
Abstract
The present report describes current concepts about the mechanism of liver cell injury caused by host immune response against hepatitis C virus (HCV) infection in human beings. This report is based on the observations from experimental studies and follow-up actions on human liver diseases. The results from different investigations suggest that liver injury depends on the presentation of viral antigen and the level of host immune response raised against HCV-related peptides. Both innate and adaptive immunity are triggered to counter the viral onset. During development of host immunity, the cell-mediated immune response involving CD4+ Th1 cells and CD8+ cytotoxic T-lymphocyte (CTL) cells were found to play a major role in causing liver damage. The hepatic Innate lymphoid cells (ILCs) subsets are involved in the immune regulation of different liver diseases: viral hepatitis, mechanical liver injury, and fibrosis. Humoral immunity and natural killer (NK) cell action also contributed in liver cell injury by antibody-dependent cellular cytotoxicity (ADCC). In fact, immunopathogenesis of HCV infection is a complex phenomenon where regulation of immune response at several steps decides the possibility of viral elimination or persistence. Regulation of immune response was noted starting from viral–host interaction to immune reaction cascade engaged in cell damage. The activation or suppression of interferon-stimulated genes, NK cell action, CTL inducement by regulatory T cells (Treg), B cell proliferation, and so on was demonstrated during HCV infection. Involvement of HLA in antigen presentation, as well as types of viral genotypes, also influenced host immune response against HCV peptides. The combined effect of all these effector mechanisms ultimately decides the progression of viral onset to acute or chronic infection. In conclusion, immunopathogenesis of liver injury after HCV infection may be ascribed mainly to host immune response. Second, it is cell-mediated immunity that plays a predominant role in liver cell damage.
Introduction
Hepatitis, an inflammation of hepatic parenchymal tissue, is caused mainly by viruses that include hepatitis viruses A, B, C, D, E, and G. The other causes of hepatitis are alcohol intake, certain medications, toxins, some specified infections, autoimmune diseases, and nonalcoholic steatohepatitis. There are at least five major types of viruses (A-E) causing hepatitis worldwide. Of them, hepatitis C virus (HCV) is a parenterally transmitted virus and spreads mainly via intravenous drugs use, unsafe transfusions, and therapeutic injections. Acute HCV infection often remains asymptomatic (65). Persistent viral infections can be latent or chronic (67). However, 60–80% patients infected with HCV become chronic where virus constantly replicates and persists for a longer duration with very little chance of its clearance (74). Chronic HCV infection induces liver fibrosis causing cirrhosis in significant number of patients in later years (23). Patients with chronic HCV infection and cirrhosis are at high risk for developing end-stage liver diseases, including hepatocellular carcinoma (HCC) (20,35). HCC caused by HCV infection, has been a major indication for liver transplant all over the world (20,23,35). Efforts have been made to develop effective prophylactic and therapeutic measures for treatment of chronic HCV infection (36).
HCV is a hepatotropic, enveloped RNA virus and belongs to Flaviviridae family. HCV-genome comprises a 9.6 kb single-stranded RNA with positive polarity. It encodes for a single polypeptide of 3,011 amino acids, which is processed by proteases from both the host cell and virus itself to give rise to three structural proteins (Core, E1, and E2) and seven nonstructural proteins (P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (2,10). Envelop protein E2 is the main viral protein which facilitates viral entry into the host cell and regulates its functions (94). Nonstructural proteins were reported to play a significant role in viral replication and assembly. Although the mechanisms of HCV clearance or persistence are not fully understood, it is assumed that level of host immune response may decide the viral escape or persistence in the infected patients.
