
Abstract
Functional immunological evidence supports the impact that the host genetic variability has on the susceptibility to develop asymptomatic or symptomatic dengue infection. Children are more prone to develop severe dengue. Thus, we have evaluated possible associations between single-nucleotide polymorphisms (SNPs) located in immune genes and the development of symptomatic dengue in children from two Colombian populations with differences in genetic backgrounds and geographical features. We genotyped 15 SNPs (in 12 genes) in 298 symptomatic children and 648 healthy controls. Ancestry proportions (APs) were inferred by genotyping 29 ancestry informative markers. We observed four SNPs associated with susceptibility to develop dengue in NOD1, RIPK2, MICB, or PLCE1 genes. Conversely, we found one SNP in TNF gene and two haplotypes in the IKBKE gene associated with resistance to develop dengue. These associations were adjusted by gender, APs, and the population of origin because the association of polymorphisms may be different in admixed populations like Colombian. To our knowledge, this is the first reported association study with dengue in IKBKE, RIPK2, and NOD1 genes. We have also confirmed previously reported associations in MICB and PLCE1 genes with dengue. Overall, our results contribute to the understanding of the genetic susceptibility/resistance to develop symptomatic dengue. Nevertheless, these associations must be validated through functional analysis.
Introduction
D
Many in vivo and in vitro functional studies have shown a variety of immune mechanisms that respond to DENV infections. During DENV infection, the intermediate form, DENV-RNA, is recognized by the endosomal Toll-like receptor (TLR) 3, which triggers the NF-κB pathway. In this pathway, IKBKE phosphorylates TBK1 and NFKBIA, activating NF-κB (10). Similar to TLR, the nucleotide-binding domain, leucine rich containing proteins NOD1 and NOD2 recognize ssRNA+ virus, like DENV, and interact with RIPK2, which induces the activation of NF-κB and mitogen-activated protein kinases signaling pathways (14). The JAK/STATs pathway activation is a potent inhibitor of DENV infection (10). These signaling pathways lead to the upregulation of pro-IL-1β, pro-IL-18, IL-6, tumor necrosis factor alpha (TNFα), IFNα/β, IL-10, and IFNγ, which results in increased vascular permeability in primary and secondary DENV infections (30).
Moreover, STAT3 phosphorylation by PLCE1 mainly mediates inflammatory cytokines (IL-6, TNFα, and IL-1β) expression and release (35). The JAK/STAT3 pathway is activated in hepatocytes infected by DENV (29). In addition to the upregulation of proinflammatory cytokines, there are other critical points involved in dengue complications. These include the apoptosis of DENV-infected cells by macrophage induction, the shift in the expression of proinflammatory cytokines (Th1 response) to anti-inflammatory cytokines (Th2 response) (17), and the activation of CD8 + T and NK cell receptors through interaction with MICB (16).
Single-nucleotide polymorphisms (SNPs) in genes encoding regulatory proteins of the antiviral immune pathways, such as JAK1 (rs11208534 and rs17127114) and IKBKE (rs1059704 and rs11118132), have been associated with dengue hemorrhagic fever (DHF) in a Brazilian population, although SNPs in IKBKE gene did not have a significant association after adjustment by the false discovery rate (FDR) method (25). Similarly, some SNPs in genes encoding cytokines such as IL10 (rs1800871) and TNF (rs1799964) genes have been associated with DHF in Indian populations (1,2). Also, the SNPs rs3132468 in the MICB gene and rs3765524 in the PLCE1 gene were associated with dengue shock syndrome (DSS) in a Genome-Wide Association Study (GWAS) in Vietnamese (13) and Thai populations (9). Conversely, the same SNPs were also associated with non-SD (33).
The associations of polymorphisms in the genes mentioned above, and other immune genes, with diseases may be different in recently admixed populations, such as Colombian. These differences can be explained by geographical heterogeneities, marked regional differences in pre-Columbian population densities, and the extent of past European and African immigration events (24). In fact, individuals with high amount of African ancestry may be less susceptible to severe forms of dengue (5). Therefore, this study aimed at discovering markers for susceptibility, or resistance, to develop symptomatic dengue. We used children from two Colombian populations, which have different ancestry and have been exposed to different environmental characteristics.
