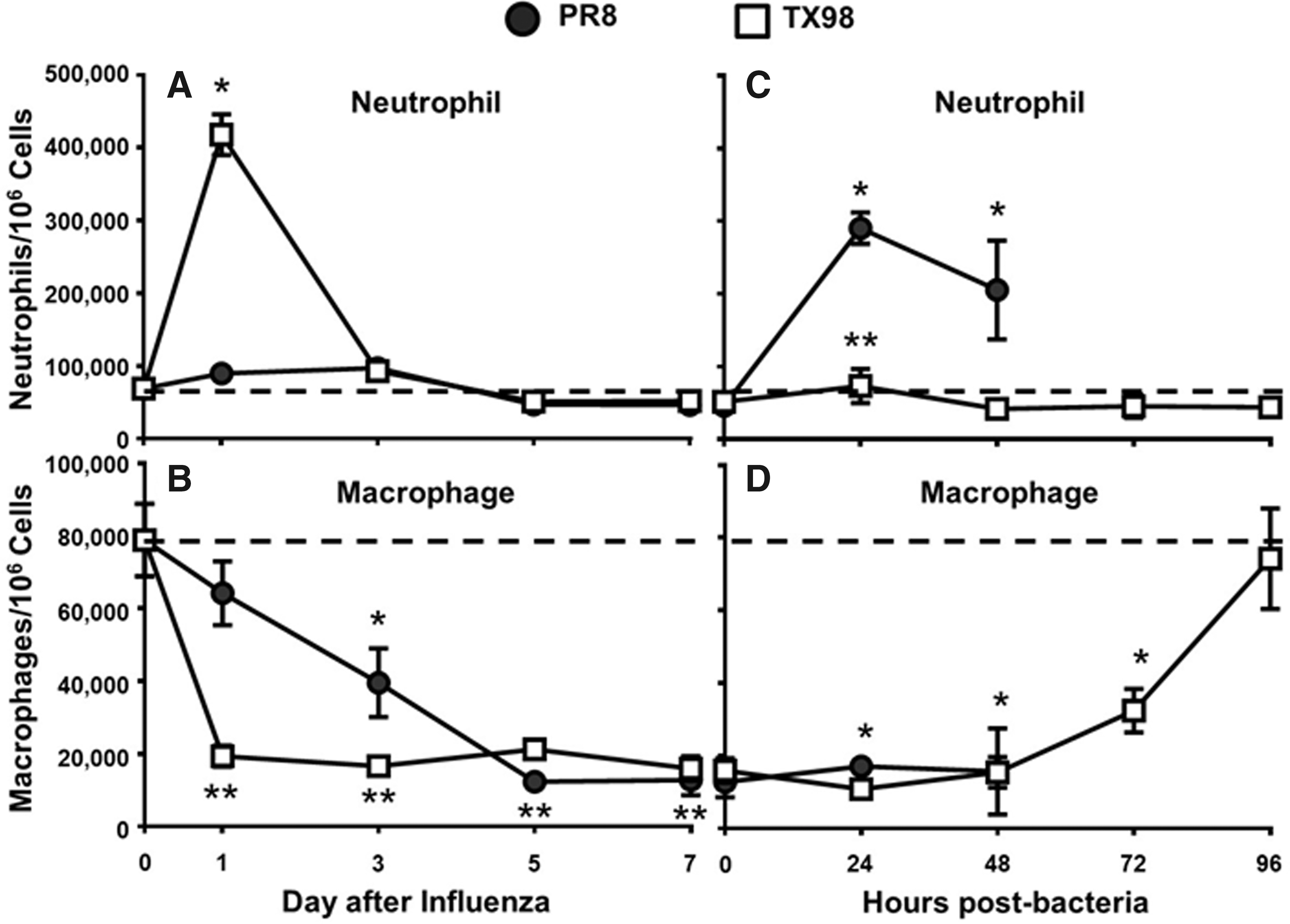
Lung macrophage and neutrophil populations during influenza virus infection and IAV:GAS superinfection. Relative percentage of cells characterized as neutrophils (CD11bhigh, Ly-6Ghigh) or macrophages (CD11chigh, Siglec-F+) from mouse lungs on days 0, 1, 3, 5, and 7 following intranasal inoculation with a sublethal (0.1 LD50) dose of influenza
Virus-infected mice were subsequently challenged with GAS as a secondary bacterial pathogen at day 7 postinfluenza virus infection, and mice that were initially infected with TX98 did not show an increase in the level of either neutrophils (Fig. 3C) or macrophages (Fig. 3D) during the first 48 h postbacteria. However, mice in the PR8-infected group showed a rapid influx of neutrophils that remained high at 24 and 48 h postbacteria (Fig. 3C). Levels of macrophages in the lungs of PR8-infected mice remained lower than their day 0 levels at both 24 and 48 h postbacterial infection (Fig. 3D), and PR8-infected mice succumbed to bacterial superinfection by 96 h postbacteria, in this study. Interestingly, macrophage levels in the lungs of mice infected with TX98 ultimately returned to their initial, baseline levels by 96 h postbacterial infection (day 11 overall), indicating that recovery from superinfection is associated with a return of macrophages to their original level.