Very little is known about host genetics and immunity to HCV infection. Several reports indicate that during chronic HCV infection, there are functionally impaired CD8+ T cells, which are not potent enough to proliferate and secrete antiviral cytokines such as IFN-γ (41,71). Such an impaired cellular immune response seems to be responsible for evasion strategy developed and used by the virus in self-defense (6,52). HCV clearance/evasion was also associated with the high polymorphism in HLA genes since HLA alleles were reported to ensure the ability of immune response against HCV infection. Studies were conducted to find out whether HLA alleles have some significance in spontaneous HCV clearance (43,53). Although such an association between HCV clearance and specified HLA alleles was noted in few studies, the same could not be validated in other reports (19,57). HCV infection causes various liver alterations, including liver inflammation. This becomes a basis for liver fibrosis which progresses to cirrhosis and finally to HCC. Once the fibrosis is set in liver, there is an increasing accumulation of high-density fibrillar collagens (for example, collagens I and III), proteoglycans, and other components of the extracellular matrix (ECM) (73). Although several other factors such as age, duration of infection, insulin resistance (IR), steatosis, and so on are also closely associated with the final outcome of the disease, all patients do not respond equally to these factors (98). This emphasizes an importance of genetic variation over environmental issues (51). Histologically, chronic HCV infection is characterized by the necroinflammatory changes in the liver with interface hepatitis, bridging necrosis, and the formation of septa in portal tract. Also, there is cell shrinkage and fragmentation of the cell nucleus in liver cells indicating apoptosis as an important step in disease causation (29).
In view of all these information available from different studies with very little coherence among their results, it appears quite appealing to have a deep insight into the progression of HCV infection to end-stage liver diseases in reference to host immune response against HCV infection. The present article was framed to discuss current concepts of HCV-related immunopathogenesis and its reaction cascade. This is also aimed to understand the mechanism of host immune response that interacts with viral proteins and shapes the course of disease causing liver damage and promoting fibrosis toward cirrhosis of the liver.
Host–Virus (HCV) Interaction
HCV traverses endothelium to come in contact with target cells, including hepatocytes. Glycosaminoglycans (GAGs) and low-density lipoprotein receptor help in concentrating HCV particles on the cell surface (3,58). It has been observed that GAGs act as the primary site for attachment of HCV particles. Viral proteins E2 and E2/E1 complex located on outer surface of HCV bind to GAGs in the first instance (3). HCV also binds to several other receptors, including scavenger receptor B1 (SR-B1), tetraspanin CD81, claudin-1 (CLDN1), occludin (OCLN), dendritic cell (DC)-specific intracellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN), epidermal growth factor receptor, and Niemann-Pick C1 like 1 (NPC1L1) to mediate viral entry into host cell (3,17,26,58,102). E1/E2 binds to cell receptors where GAGs and CD81 play a major role. After interaction with HCV E2, CD81 activates Rho GTPase family that remodel actin allowing the E2-CD81 complex to contact CLDN1. This CD81-CLDN1 complex helps the virion to translocate to a tight junction for its clathrin-mediated endocytosis (102). Following internalization, HCV fuses with endosome and the viral genome is released into the cytosol to start the replication process. A membrane-associated replication complex composed of nonstructural proteins, host protein, and viral RNA is formed in the endoplasmic reticulum membrane. Replicated RNA and core protein are recruited to surface of lipid droplet (26). E2/CD81 binding also stimulates RANTES (Regulated Upon Activation Normal T Cell Expression and Secretion) secretion in CD8+ T cells altering migration of immune competent cells (61). E2/CD81 interaction secretes MMP2 (Matrix Metalloproteinase-2) (54), which disrupts normal basement membrane to activate hepatic stellate cells. MMP2 facilitates migration of inflammatory cells at the site of liver injury.