Materials and Methods
Subjects
We used a case–control design including samples from two Colombian administrative departments (i.e., states) with many known cases of DENV. (a) Huila: included 176 dengue cases (DG) and 311 healthy controls (HC), sampled from 2010 to 2012. (b) Antioquia: included participants of two outbreak periods 2006–2009 (61 cases and 176 controls) and 2010–2012 (60 cases and 161 controls). These departments are located southwest (Huila) and northwest (Antioquia) of the Colombian Andean region and have different climatic features. All participants (DG and HC) were Colombians by birth and with ancestors from Antioquia or Huila, respectively, at least three generations back. The participants were not related to each other. DG included pediatric patients from the Hospital Universitario Hernando Moncaleano Perdomo (UHHMP) in Neiva (Huila) and from several medical centers in Medellin (Antioquia): Hospital General de Medellin, Hospital Pablo Tobón Uribe, Hospital Marco Fidel Suarez, Hospital San Vicente Hospital, and Sura EPS.
Dengue specialists classified patients younger than 15 years as cases in the Colombian Institute of Tropical Medicine of Medellin and the UHHMP of Neiva, following the latest criteria of the WHO 2009 (34). All cases were either IgM or IgG positives. In Huila, 95.3% were IgM positive and 72.2% were IgG positive, whereas in Antioquia, 97.5% were IgM positive and 25.2% were IgG positive. Moreover, all DG were diagnosed by a clinical examination, with DENV detected in some of them. Immunological tests were performed in the outbreak period. Serological confirmation was performed using IgM- and IgG-specific ELISA (Kit Panbio® Dengue Duo IgM and IgG Capture ELISA; PanBio Diagnostics). Most children DG patients in Antioquia were classified as DWWS (57.4%), followed by SD (7.4%), and one case with DWOS. Meanwhile, in Huila, 58% of children DG patients were classified as DWWS, followed by SD (35.2%), and two patients with DWOS. The 34.4% and 5.0% of DG cases in the samples of Antioquia and Huila, respectively, could not be classified by lack of clinical information and were confirmed with laboratory tests.
HC between 10 and 40 years were residents in Huila and Antioquia during previous dengue epidemics. HC also have grandparents from the region and were not related to each other or the study cases. We included some adults as HC to complete a minimum number of HC (not enough HC children were permitted by guardians to participate in the study), with the added benefit that HC adults have been more exposed to DENV than the HC children and cases in these endemic areas. The healthy condition of these adults is an excellent source of validation of the impact of the genetic makeup in the resistance to develop the disease. We would like to note that all four DENV serotypes were reported in the study areas during the sampling time frames.
Serological confirmation was not performed in HC. However, all HC were requested to report any bleeding or other known severe symptoms of dengue during their lifetime. Besides, they had to indicate the existence of any consultation or hospitalization that they have had in suspicion of dengue. There were no positive reports to these inquiries. Thus, no HC has a record of admission for dengue during their lifetime.
DNA extraction and SNPs genotyping
DNA extraction was performed from white cells by either salting out or phenol–chloroform methods. DNA quality and quantity were assessed using NanoDrop 2000c (ThermoFisher Scientific). For the genetic ancestry determination, samples were then genotyped with 29 ancestry informative markers (AIMs) sets using capillary electrophoresis and the polymerase chain reaction-restriction fragment length polymorphism technique (PCR-RFLP) with conditions as described in another Colombian study (5). For the association study, candidate SNPs were obtained from the Human Genome Diversity Project, International Human Haplotype Map Project, OMIM, and PubMed. We considered the localization (promoters, 3′/5′ UTRs, introns/exons of genes for immune response) and previous genetic associations with dengue, viral infections, or acute inflammatory responses. As a result, we selected two SNPs located in exons, nine located in introns, and four in regulatory regions (Supplementary Table S1; Supplementary Data are available online at
We included 15 SNPs in 12 genes related to different functionalities: cytokines (TNF: rs1799964; IL8: rs4073, rs2227307; IL10: rs1518111; IL18: rs5744247, rs1834481), viral recognition (NOD1: rs5743336), activation of immune response pathways (IKBKE: rs1059704, rs11118132; JAK1: rs11208534), migration of dendritic cells (CCR7: rs2023906), and immunopathological responses inducing SD manifestations (MICB: rs3132468; RIPK2: rs40457; CSF3: rs2227319; PLCE1: rs3765524). The SNP genotyping was performed by PCR-RFLP using Taq DNA polymerase (Fermentas) and the protocol described by the manufacturer. The polymerase chain reaction products were digested by restriction endonuclease enzymes indicated in Supplementary Table S1.