Host cytokine responses after PR8 and TX98 infection and bacterial superinfection
Focusing our attention on the period of equivalent virus titers (day 3) and the time of secondary bacterial infection (day 7), we measured IL-4, IL-6, IL-13, IL-17, IL-22, TNF-α, and IFN-γ in lung homogenates. Compared with the TX98-infected mice, at day 3 after virus infection, levels of IL-6, IL-13, IL-22, and IFN-γ were significantly increased after PR8 infection (Fig. 4). Alternatively, TNF-α levels were significantly increased after TX98 infection, compared with PR8. Expression of IL-4 and IL-17 did not differ between PR8 and TX98 at day 3 postinoculation. At day 7 postvirus infection, levels of IL-6, IL-13, and IL-22 were lower than at day 3 postinfection, but remained increased in PR8-infected animals compared with TX98-infected mice. Expression of IL-4, IL-17, TNF-α, and IFN-γ did not differ between the PR8 and TX98 infection groups at day 7, which was when bacteria were introduced.
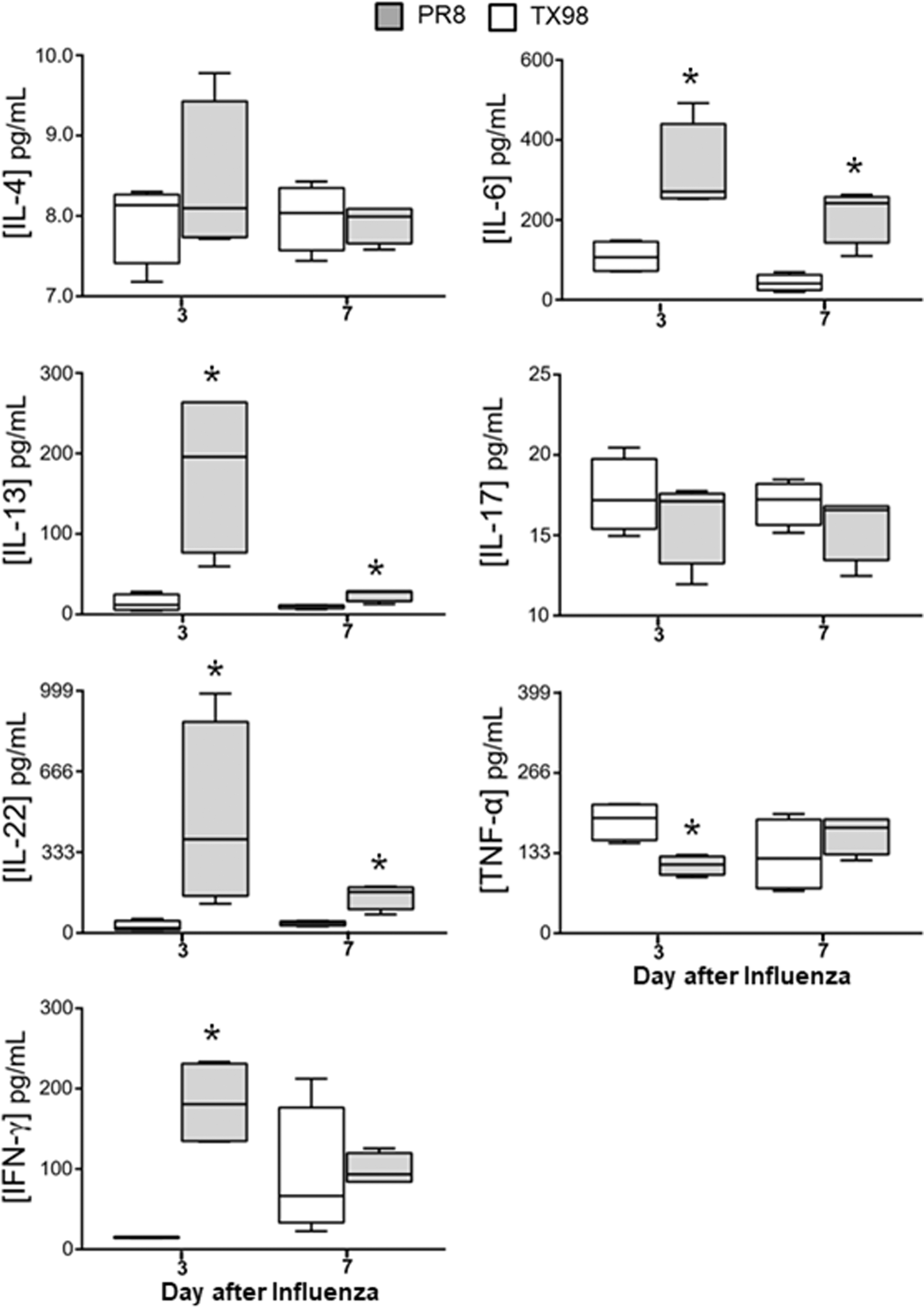
Cytokine expression after influenza virus infection. Mice were inoculated on day 0 with a sublethal (0.1 LD50) dose of either PR8 or TX98 and lungs were collected from mice on days 3 and 7 after influenza virus infection. White (TX98) and gray (PR8) boxes represent the cytokine levels for each group (n = 4 mice per group), with the mean represented by the horizontal line and the whiskers showing the range of values. Groups were compared using an unpaired Student's t-test, with significant difference indicated when p < 0.05. *Denotes a significant difference when compared with TX98.