Effect of HCV on Host Innate Immunity
Various components of HCV, including RNA and its translated proteins, are recognized as a foreign antigen by the host immune system. HCV induces production of type-I, II, and III Interferons (IFN-α, β, γ, λ1–4), which are assumed to be critical for host defense against the viral onset. These IFNs help in phosphorylation of STAT-1 and STAT-2 leading to the formation of transcription factor ISGF-3 (Consisting of pSTAT-1, pSTAT-2, and IRF9), which translocates to the nucleus and modulates production of interferon stimulated genes (ISGs) (83). The products of ISGs suppress viral replication and promote the infected cells to apoptosis (56). IFN-α and β also help hepatocytes in presenting antigen to CD8+ T cells (80). A defect in type-I and III IFN signaling allows the hepatocyte to support continued presence and replication of HCV. Hepatocytes sense the intracellular presence of HCV by Toll-like receptors (TLRs), retinoic acid-inducible gene-1 (RIG-1), melanoma differentiation-associated gene-5 (MDA-5), and so on in the cytoplasm. Binding of HCV RNA with RIG-1 and MDA-5 activate signals to produce IFNs. HCV influences predominantly the production of IFN-λ4, which induces ISGs (77). During infection, pathogen-associated molecular patterns (PAMP) signaling via TLR-3 recruits TIR domain-containing adaptor protein inducing IFN-β (TRIF) to cause phosphorylation of interferon regulating factor-3 (IRF-3) by protein kinases 1-κB kinase-ɛ (IKK-ɛ) & tumor necrosis factor receptor (TNFR)-associated NF-κB. TRIF also activates transcription factor NF-κB. Ultimately, IRF3 and NF-κB factors induce production of IFN type-1 and IFN-inducible genes, which mount antiviral actions. NF-κB simultaneously regulates expression of chemokines and inflammatory cytokines, which mediate inflammatory response to HCV. NF-κB is also involved in hepatocellular apoptosis (87). HCV proteins NS3/NS4A cleave TRIF (46) defending virus against immune response via disruption of PAMP-signaling. Disruption of these pathways supports viral persistence. To block the cellular response to type-I and III IFNs production, HCV core upregulates suppressor of cytokine signaling (SOCS-3) (99), which is a negative regulator of STAT phosphorylation and also inhibits ISGF3 activity (48). Based on these results it appears that HCV pursues many strategies to disrupt not only induction and production of IFN type I and III but also check ISGs expression. HCV is eliminated mostly by Interferon-γ driven ISG expression in hepatocyte (15,89). However, ISG expression could not be noted in lymphocytes and monocytes (13). ISG induction is largely determined by Interferon-λ4 genotype (14,88) although IFN-α does not induce ISG expression (18,75). Infected hepatocytes or IFN-producing immune cells are considered to be the best source of IFN-λ4 production and ISG expression. HCV uses some escape mechanism which allows it to persist despite ISG expression. Furthermore, an interaction between HCV and immune stimulatory pathways inhibit IFN-signaling via JAK/STAT pathway and IFN- inducible proteins using different mechanisms (27). Activation of IL-8 by HCV NS5A is significant since it inhibits IFN activity (68), both in vivo and in vitro, and acts as proinflammatory chemokines and cytokines to inflammatory cells. In combination with other growth factors, IL-8 also activated mesenchymal cells producing ECM.
It has been observed that natural killer (NK) cells play a significant role as a first line defense against HCV, both during acute as well as chronic diseases (28). They lyse infected cells and secrete inflammatory cytokines. NK cells inhibit viral replication and produce a link between innate and adaptive immune responses (Fig. 1). The NK cells are critical in initial antiviral action and strongly help in HCV elimination (40). The regulation of NK cell activity results from an interaction between activating and inhibiting receptors with their ligands. The possible ligands include HLA-A, -B, -C, and -E, which bind to killer cell immunoglobulin-like receptors and CD94-NKG2A/C complex receptors. Similarly, the ligand MIC-A and MIC-B bind with NKG2D receptor (59). This ligand binding with receptors reduces the cytolytic activity of NK cells and also reduces the level of CD4+ Th1-cells. As a result, the failure of NK cytolytic activity contributes to virus persistence in chronic HCV infection. Simultaneously, there is an increased secretion of IL-10 and TGF-beta by the NK cells. These cytokines stimulate hepatic stellate cells leading to an increased fibrosis in chronic HCV infection (66). There is increasing evidence showing that both HCV genotypes and viral genetic variation arising during infection encounter a continuous immune pressure, and so, viral escape can be determined by the collective impact of viral genotypes and host immune pattern (21,70).