Statistical analyses
Individual ancestry proportions (APs) and the average for each sample were estimated using the allele frequencies of the 29 AIMs and the allele frequencies of ancestral populations: CEU, CHB, and YRI as European, Amerindian, and African ancestries, respectively. Ancestral allele frequencies were taken from the 1000 Genomes Project (
The association with symptomatic dengue was performed with unconditional multiple logistic regression using only SNPs in Hardy–Weinberg equilibrium (χ2 test), and SNPs with <30% missing values in the HC group. Confounding continuous variables such as age and European, Amerindian, and African APs (Eur-AP, Ame-AP, and Afr-AP) were compared between the DG and HC groups by the Mann–Whitney test, and possible differences in gender were assessed with Fisher's exact test. The adjusted regressions by confounding variables were presented like adjusted odds ratio (aOR) and confidence interval (CI). Allelic, genotypic, and allele carrier regressions were adjusted for multiple comparisons by FDR. Haplotype, unconditional multiple logistic regression analysis was performed with associated SNPs in linkage disequilibrium (LD), as given by the r2 and D′ statistics and χ2 tests in the HC group. The allelic phases were estimated by the expectation–maximization method, and the analysis of haplotype associations was performed with the haplo.stats package of R.
A posteriori power calculation was performed considering the (a) weighted average prevalence of dengue in Antioquia and Huila during the respective years of sampling, calculated based on data from the SIVIGILA, Instituto Nacional de Salud of Colombia* (b) population sizes of the departments, according to DANE** (c) assumption of additive genetic model; (d) minor allele frequency (0.175–0.40); (e) D′ = 0.95 between markers and SNPs associated with disease; and (f) cases–controls proportions in each sample (Fig. 1). The power calculation was performed in the Genetics Design package of R (32). All statistical analyses were performed in the R software v.3.4.1. †
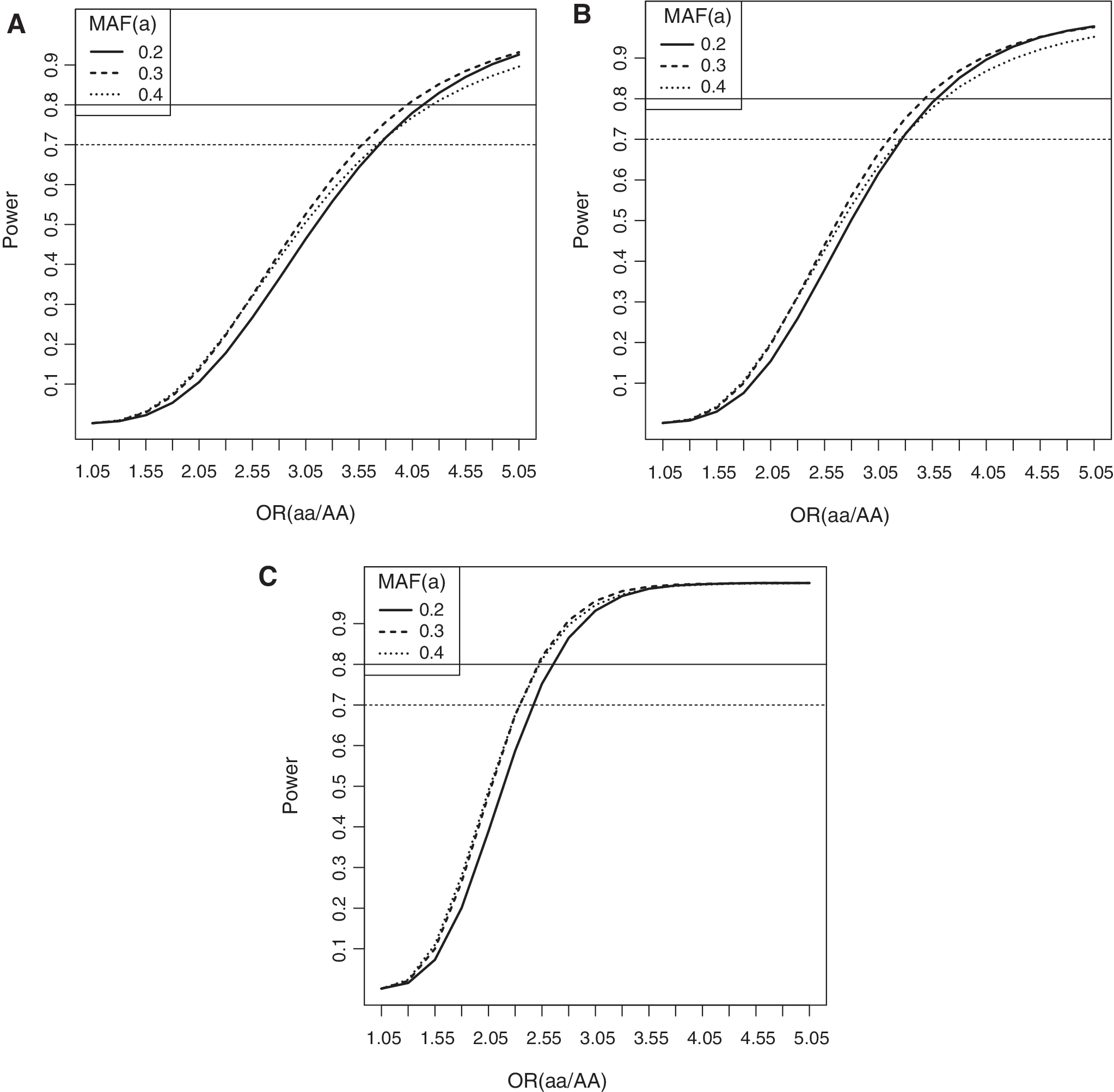
Power statistical estimations for association tests in the samples:
Ethical considerations
The study was performed following the ethical standards of the Declaration of Helsinki. The ethical approval was obtained from the Research Unit and Bioethics Committee of the University of Antioquia, Colombia (act no. 09-12-225). The study was classified as of minimal risk (Art. 11, Resolution 008430-1993, Colombian Ministry of Health). All the subjects and their legal guardians were informed of the study and gave their written informed consent to participate in the study.