After inoculation with bacteria on day 7, PR8-infected mice showed a significant increase in the expression of the cytokines IL-4, IL-6, TNF-α, and IFN-γ within 24 h postbacteria (Fig. 5), and the levels of these cytokines remained high throughout the duration of the superinfection for the PR8-infected group. This increase in cytokines coincided with an influx of neutrophils into the lungs of PR8-infected mice (Fig. 3C). Levels of the cytokines IL-13 and IL-22 were also elevated in PR8-infected animals with statistically significant levels at 48 h postbacteria. Cytokine detection at 96 h postbacteria could only be performed for TX98-infected mice, as all of the PR8-infected mice had succumbed to bacterial superinfection by this point in the study. Our results show that host cellular and molecular immune responses against PR8 and TX98 contribute to the differential outcomes observed in a superinfection model, and our next goal was to identify virus genes that may contribute to increased severity of a bacterial superinfection.

Cytokine expression after influenza virus and secondary bacterial infection. Mice were inoculated on day 0 with a sublethal (0.1 LD50) dose of either PR8 or TX98 and inoculated on day 7 with a sublethal dose of Streptococcus pyogenes. Lungs were collected from mice at 24, 48, and 72 h for all groups, and at 96 h postbacteria for mice in the TX98-infected group. White (TX98) and gray (PR8) boxes represent the cytokine levels for each group (n = 4 mice per group), with the mean represented by the horizontal line and the whiskers showing the range of values. Groups were compared using an unpaired Student's t-test, with significant difference indicated when p < 0.05. *Denotes a significant difference when compared with TX98. NA is indicated because all mice had succumbed to infection.
Characterization of TX98 and PR8 reassortants
To determine the individual virus gene segments that may contribute to survival, reverse genetics (23,25,45) was used to create reassortant viruses containing gene segments from both TX98 and PR8 viruses in the combinations presented in Figure 6. Viruses created by reverse genetics were characterized in both MDCK cells and mice, with TCID50 and LD50 results reported (Table 1). Our reassortant viruses grew in MDCK cells with values ranging from 106.38 to 109.17 TCID50/mL, indicating a range of infectivity that corresponded with propagation of the parental viruses in tissue culture. Similarly, when we evaluated individual LD50 values, our results show that all of these reassortant viruses could cause a lethal infection in mice, with a range similar to that established by the TX98 (106.625 TCID50) and PR8 (102.55 TCID50) parental viruses. Since all of these viruses established a lethal infection in mice, we evaluated individual genes that modulate superinfection severity in our model.

Reassortant viruses created to test single gene segment contribution to survival in influenza–GAS superinfections. White (TX98) and gray (PR8) boxes indicate the source of virus gene segments.
Contribution of the HA gene segment toward survival after superinfection
When we tested the TX98 virus expressing the PR8 HA gene, we observed LD50 values that were similar to that of the wild-type PR8 virus (Table 1). Similarly, when included in our influenza virus–bacteria superinfection model (Fig. 7), both the weight loss and survival profiles for this virus were similar to those observed with wild-type PR8 (Fig. 1). To further demonstrate that the HA gene can contribute to the outcome of a superinfection, we created a PR8 virus that expressed the TX98 HA on its surface. This virus had an LD50 value that was similar to TX98 (Table 1), and induced a superinfection resembling what was observed after TX98 (Fig. 1). Furthermore, when lung virus titers were detected on day 7 after virus infection (Fig. 8), the TX98-HA virus showed reduced titers at day 7, which was similar to the wild-type TX98 virus (Fig. 2). Alternatively, the PR8-HA virus (Fig. 8) had significantly increased lung virus titers that were similar to the parental PR8 virus (Fig. 2). These data indicate that the HA gene alone can confer the lethal (PR8-like) or survival (TX98-like) phenotypes observed during a superinfection, with similar weight and virus titer kinetics observed.