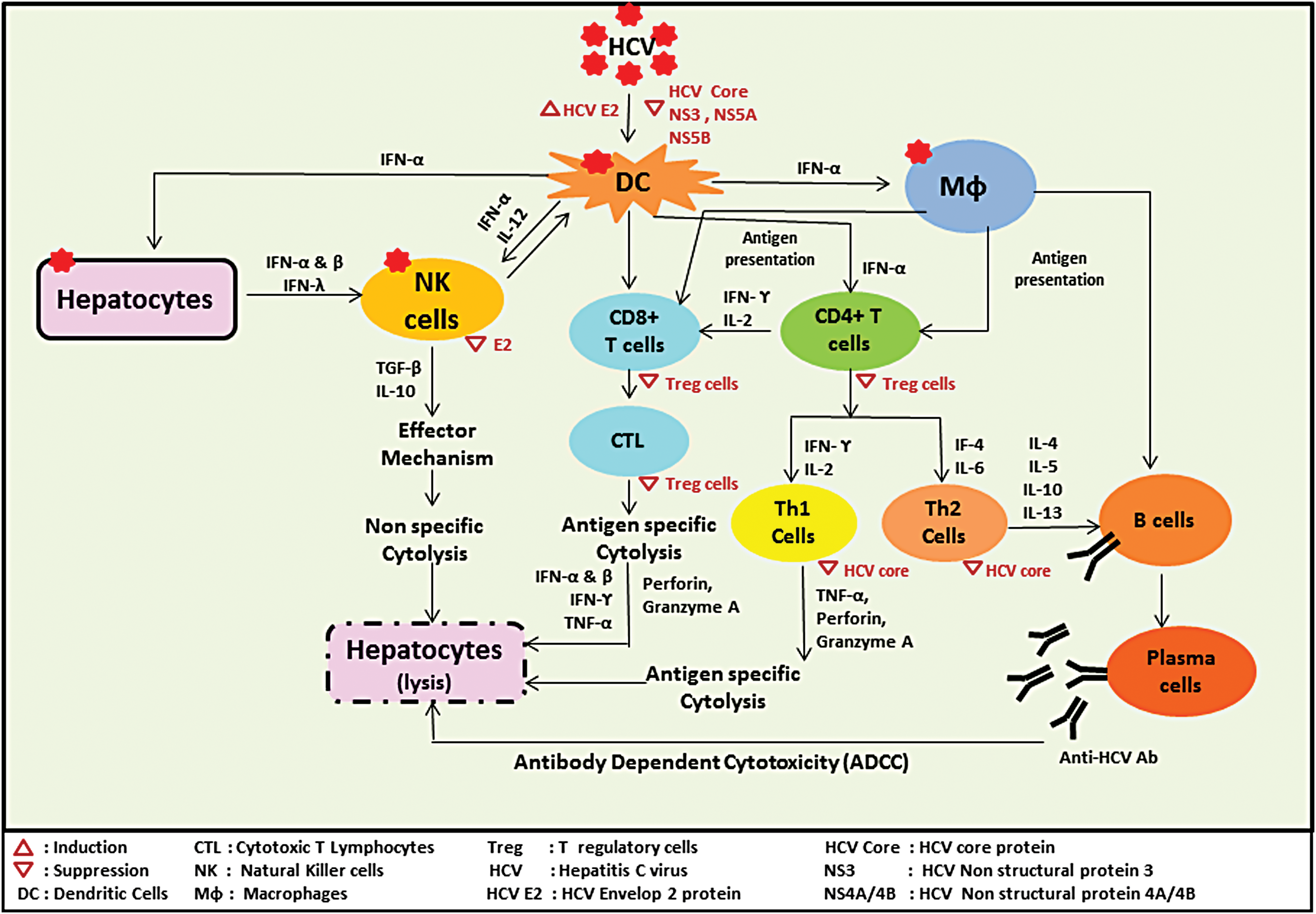
Immune reaction cascade for HCV-mediated hepatic cell lysis. HCV, hepatitis C virus. Color images are available online.
Effect of HCV on Host Adaptive Immunity
Cell-mediated immunity
T cell-mediated immune response involves CD8+ cytotoxic T-lymphocyte (CTL) cells and CD4+ T helper cells, which recognize viral antigen on infected cells via MHC-I and MHC-II, respectively. Cell-mediated response is assumed as a key player in resolving HCV infection (5). After HCV infection, specific CTLs (25) and CD4+ Th1 cells limit the course of the disease. Cellular immunity is seen persisting for long in HCV-resolved patients, indicating that cellular response controls the HCV infection (42). Despite 40–80% of liver-infiltrating CD4+ cells, only 1% of them are HCV specific. These cells may be involved in host protection rather than liver injury (44). Simultaneous with facilitating viral clearance (93), CD4+ T cells carry Perforinlow, fail to secrete IFN-γ, and thus exert ex-vivo killing of infected cells. CD4+ T helper cells also secrete anti-inflammatory cytokines (92). Thus, a balance of these cells becomes a key issue in HCV immunopathogenesis in addition to various other developments as the emergence of T cell escape mutants, lack of T cells, or T cell exhaustion in the course of HCV-related diseases. HCV has a high mutation rate as RNA-dependent RNA polymerase does not have proofreading ability. This gives rise to several related HCV variants in an infected person. These molecular species have been classified under seven major genotypes (G1–G7) (82) and much more their subtypes. As described above, these genotypes behave differently in inducing host immune response.