Results
Populations of study and environmental features
We sampled 298 symptomatic children (DG) and 648 HC with the following characteristics (Table 1), briefly: age was different between DG and HC in the Antioquia and Huila samples departments (p < 0.0001 for both). Age was not used as a confounder because 75.7% of HC were adults. The proportion of males/females was significantly different between the DG and HC groups in both Antioquia and Huila (p = 0.0003 and p = 0.0278, respectively). There were significant differences among the ancestral proportions between the DG and HC in Antioquia (Eur-AP: p < 0.0001; Ame-AP: p < 0.0001; and Afr-AP: p = 0.0096). In Huila, only the Eur-AP was different between the DG and HC (p < 0.0001). Due to differences in gender and ancestral proportions, these variables were used as confounders in the association analysis. We observed that the Eur-AP was similarly high in the samples of Antioquia and Huila. The second largest component was Afr-AP in Antioquia, whereas in Huila, the second one was Ame-AP (Table 1).
Gender (male/female) is presented in absolute values. Statistical significance was set at p < 0.05.
Continuos variables presented in median (interquartile range: Q3–Q1).
Mann–Whitney test.
Fisher's exact test.
HC, healthy controls; DG, dengue cases; Eur-AP, European ancestry proportion; Ame-AP, Amerindian ancestry proportion; Afr-AP, African ancestry proportion.
Samples were taken from two dengue endemic areas, Huila and Antioquia departments. The sampling included one outbreak in Huila and two in Antioquia. Then, we evaluated potential differences in ancestry, the frequency of genotypes, and dengue severity between two outbreak periods. We observed differences only in European and African APs (Supplementary Tables S2, S3, and S4). However, the adjusting for APs had already been taken into account. Moreover, association analyses with the sample of Antioquia adjusted by outbreak or not yielded similar results (Supplementary Table S5). Therefore, the sampling in two periods in Antioquia does not represent a variable to consider like a confounder. There are climatic variations between Antioquia and Huila. The average precipitation was higher in Medellin, Antioquia, than in Neiva, Huila (2,050 and 1,712 mm, respectively). The average annual temperature was lower in Medellin than in Neiva (22.7°C and 27.7°C respectively), during the respective periods of sampling (
SNPs associated with the development of symptomatic dengue
The allele and genotype frequencies of 15 SNPs (Supplementary Table S6) were assessed in 298 symptomatic children and 648 HC from two different Colombian populations. The associated SNPs with symptomatic dengue in the pooled sample (Antioquia and Huila) and in the populations separately are presented in Tables 2, 3, and 4, respectively. The genotype frequencies of all the SNPs were in the Hardy–Weinberg equilibrium in the HC samples (Tables 3 and 4). SNPs with at most 30% missing genotypes (as a cutoff) in the HC group were included in further analysis. Statistical power estimations allowed the acceptance of the no-association hypothesis (>80%), with ORs ≥4.3 or ORs ≤0.23 in the sample of Antioquia (Fig. 1A), ORs ≥3.55 or ORs ≤0.28 in the sample of Huila (Fig. 1B), and ORs ≥2.5 or ORs ≤0.4 in the pooled sample (Fig. 1C). We observed four SNPs associated with the development of dengue symptoms in NOD1, RIPK2, MICB, or PLCE1 genes in Colombian populations. Conversely, we observed one SNP in TNF gene and two haplotypes in the IKBKE gene associated with resistance to develop symptomatic dengue. All associations were corrected for gender and APs and adjusted by FDR. We do not observe associations between symptomatic dengue and SNPs in JAK-1, CSF3, CCR7, IL-18, IL-8, and IL-10 genes (Supplementary Table S6).
aOR values adjusted by gender and ancestry percentages. p-Values adjusted for multiple comparisons by FDR. FDR values in bold indicate significant association (FDR <0.05) or borderline association (FDR within 0.05–0.1).