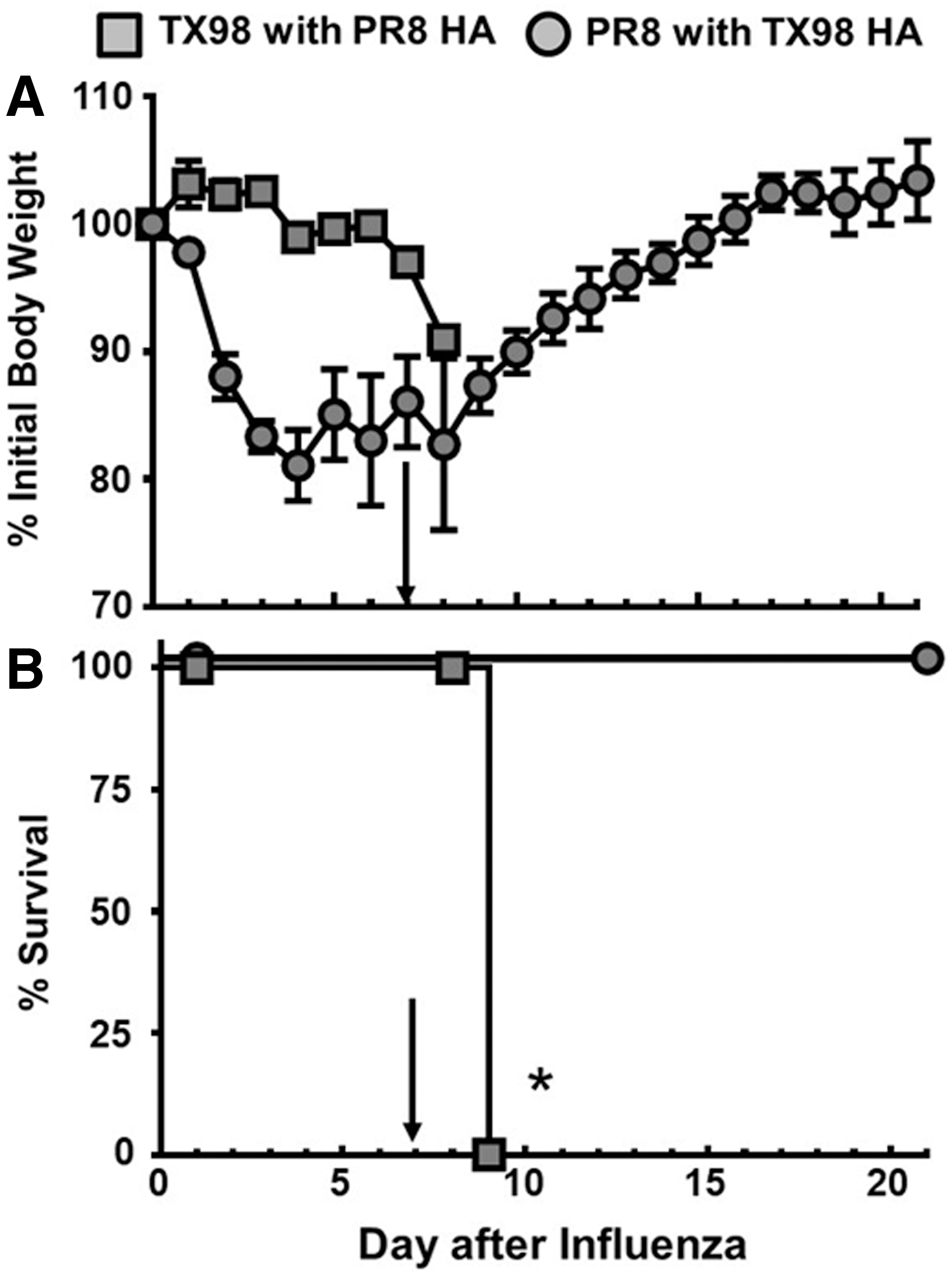
Morbidity and mortality for HA reassortants in IAV:GAS superinfections. Groups of mice were inoculated with either TX98 7:1 PR8 HA (n = 8) or PR8 7:1 TX98 HA (n = 4) at day 0, and subsequently inoculated with MGAS315 at day 7 (marked by arrow). Weights
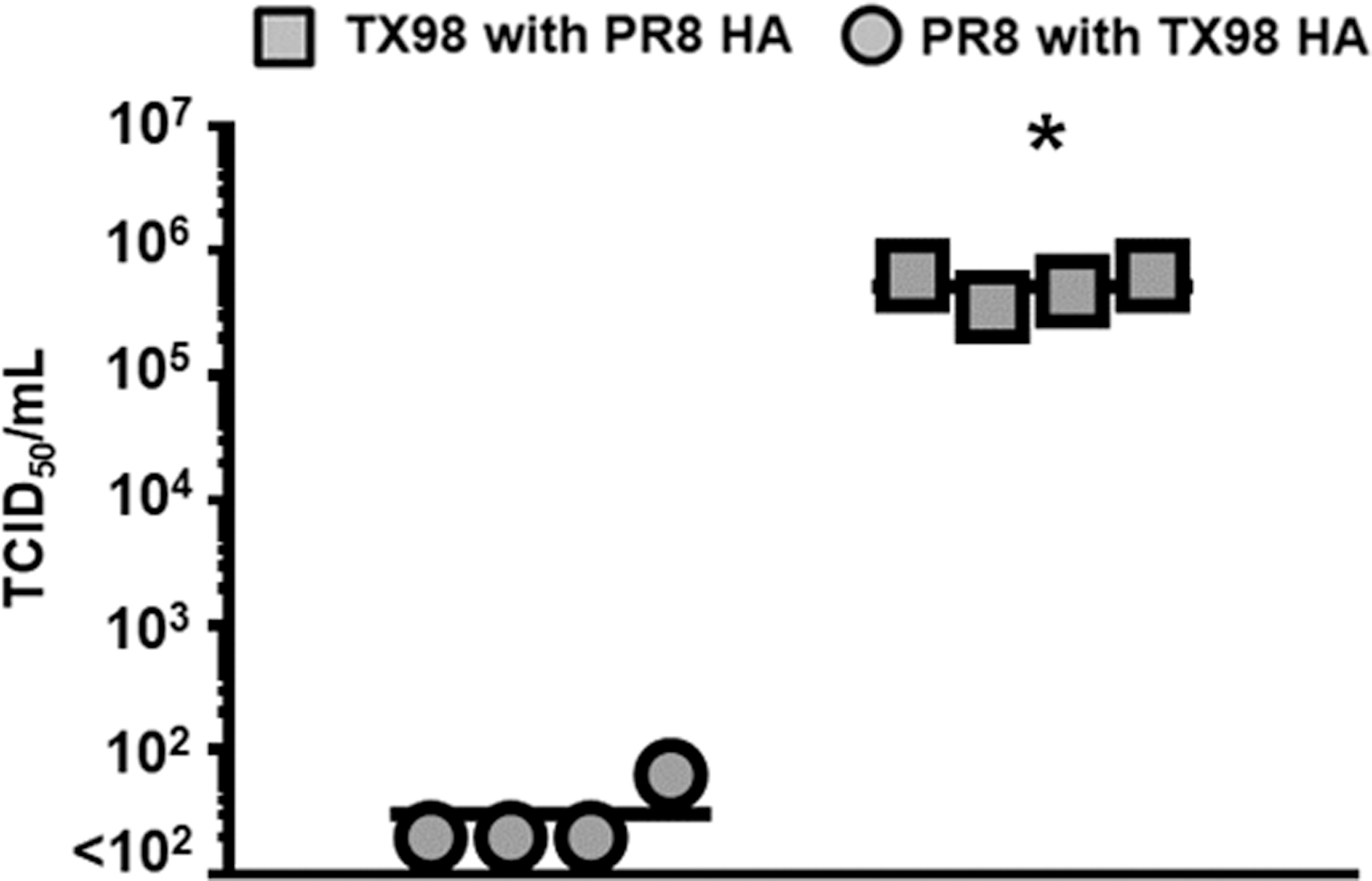
Lung viral titers in PR8- and TX98-HA reassortant viruses in IAV:GAS superinfections. Lung viral titers were measured on day 7 after infection with either TX98 7:1 PR8 HA or PR8 7:1 TX98 HA using TCID50. Each data point represents the lung viral titer from an individual mouse, and the mean viral titer for each group is represented by a horizontal bar. All groups contained four mice. *Indicates a significant difference between the titers (p < 0.05) using one-way ANOVA followed by Tukey's multiple comparison test. ANOVA, analysis of variance; TCID50, 50% infectious dose in tissue culture.
Non-HA virus gene contributions to superinfection
To complete our evaluation of reassortant viruses within our superinfection model, we tested additional individual gene reassortants (Fig. 6 and Table 1). Interestingly, viruses that expressed either the PR8 NS gene or the PB1 gene, on the TX98 background, induced significant weight loss in mice after infection (Fig. 9). The virus expressing the NS gene induced a significant mortality after superinfection (20% survival, Fig. 9B), whereas the virus expressing the PR8 PB1gene yielded 75% survival (Fig. 9D). The remaining individual TX98 gene reassortant viruses expressing the PB2, PA, NP, NA, and M segments from PR8 yielded at least 75% survival, which was more like the wild-type TX98 virus than the wild-type PR8 virus (Fig. 1).