Binding of the viral E2 protein to DC induces their maturation, although at the same time, HCV core, NS3, NS5A, and NS5B have been reported to inhibit the maturation and function of DCs (100). The response of DC to HCV infection is not only crucial to determine the course of the disease but also necessary for activation of Th cells and CTL. DCs respond to HCV viral protein and induce HCV-specific T cells. It has been seen that usually strong T cell response arises during acute HCV infection (81). E2 protein, which is most outer protein on virus surface, encounters DC at first after infection. Upon maturation, DC upregulates expression of their surface proteins and increases antigen presentation abilities. Simultaneously, DC increases their capability to produce more cytokines such as IL-12. As a result, DC brings about enhanced activation of T cells, which in turn produce more IFN-γ. This induces development of the Th-1 type of immune response, which regulates CTL activation. It has been found that Th-1 response is essential for successful clearance of HCV in acute infection (1). Patients having impaired IL-12 production or less IFN-γ production allow the virus to persist and develop a course of chronic disease (9).
HCV induces both humoral and cellular immunity in the host. Presence of CD4+ and CD8+ T cells can be demonstrated in the blood of infected patients. However, it is worth to record that cellular rather than humoral immunity plays a more important role in controlling HCV infection (37,38). The virus-specific CTL damage the infected liver cells and check viral replication by secreting IFN-α, β, γ, and TNF-α. These cells also produce an antiviral environment in infected cells. Deficient or impaired host response progresses to viral persistence and development of chronic infection. The failure of host response may be responsible for the emergence of escape variants due to high mutation rate in HCV genome or stunning of HCV-specific CTL. To understand the basic causes of impaired immune response, including defective cellular and humoral immune response, several theories have been suggested. One such theory states that there is a high frequency of CD4+CD25+ regulatory T cells in the blood of chronic HCV-infected patient (8). Furthermore, it was found that HCV-infected liver also shows the presence of CD4+FoxP3+ T cells (97). These regulatory cells suppress HCV-specific CD4+ and CD8+ T cells proliferation and IFN-γ secretion. T reg cells secret IL-10 and TGF-β that suppress virus-specific T cell responses (8). CD4+CD25+ T reg cells do not only suppress the T cell response in chronic infection but also control the memory cells. In this way, regulatory CD8+ T cells play an important role in chronic HCV infection. HCV-specific CD4+CD25+/FoxP3+ T cells suppress T cells response via TGF-β secretion (47). T regulatory cells in the liver also protect the host from tissue damage. All the details related to immune-mediated cytolysis of hepatic cells are described in Figure 1.
Innate lymphoid cells (ILCs) are a recently identified group of heterogeneous innate immune cells that are distinguished from B and T cells. ILCs are divided into three types: group 1 ILCs (ILC1s + NK cells); group 2 ILCs (ILC2s); and group 3 ILCs [ILC3s + lymphoid tissue inducer (LTi) cells], based on their ability to produce cell-associated cytokines type 1, type 2, and Th17 (79). Dysregulation of ILCs can result in inflammatory disorders. Current studies show that ILCs play important roles in the development of chronic liver diseases. Hepatic ILCs are involved in the immune regulation of diferent liver diseases such as viral hepatitis, mechanical liver injury, and fibrosis. For example hepatic ILC1s play an important role in maintaining liver tolerance during HCV infection. Hepatic viral infection increases NKG2A expression on ILC1s and leads to inhibition of CXCL9 expression and accumulation of IFN-γ+CD49b+ NK cells (cNK cells). The result is loss of IFN-γ production, important for the enhanced priming of CD8+ T cells (49).