FDR, false discovery rate; SNP, single-nucleotide polymorphism.
aOR values adjusted by gender and ancestry percentages. p-Values adjusted for multiple comparisons by FDR. FDR values in bold indicate significant association (FDR <0.05) or borderline association (FDR within 0.05–0.1).
HWE, Hardy–Weinberg equilibrium.
aOR values adjusted by gender and ancestry percentages. p-Values adjusted for multiple comparisons by FDR. FDR values in bold indicate significant association (FDR <0.05) or borderline association (FDR within 0.05–0.1).
Regarding the association with the development of symptomatic dengue (Table 2) in the NOD1 gene (related to pathogen recognition), we observed the rs5743336 TC genotype and C-allele carriers associated with the development of symptomatic dengue only in the pooled sample (aOR = 1.95, FDR = 0.0076 and aOR = 1.58, FDR = 0.0473, respectively). In the MICB gene, we observed the MICB-rs3132468 GG genotype associated with the development of symptomatic dengue in the pooled sample (aOR = 3.01, FDR = 0.0198). In the RIPK2 gene, the rs40457 AG genotype and G-allele carriers were associated with the development of symptomatic dengue (Table 3) in Huila (aOR = 1.96, FDR = 0.0242 and aOR = 1.97, FDR = 0.0193, respectively). The effect size was similar to those G-allele carriers associated in the pooled sample (aOR = 1.5, FDR = 0.0473, Table 2). Finally, in the PLCE1 gene, the rs3765524 CT genotype and C-allele carriers were associated with developing symptomatic dengue in the pooled sample (aOR = 1.78, FDR = 0.0187 and aOR = 1.62, FDR = 0.0421, respectively, Table 2) and in Huila (aOR = 4.0, FDR <0.0001 and aOR = 3.8, FDR <0.0001, respectively, Table 3).
Furthermore, the associations with the resistance to develop dengue were observed only in two genes. Among the genes related to cytokine induction, we observed in IKBKE gene the rs1059704 C-allele carriers associated with the development of symptomatic dengue in Antioquia (aOR = 2.0, FDR = 0.0476). Conversely, we observed that rs1059704 C-allele carriers and the rs11118132 CC genotype were borderline associated with resistance to develop dengue (FDR = 0.0769 and FDR = 0.0904, respectively, Table 3) in Huila, where these SNPs were not in LD (D′ = 0.0518, p = 0.5539). Then, two haplotypes were reconstructed and showed association with resistance to develop dengue (Table 5): the rs1059704C-rs11118132G haplotype (aOR = 0.27, p = 0.001) and rs1059704T-rs11118132C haplotype (aOR = 0.46, p = 0.0015, Table 5). Another association with resistance to develop dengue included the TNF gene, the rs1799964 TC genotype and C-allele carriers, in the pooled sample (aOR = 0.48, FDR = 0.0017 and aOR = 0.51, FDR = 0.0017, respectively, Table 2) and Antioquia (FDR = 0.0476) (Table 3***).
Adjusted model by gender, ancestral components, and population. p-Values in bold indicate significant associations (p < 0.05).
The SNPs rs11208534 (JAK1), rs2227319 (CSF3), rs2023906 (CCR7), rs1834481 and rs5744247 (IL18), rs4073 and rs2227307 (IL8), and rs1518111 (IL10) showed no association in this study. We performed the analysis of association with severity (DWOS+DWWS vs. SD) adjusted by gender and AP, but only one SNP was associated with severity. In JAK1 gene, the rs11208534 AG genotype and G-allele carriers were associated with resistance to dengue severity in Huila population (aOR = 0.33, p = 0.0101 and aOR = 0.36, p = 0.0095, respectively).