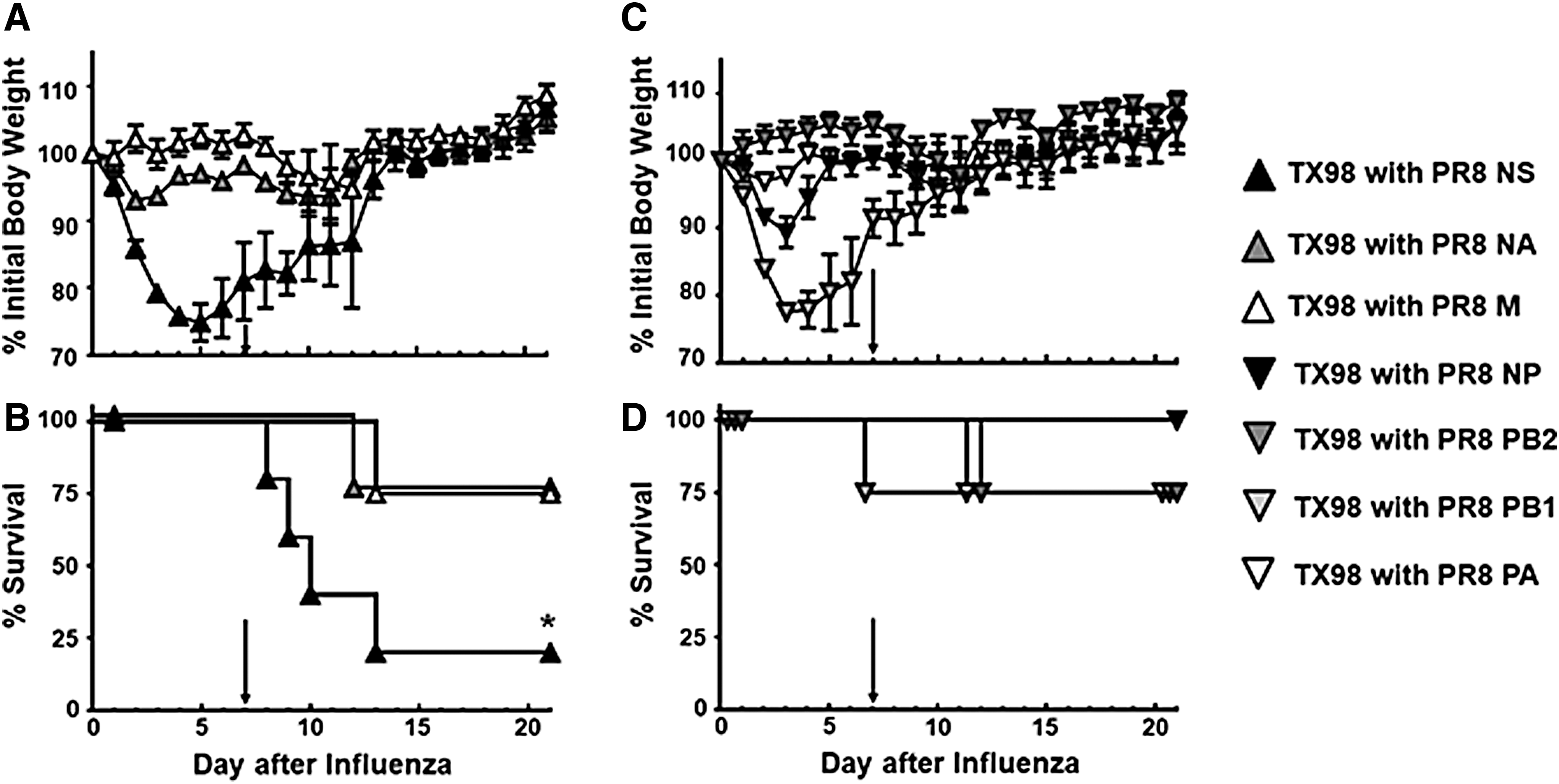
Morbidity and mortality for TX98 reassortant viruses expressing individual PR8 genes in our influenza virus–GAS superinfection model. Groups of mice were inoculated with 0.1 LD50 of TX98 viruses expressing either PR8 PB2 (n = 4), PR8 PB1 (n = 4), PR8 PA (n = 4), PR8 NP (n = 4), PR8 NA (n = 4), PR8 M (n = 4), or PR8 NS (n = 15) in 100 μL i.n. on day 0. Seven days later, mice were inoculated with 0.1 LD50 MGAS315 in 100 μL i.n. (marked by arrow). Weights
Discussion
We recently showed that infection with the primary swine influenza virus isolate TX98 is associated with a less severe secondary bacterial infection than is observed with the PR8 influenza virus (81). Based on this observation, we now present data related to pathogen load, host lung cellular infiltrates, cytokines, and viral genes that correspond with susceptibility to secondary bacterial infections. Specifically, in this study, we designed experiments to identify host immune responses as they relate to virus and bacteria titers in lungs and spleens of mice after infection, and identified the influenza virus HA as a contributor to the phenotype observed. In this study, we show that during the course of a PR8 superinfection, there were delays in virus clearance that were associated with GAS in the lungs and systemic spread of bacteria into the spleen. Alternatively, TX98 superinfections were characterized by early viral clearance and a relative lack of GAS in the lungs and spleen after superinfection. These pathogen levels corresponded with differences in both the cellular infiltrates and the cytokine profiles that may ultimately hint at mechanisms of pathogen clearance. We will discuss our findings of viral genes and host responses that contribute to superinfection severity, with emphasis on the contributions of the HA gene toward the phenotypes observed.