Humoral immunity
Anti-HCV antibodies appear in the blood within few weeks after HCV infection. While the presence of antibody in serum is quite helpful for the diagnosis of HCV infection, their role in the protection of host against infection or in immunopathogenesis is still controversial. Usually, anti-HCV antibodies do not protect the host against reinfection. At the same time, there are reports demonstrating that HCV infection gets resolved in certain cases even without the appearance of antibodies (69,71). It may be ascribed to a frequent mutation in hypervariable region (HVR1) on E2 protein, which is a target for neutralizing antibodies. This gives rise to a postulation that HCV adapts immune selection pressure exerted by anti-HCV antibodies via escape mutation. The protective antibodies have been noted infrequently in patients with self-limited disease (50). Despite all these reports, few other studies have shown that antibody response is linked to liver injury (22) and involvement of E2 antibodies in liver damage via ADCC (62) (Fig. 1). E2 antibodies are not involved in viral clearance (32). Under these circumstances, it is difficult to exactly evaluate the contribution of HCV antibodies in either HCV virus elimination or liver damage. But antibodies against HCV proteins do not appear to be protective antibodies.
Host Immune Response in Acute and Chronic HCV Infection
Acute HCV infection
The acute HCV infection induces a specific T cell response and the strength of these T cells decides the outcome of acute infection. Persistent and strong CD4+ and CD8+, IFN-γ+ T cell response to HCV nonstructural proteins is usually associated with resolution of acute HCV infection. On the contrary, a weak or transient response helps the viral persistence (86). This is supported by the studies conducted in experimental chimpanzees infected with HCV, where T cell response in the liver had a close relationship with virological outcome in the blood of patient (11,103). Other studies clearly demonstrate that both CD4+ and CD8+ T cells are needed for efficient viral clearance (39,90). It was also observed that in the initial phase of acute HCV infection, the proliferating capacity of CD4+ T cells also influences conversion of stunned HCV-specific CD8+ T cells to the ones with strong antiviral activities (78) and increased cytotoxicity to infected hepatocyte (34). Similar to T cell function, some studies have shown the impact of NK cell activation on HCV clearance. They reported that HCV dysregulates NK cell function. Active cytotoxic NK cells eliminate infection in patients with HCV (30). However, this is not a universally established fact. On the similar line, many reports are available showing that neutralizing antibodies detected 6–8 weeks after infection prevent viral binding to targets such as CD81, LDL-R, SRB1, and claudin-1 (64) in early acute HCV infection. This helps in resolution of infection in some cases (101). This concludes that in the acute phase of infection, T cells, NK cells, and anti-HCV antibodies show varied roles.
Chronic HCV infection
Lymphocytic infiltrates in the liver is a sign of chronic HCV infection. While during the acute phase, intrahepatic lymphocytes are involved in HCV elimination, their presence during chronic disease results in progression of the infection. HCV-specific CTLs are involved in cell killing via perforin, Fas/Fas ligand, and TNF pathways (33). An increased level of proinflammatory cytokines has also been recorded in chronic HCV infection (45). This was related to enhance portal inflammation and liver fibrosis (55). Inflammatory cytokines in chronic disease recruit chemokines receptor, CXCR3 expressing NK, γ δ T cells, and CD8+ T cells (72). Besides, these chemokines also modulate liver fibrosis (4). These cells ultimately get activated and release perforin damaging target cell membrane followed by the action of granzyme A, which cleaves caspase-8 inducing apoptosis (76). The cellular immune response in chronic cases also expresses Fas ligand and secretion of TNF. In this pathway, activated caspase-8 cleaves proapoptotic protein known as Bid, which induces mitochondria to release cytochrome c into the cytoplasm (24). This cytochrome c binds APAF-1, which in turn activates caspase-9 and the downstream effector caspases leading to programmed cell death. An antiapoptotic molecule-like Bcl-2 blocks cytochrome c release and protection against liver damage (85). HCV proteins interact with apoptotic pathways and the pro- and antiapoptotic factors (63). Like Fas ligand, there is another death factor called TRAIL (TNF-related apoptosis-inducing ligand) that upregulates during chronic HCV infection (84). IFN type I and II induce expression of TRAIL and its receptors (84). In chronic HCV infection, it may also promote steatosis (60). However, all the functions of the TRAIL system are not fully understood. During chronic HCV infection, the sustained antigenic stimulation induces some inhibitory coreceptors or checkpoint molecule on HCV-specific CD8+ T cells, which develop functional hyporeactivity called anergy. These inhibitory checkpoint molecule are part of immune regulation which includes programmed death-1 (PD-1), cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), programmed cell death ligand 1 (PD-L1), lymphocyte-associated gene-3 (LAG-3), killer cell lectin-like receptor subfamily G member 1 (KLRG1), and NK cell receptor 2B4 (CD244) associated with the suppression of antitumor immune responses in solid tumors. Moreover, these immune checkpoints are very important therapeutic target for liver cancer. For example, the combination of anti PD-1/PD-L1 with anti CTLA-4 antibodies are used for the treatment of HCC. All these information demonstrate that the introduction of HCV-related proteins with different pathways help the virus to persist and cause chronic liver disease.