Discussion
Effect of vector and viral factors on genetic associations
The genetic associations with dengue disease may be influenced by variation in dengue outcomes, the vector, viral, and host factors (7), which may be changing across the study regions. The vector factors are related to environmental features that determine the rates of reproduction and survival of mosquitoes. For instance, the differences in the temperature and the precipitation in specific regions could affect the incidence of dengue (4). We found that the average prevalence was lower in Antioquia than in Huila (0.18 and 3.4 cases/1,000 person-years, respectively). The increased incidence of dengue is related to an increased viral transmission, promoted by the effect of high temperature and frequency of A. aegypti bites. Similarly, increased precipitation leads to an increase in mosquito breeding sites and survival of adults. The difference in the incidence of dengue found between the Colombian populations could be related to the higher average precipitation in Medellin (Antioquia) in comparison with Neiva (Huila), and/or a higher average annual temperature in Neiva than in Medellin during the respective periods of sampling. In fact, the departments with precipitations higher than 3,000 mm/year or with an average annual temperature higher than 25°C had the highest incidence of dengue in Colombia. Huila is included due to its high temperature (4).
Concerning viral factors, there have been reports that the circulating serotypes in Antioquia were DENV 1, DENV 2, and DENV 3 during the 2008–2009 period (3), and all serotypes were present during 2010–2011 period (22). In Huila was reported the circulation of DENV 1 and DENV 4 in 2010 (11). Although no other reports were found describing the serotypes circulating in Huila region, all four serotypes have been circulating in Colombia since 2006 until 2012 (6). We do not know the frequency of each serotype in each area, and how it could have influenced the incidence of dengue disease in these regions. For these reasons, there would be expected differences in the genetic associations with dengue between analyzed regions.
We have reviewed that differences in dengue incidence could influence the genetic associations, and there is a relationship between the age and the dengue severity. In fact, from 2004 to 2013, there has been reported that the proportion of classic dengue fever (DF) (mild disease) varied between 86% and 93.6% in Colombia, and the studied departments were among the top 10 with the higher incidence of dengue (4). Moreover, these researchers found the highest proportion of the DF cases arose in the adult group (15–45 years), but the highest percentage of cases of DHF was observed in children (<15 years) (4). In summary, the highest proportion of DG in Colombia during the sampling period were adults, then the use of adults in the HC group may have not negatively impact on our study of association with the development of dengue.
Polymorphisms associated with symptomatic dengue
In the following sections, we present the discussion in the possible order in which the proteins (encoded by the associated genes) participate in the immunopathogenesis of dengue.
Nucleotide-binding oligomerization domain-containing protein 1
The SNP rs5743336 (796 T/C) was related to NOD1 gene regulation. The 796 A-allele reduces by 50% the NOD1 expression and the ability to activate NF-κB, in comparison with 796 G-allele (27). The decrease in NOD1 expression and NF-κB activation may be a protective response because the NOD1-forming inflammasomes can lead to the upregulation of IL-1β and IL-18. These cytokines lead to the activation of NF-κB by binding their cognate receptors, IL-1R and IL-18R. Excessive production of IL-1β and IL-18 can lead to inflammatory apoptosis (12), which may lead to extravasation and hemorrhages. Therefore, the association with the development of symptomatic dengue that we observed in both the heterozygote genotype and the C-allele carriers may be related to overactivation of NF-κB and their proapoptotic functions. NOD1-rs5743336 has not been associated with dengue yet; however, NOD1-rs5743336 GG genotype has been borderline associated with an increased risk of atrophic gastritis (15). This association supports the importance of NOD1-rs5743336 C-allele in the exacerbated inflammation and dengue development.
Inhibitor of kappa light polypeptide gene enhancer in B cells kinase epsilon
IKBKE-rs11118132 has been reported to be associated with DHF in Brazil (25). We observed IKBKE-rs1059704 C-allele carriers related to the development of symptomatic dengue in Antioquia. These results are probably related to the IKBKE upregulation in peripheral blood mononuclear cells of children patients with DHF in Thailand (30). Conversely, we observed the rs1059704C-rs11118132G haplotype with higher protective effect than the rs1059704T-rs11118132C haplotype in Huila. Although the biological meaning of these SNPs is unknown, we propose that the protective effect of the rs11118132 G-allele in the first haplotype may be explained through the linkage with the IKBKE-rs12142086 (C/T). The rs12142086 C-allele disrupts the binding of the splicing factor 1 with the pre-RNA of IKBKE gene. This allele can also influence IKBKE transcriptional regulatory function over NFKBIA (as reported in a Swedish population), where the rs12142086 (C/T) and rs11118132 (C/G) are in LD (31). Thus, the C-allele of these two SNPs would be related to the impairment of IKBKE function. Therefore, the rs11118132 G-allele may be associated with the proper protein functioning of IKBKE and, in consequence, with the activation of NF-κB. We could not reconstruct haplotypes for the other genes because we do not observe other genes besides IKBKE with more than one variant associated.