Results from our evaluation of host responses against TX98 and PR8 indicate that host neutrophils are recruited to the lung more rapidly in response to infection with the TX98 virus, compared with the PR8 virus. This observation may explain the differences in pathogen clearance kinetics and, ultimately, the severity of secondary bacterial infections. Specifically, we interpret these findings to indicate that early recognition of influenza viruses, presumably at the stage of HA interaction with host cells, can have profound effects on superinfection outcomes. One possible explanation for this finding could be that influenza viruses with increased N-linked glycosylation sites appear to be more prone to recognition by carbohydrate-binding proteins, such as C-type lectins (20,60). It has been reported that increasing the number of glycosylation sites on HA, which we observe with TX98, can mediate early host detection through lectin binding, subsequent neutrophil recruitment, and incidental lung damage during early host responses (4,46,60,77). The TX98 HA has a predicted seven potential glycosylation sites, which may allow viruses expressing this HA to be more readily recognized by C-type lectins. This recognition could potentially lead to virus aggregation, complement fixation, opsonization, phagocytosis, and the induction of inflammatory responses as it is recognized by cells of the innate immune system (4 60,72). Alternatively, the PR8 HA only expresses five potential sites for N-linked glycosylation, which may allow it to infect a host and replicate with delayed recognition by the innate immune system (43). Both our cellular and molecular (MPO) data show that early neutrophil recruitment occurs after TX98 infection, but not after PR8 infection, further hinting that these additional sugar moieties may play a role in early immune responses. Future work will evaluate interactions between the HA expressed by these viruses, and the subsequent immune response induced, with consideration for the glycosylation state of the HA expressed, which we plan to define using point mutations within the individual HA genes (77).
Separate from glycosylation, the H3 of TX98 is derived from a wild-type influenza virus, whereas the H1 of PR8 comes from a mouse-adapted, laboratory strain (81). Serial passaging of influenza viruses can increase the affinity of the HA for the sialic acid residues present in the host, and it is likely that the mouse-adapted PR8 HA has a higher affinity for murine sialic acid receptors, compared with the swine-origin TX98 HA (15,32). Despite this, increased receptor binding may not fully explain the observed delay in clearance of viruses possessing the PR8 HA or their contributions to superinfections. Differences between these viruses in the fusion events mediated by the stalk region of the HA should be considered, especially since we know that this region can contribute to virus infectivity and host cell tropism by altering the pH of fusion (19,32,36). Previous studies have shown PR8 HA fusion to be activated below pH 5.8, and recent work in classical swine HAs isolated before the 2009 pandemic suggest that the TX98 H3 has a fusion pH between 5.5 and 5.8 (10,34,64). We have initiated studies to determine the specific contributions of the globular head and the stalk domains of the influenza virus HA toward superinfection outcomes.
As evidence of the impact that HA can have on host immune responses against an invading pathogen, we observed an early influx of neutrophils into the lungs of mice infected with TX98 that we did not see after PR8 infection. This increase in neutrophils is transient during the initial virus infection, but induction of this cell population, and the cytokine environment that develops, has apparent effects on severity of a secondary bacterial infection. This is evidenced by the absence of neutrophils in the lungs of TX98-infected mice after inoculation with bacteria, and the rapid clearance of bacteria from the lungs. Alternatively, PR8-infected mice do not have a neutrophil response after virus infection, but suffer from rapid neutrophil influx after bacteria inoculation that is associated with death after secondary infection. Since these changes in host cell and cytokine populations are observational at this stage, we can only speculate that these specific host responses dictate severity of a secondary bacterial infection through fine-tuning of the host response as early as 24 h after virus infection.