Viral Escape from Host Immunity
The exact mechanism behind evasion process is not very clear. Current reports that indicate that defect at multiple levels, including activities of antigen presenting and effector cells, are responsible for evasion. It may be illustrated by individual steps of an immune response. The first example is DC, which has a critical role in immune development against any infection, including HCV. These cells act as antigen presenting cells and so lack of interaction of DC and T cells can impair the host to clear the infection. Several studies have reported that HCV impedes DC functions toward adaptive immunity with alteration in CD4+ T cells and CD8+ T cells response. It appears to be due to altered cytokines release leading to defective T cells functions (12,91). Similarly, the defect in CD4+ CD25+ T regulatory cell causes the alteration in the production of humoral factors or their negative effect on T cells function (16). HCV also disturbs humoral response enabling evasion of the virus. Antibodies clear pathogen by their neutralizing actions, opsonization, and antigen degradation. In case of HCV, antibodies do not seem to clear HCV. HCV E2 protein binds virus to hepatocytes. A high rate of mutation in E2 HVRs (96) produces HCV variants that impair the binding of neutralizing antibodies. Regarding the role of CD4+ and CD8+ T cells, they have a major role in combating HCV infection (7). Their depletion results in low CD8+ effector cell response causing a continued viremia (31). Similarly, CD4+ helper T cells, Th1, and Th2 play important roles in HCV infection. Th1 conduct cell-mediated response evoked by IL-2, IL-12, and IFN. Th2 promote humoral response evoked under the effect of IL-4, IL-6, and IL-1. HCV core reduces production of IL-12 and so impaired Th1 and Th2 functions (95). Mutation in viral epitopes results in diminished CD4+ T cell response. While CD4+ T cells coordinate immune response, CD8+ T cells mediated cytotoxicity. HCV impairs the functionality of the CD8+ T cells. These effects in CD8+ T cells include, their immaturation, low cell-killing capacity, decreased TNF- α secretion, low perforin and granzyme A level, low expression of Fas, and so on. This concludes that HCV often disturbs Ag-presentation, humoral immune response, cell-mediated immune response, apoptosis in the favor of viral persistence, and development of chronic HCV infection.
Conclusion
This study concludes that HCV, which is an RNA virus, is noncytopathic in nature and does not integrate with the host genome. Liver damage resulting from HCV infection is caused by a reaction cascade triggered by host immune response against HCV-related components. Both innate and adaptive immune responses develop against HCV infection. While innate response works along the lines of interferon-induced gene expression (ISG) and NK cell activation against HCV-infected cells, adaptive response focuses mainly on cell-mediated cytotoxicity with the predominant role of CD4+ Th1 cells and CD8+ CTL cells. Humoral response where anti-HCV antibodies are produced few weeks after infection act as damaging antibodies rather than protective antibodies against HCV infection. These antibodies act via ADCC to cause liver injury. Immune response against HCV faces a barrel and check-like regulation starting from virus–host interaction to the immune-mediated reaction cascade causing infected cell damage. A final balance between HCV-induced cellular damage and a check on these reactions decides the viral elimination and recovery from the disease or persistence of virus causing chronic infection. HCV-induced immunopathogenesis is a complex phenomenon, not fully understood as yet, and still needs many more investigations to resolve various issues related to it.
Footnotes
Acknowledgment
We appreciate the infrastructure provided by All India Institute of Medical Sciences, New Delhi, India, for conducting this study.
Authors’ Contributions
All authors have made equal contributions in the performance of study, compilation of data, and preparation of the article.
Author Disclosure Statement
The authors declare that there is no conflict of interest among them related to this study.