Major histocompatibility complex class I polypeptide-related sequence B
The MICB-rs3132468 (A/G) was reported to be associated with DSS according to a GWAS carried out with the Thai and Vietnamese populations (7,11). This SNP was also associated with less severe forms of dengue in another independent Vietnamese population (33). Our findings of the MICB-rs3132468 GG genotype related to the development of symptomatic dengue is consistent with the reported results. The DSS risk rs3132468 C-allele was significantly associated with lower mRNA expression of MICB (9). The proteolytic cleavage of MICB on the surface of the cell membrane produces soluble MICB (sMICB), which blocks the activation signal generated by the interaction of MICB and NKG2D (NK receptor), reducing the NK cell activation (16). In particular, there has been a report of increased circulating levels of sMICB during acute primary DENV infection in symptomatic children (16). Therefore, the association of MICB-rs3132468 GG genotype with the development of symptomatic dengue that we have observed may be related to MICB downregulation and increasing levels of sMICB. Higher sMICB levels impair NK cells activation, and in consequence, a reduction of the antiviral response.
Tumor necrosis factor alpha
We observed the TNF-rs1799964 TC genotype and C-allele carriers associated with resistance to develop dengue. Conversely, in another study, the rs1799964 CC genotype frequency was higher in both DHF and DF patients than in HC in Indian populations (1,2). The C-allele risk of dengue can be supported by (a) the association of rs1799964 CC-genotype with TNF upregulation (26); (b) TNFα triggering of endothelial cells apoptosis and plasma leakage (36); and (c) higher serum levels of TNFα in DHF than in healthy children in Huila, Colombia (11). The contrasting results of rs1799964 CC genotype associations between the Colombian and Indian populations are probably due to baseline differences in allelic frequencies between them. The rs1799964 C-allele frequency is higher in an Indian population [Indian study: cases C = 0.35, HC C = 0.29 (1); 1,000 genomes, BEB-Bengali-India: C = 0.43] than in a population of Antioquia, Colombia (our study: DG C = 0.13, HC C = 0.20; 1,000 genomes, CLM-Medellin-Colombia: C = 0.18). Considering that TNF-rs1799964 C-allele was associated with TNF overexpression, it may be possible that the cases in the Indian population have a higher average TNF expression than those in Colombia. In consequence, the low frequency of the rs1799964 C-allele in the Colombian population may cause the weak observed effect on the increase of the macrovascular permeability. Then, there would be a lower possibility of developing hemorrhages in Colombia in comparison with the Indian populations.
Receptor-interacting serine/threonine kinase 2
Concerning dengue, an increased RIPK2 mRNA expression in DENV-infected HepG2 cells and an induction of apoptosis by RIPK2 mediation have been observed (20). Although the rs40457 heterozygote genotype and G-allele carriers have not yet been associated with dengue, we have observed them to be related to the development of the disease. Moreover, the rs40457 G-allele and the rs40457G-rs42490A haplotype were associated with an increased leprosy risk by GWAS in Eastern China (37) and India, respectively (18). Even though the functional effect of RIPK2-rs40457 is still unknown, the relationship between Mycobacterium leprae infection and dengue disease with RIPK2 overexpression suggests that RIPK2-rs40457 G-allele may alter the immune response. The rs40457 G-allele may increase apoptosis and bleeding.
Phospholipase C epsilon 1
The PLCE1-rs3765524 (C/T) SNP causes a Thr1777Ile substitution that alters the protein stability and function (28). Moreover, the PLCE1-rs3765524 C-allele has been associated with PLCE1 upregulation (9). PLCE1 is an upstream regulator of NF-kB activation (8), and PLCE1-knockout cells have a decreased expression of IL-6, TNFα, and IL-1β (35). Consequently, the presence of PLCE1-rs3765524 C-allele may increase cytokine expression and the risk of bleeding. Similar to our results of C-allele association with dengue susceptibility, this allele was related to DSS in Thai children (9). Moreover, the rs3765524 T-allele was associated with protection to DSS by a GWAS with Vietnamese children (13). The associations mentioned above suggest that the PLCE1 overexpression in cells, given by the effect of rs3765524 C-allele, may increase the inflammatory cytokines expression. In consequence, it may raise the plasma leakage.