These host–virus interactions correlate well with previous research into the impact of IL-13 and type I IFN expression on superinfection outcomes (13,65), as there appears to be an impact on superinfection outcomes that relies on interactions between the host and the virus well before bacteria induce their effects. Along with our previous work comparing PR8 and TX98 (81), in this study we show that the virus used to infect the host can influence the severity of a secondary bacterial infection. Our initial evaluation of TX98 showed that survival after secondary infection with S. pyogenes could be observed even when bacteria were delivered at 5 days postinfluenza virus infection, which is important because this is a time when both virus and bacteria were present in the lungs of infected animals (81). Interestingly, work by our group has shown that protective effects can be observed when S. aureus bacteria are delivered at day 3 after PR8 infection (66,68), but that this protection is lost by day 5 for both S. aureus and S. pyogenes (81). This implies a potential need for future research to identify therapeutic, antiinflammatory interventions that can be administered to extend the window of protection and, ultimately, limit secondary bacterial infections.
The contribution of host macrophages toward severity of a secondary bacterial infection has been studied for decades (5,47,48,79,82), and this cell population appears to be strongly influenced by the cytokine environment after influenza virus infection (6,29,30,62,70). Specifically, a loss of macrophage cell function has been observed historically within superinfection models (5,28,78), with evidence of defects in phagocytosis of bacteria after PR8 infection (70). Interestingly, both PR8- and TX98-infected mice had similar levels of macrophages in their lungs at the time of secondary bacterial infection, hinting that the functionality of these cells contributes to the severity of these secondary infections. We observed an increase in IL-27 early after TX98 infection (data not shown), which can induce expression of suppressor of cytokine signaling (SOCS) by host cells, a host factor that might dampen global cytokine responses against TX98 (52,59,71) and aid in recovery from primary virus infection. In fact, our data show that PR8 induces a strong, progressive, proinflammatory cytokine response that is not seen after TX98 infection, which may be due to an absence of SOCS induction. Thus, it is possible that early IL-27 expression may contribute to both rapid virus clearance and prevention of secondary bacterial infection in a way that could be used to improve clinical outcomes. It is worth noting that both cytokine and host cell contributions during influenza virus-associated secondary bacterial infections in pigs show that severe pulmonary lesions develop as a consequence of proinflammatory responses (31,33,58). Thus, additional work is needed to define the differential host responses that direct outcomes in our model, and findings in pigs support further evaluating the pathogenesis of secondary bacterial infections beyond the mouse model (40,53).
In this study, we provide evidence that influenza virus infections are complicated by secondary bacterial species due to host cell and cytokine responses that develop after influenza virus inoculation. Our data show that a neutrophil influx early after TX98 infection is associated with rapid clearance of virus from an infected host, and reduced severity of a secondary bacterial infection. Interestingly, the similarities between the host neutrophil and macrophage populations at the time of inoculation with bacteria, and subsequent differences in responses to the bacterial invader, indicate that cytokine environments and cellular functionality may influence the severity of a secondary bacterial infection. Thus, we conclude that the immune environment established by TX98 reduces the proinflammatory response that develops against a subsequent GAS infection, and allows mice to survive a superinfection. While we have largely focused our discussion on HA contributions in this study, our data with the NS and PB1 reassortant viruses (Fig. 9) indicate that HA is not the only virus gene that can contribute to illness and susceptibility to secondary infection. Future work will explore the contributions of NS1 and PB1, including PB1-F2 (81), toward host immune responses associated with severe secondary bacterial infections. Moreover, we will work toward understanding virus-induced immune responses to identify anti-inflammatory therapies that can be used to treat PR8-like superinfections so that they yield TX98-like outcomes.
Footnotes
Acknowledgments
The authors thank Richard J. Webby (St. Jude Children's Research Hospital, Memphis, TN) for providing both the plasmids and the viruses that were used in this study. The work presented was funded by start-up funds from by the Division of Basic Biomedical Sciences at USD (V.C.H.), the USD Foundation (V.C.H.), the Sanford School of Medicine Research Committee (V.C.H.), the Sanford School of Medicine Medical Research Committee (J.M.K. and T.W.), a New Faculty Development Award through the Office of Research and Sponsored Programs (ORSP) at USD (V.C.H.), the U. Discover program at USD (T.E.B., T.W., and J.M.S.), and an Inside TRACK award from the ORSP at USD (V.C.H.). Further support was provided by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) (P20GM103443, V.C.H. and A.B.), the National Center on Minority Health and Health Disparities of the NIH (P20MD001631-06, T.J.G.), the National Institute of Allergy and Infectious Disease of the NIH (R15AI094437-01A1, M.S.C. and R44AI117976-01A1, V.C.H.), the BioSNTR which is supported by the National Science Foundation Established Program to Stimulate Competitive Research (NSF-EPSCoR) under grant number IIA-1355423, and the Governor's Office of Economic Development of the state of South Dakota (V.C.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.