Ancestry and dengue susceptibility
There is a consensus on which the differences in ancestry composition among populations could affect the SNP allelic frequencies and the possible associations to diseases. Moreover, the hypothesis of the African ancestry protection against dengue severity has been supported by epidemiological and genetic studies (5). In our case, we observed that Eur-AP was the highest AP of the two Colombian populations, but the Afr-AP was higher in the sample of Antioquia than in the one from Huila. These ancestral compositions were similar to the ones estimated in other three association studies carried out in Antioquia and Huila (3,4,21). Considering the Huila's higher dengue prevalence and the observed higher number of SD cases in Huila, compared to Antioquia, we could strengthen the hypothesis of the African ancestry protection against dengue severity. However, this statement should be taken with caution because the convenience sampling (in a case–control study) that we used is not appropriated to analyze possible relationships between ancestry and associated SNPs with dengue.
An approximation using previously reported data could support the connection of African ancestry with the protection against dengue severity. We compared the allele frequencies of associated SNPs, from two Colombian populations (Antioquia and Huila), and the allele frequencies of the populations CEU, CHB, and YRI (used as ancestral references) taken from the 1000 Genomes Project. We can observe similar allele frequencies between the actual Colombian and ancestral populations. Three SNPs in IKBKE, PLCE1, and RIPK2 genes seem to be related to the Amerindian ancestor (CHB). SNPs in TNF, MICB, and NOD1 genes appear with similar frequencies in the African ancestor (YRI). One SNP in IKBKE may be connected with the European ancestor (Supplementary Table S7). Some of these observations agree with the results of Rishishwar et al. (23). These researchers developed a method to search for genomic regions with anomalous patterns of admixture. They used different retention patterns of chromosomal segments from specific ancestral populations, in the modern admixed population. They analyzed a sample of Medellin (Antioquia), and they found that the IKBKB (belonging to the same family of IKBKE gene), PLCL (or PLCE1), and RIPK2 genes are located in the Asian-ancestry-enriched segments. Also, the MICB and TNF genes were located in the African-ancestry-enriched segments, whereas the NOD1 gene was not found in any enriched segments (Supplementary Table S8, 23).
In our study, we observed that the SNP in the TNF gene was associated with protection against the development of dengue. Therefore, the previous observations about the African origin of TNF gene (23) support the hypothesis that the African ancestry provides a protective effect against developing SD. Noteworthy, other ancestries could be conferring protection as well, such as the Asian ancestry (Amerindian ancestry in our study, Ame-AP). This observation is supported by the Asian ancestor described for the IKBKB gene (23) and our results of the associated haplotypes in the IKBKE gene with protection against symptomatic dengue. Also, we observed that more SNPs were associated with the development of symptomatic dengue in Huila than in Antioquia, which is probably connected with the low Afr-AP observed in Huila. Besides, some observed differences in SNP–disease associations between the two samples may be related to the higher statistical power achieved for the sample of Huila, in comparison with Antioquia.
The observed SNPs associated with the development of symptomatic dengue in the analyzed Colombian populations suggest that the altered expression of immunomodulatory and proinflammatory genes might be involved in the susceptibility to develop/manifest dengue. Even more, they might also be implicated in the development of severe hemorrhagic forms in the studied Colombian children.
Populations from different regions of a country may differ in their allele frequencies, and this is particularly evident in Colombia, where nonadapted immigrants have not been previously exposed to new and diverse pathogenic microbes in a variety of environments. We observed fewer and lower strength associations of alleles in immune genes with symptomatic dengue in Antioquia than in Huila, probably due to differences in the genetic diversity, the ancestry mixtures, the different geographical regions and climates, and in the outbreaks history of dengue between the analyzed populations.
Conclusions
Associations of symptomatic dengue with SNPs within MICB and PLCE1 genes in the Southeast Asian populations were confirmed in our study. Noteworthy, SNPs in IKBKE, RIPK2, and NOD1 genes were associated with the development of symptomatic dengue herein for the first time. Moreover, we observed that SNPs in MICB, PLCE1, and IKBKE genes had a strong association with resistance or susceptibility to dengue disease. Nevertheless, studies in other Colombian populations are necessary to reveal the importance of these and other SNPs, or haplotypes, as markers for the development of dengue disease in the country. Additionally, associated SNPs should be assessed through functional analyses to confirm their relevance in dengue pathogenesis, their possible use as prognostic biomarkers, and for their use in the development of dengue therapies.
Footnotes
Acknowledgments
This work was supported by the Departamento Administrativo de Ciencia, Tecnología e Innovación of Colombia, COLCIENCIAS (project 1115-493-26145; contract number 441.2009) and by the Programa de Doctorados Nacionales scholarship (528-2011-52471285). We thank participating hospitals: Hospital Universitario Hernando Moncaleano Perdomo, General Hospital of Medellín, Pablo Tobón Uribe Hospital, Marco Fidel Suarez Hospital, San Vicente Hospital, and Sura EPS. Finally, we thank Carolina DA for reviewing the translation.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.