
Abstract
Influenza A viruses (IAVs) can be classified into dozens of subtypes based on their hemagglutinin (HA) and neuraminidase (NA) proteins. To date, 18 HA subtypes and 11 NA subtypes of IAVs that spread in animals and humans have been found. Following infection, the IAV first induces the innate immune system, which can rapidly recruit innate immune cells and cytokines to the site of infection. Influenza-induced cytokine storms have been associated with uncontrolled proinflammatory responses, which may lead to significant immunopathy and severe disease. Cytokine storms are complicated by several types of cytokines and chemokines that have various activities. In addition to their direct effects, their crossregulation causes cytokine networks to form; these networks determine the outcome of viral infections. In this review, we focus on cytokine storms and their signaling pathways that are triggered by the different subtypes of IAV.
Introduction
They are divided into four genera (types A, B, C, and D), of which influenza A virus (IAV) can infect a wide range of animal species (9,77). IAV can be further classified according to the molecular structure and genetic characteristics of hemagglutinin (HA) and neuraminidase (NA) proteins. To date, 18 HA subtypes and 11 NA subtypes have been identified and are circulating in animals and humans (65). HA is the most abundant surface glycoprotein of the IAV that has the ability to attach the host cell, causing cellular fusion and viral entry. The high variability of HA allows IAVs to escape from host immune surveillance and results in influenza seasonal epidemics. NA, the second most abundant glycoprotein, cleaves sialic acid (SA) moieties, promotes the release of nascent virions, and facilitates IAV dispersion. NA also plays crucial roles in the viral infection and HA-mediated membrane fusion by binding to SA receptors. IAVs infect the trachea, bronchi, and alveoli, as well as destroy epithelial cell. Therefore, the ability of the virus to spread to the lower respiratory tract and cause tissue damage are key elements in fatality. The majority of fatalities also developed bacterial pneumonia. It is recognized that a major cause of respiratory failure is coexistent bacterial pneumonia leading to acute respiratory distress syndrome (ARDS). This is characterized by damage to the endothelial–epithelial barrier of the alveoli, resulting in fluid leakage and accumulation in the alveolar lumen inhibiting gas exchange.
Seasonal human IAVs usually preferentially attach to the larynx, trachea, and bronchial epithelium. In contrast, IAVs that are highly pathogenic to humans, such as the H1N1 and H5N1 viruses, colonize the bronchioles and alveolar epithelium (17,35,36,64,69). IAVs infect, replicate, and proliferate in a variety of host cells. Although monocytes/macrophages and other white blood cells can also be infected, the lung epithelial cells are the main sites of replication for the IAV. Two different subtypes of the IAV (H1N1 and H5N1) from different species that are not pathogenic to mice were forced to evolve through successive passages in the lungs of mice. The two adapted viruses that were obtained showed almost the same level of virulence and had similar LD50 values. A common feature of both viral subtypes is diffuse alveolar damage, which is dominant in both histopathological features (35,45).
Innate immunity is the first line of defense against pathogens, such as the flu virus. Innate immunity has a wide range of effects, is not specific, reacts quickly, and recruits immune cells to the infected site by producing cytokines. The innate immune response has no immune memory, so if the body is exposed to it in the future, it will not recognize or remember the same pathogen (6,39,66). Many cells are involved in innate immune responses, such as phagocytic cells, mast cells, basophils, eosinophils, and natural killer (NK) cells. The rapid recruitment of immune cells to the site of infection and inflammation by the production of cytokines is an important function of innate immunity. The production of cytokines mobilizes many defense mechanisms throughout the body and activates the response of local cells to infection or injury. These cytokines are essential for triggering cell recruitment and local inflammation (63). To date, influenza-induced cytokine storms have been associated with uncontrolled inflammatory responses, which have led to significant pathological consequences and serious diseases (10,19,34,60).
As we gain a deeper understanding of the different roles of individual cytokines, the concept of cytokine storms becomes more complex. Their crossregulatory function in cytokine networks has important implications for infection outcomes (52). Severe cytokine release can circulate and cause systemic cytokine storms, and this can lead to multiple organ dysfunctions (12). In this review, we focus on cytokine storms and their signaling pathways that are triggered by the different subtypes of IAV.
The Role of Innate Immune Cells in the Pathogenesis of Influenza A Infections
Macrophages
Alveolar macrophages play an important and recognized role in the innate immune response. Although alveolar macrophages are the most prominent immune cells, they are usually present on the mucosal surface of the respiratory tract and can promote or inhibit airway inflammation. A previous study has suggested that macrophages of different origins or phenotypes may have protective effects but may be harmful during the different stages of IAV infection. IRF5+ (the interferon [IFN]γ-activated transcription factor IFN regulatory factor 5) macrophage phagocytosis and antigen presentation may be useful for the initial response after allergen exposure, while the enhanced tissue repair function of Ym1+ (M2 marker) macrophages may be useful at a later point in time (43). Under steady-state conditions, accomplishing these tasks should help eliminate inflammation. However, in IAV infections, either or both of these macrophage phenotypes remain active, and this results in chronic inflammation and further damage to the airways.
Neutrophils
Neutrophils are the earliest effectors of the immune response during a lung infection and are a prominent early feature during an IAV infection (48); they are involved in the targeted killing of pathogens (55), and excessive neutrophil responses lead to tissue damage, so neutrophils can also cause pathological damage in the lungs after an IAV infection (47). Neutrophils also release chemokines and preformed granule proteins that attract monocytes and/or macrophages to the site of infection, thereby forming an immune infiltration of the lungs (42). Neutrophils in the lungs combined with influenza-infected cells strongly induce the formation of neutrophil extracellular traps (NETs), which also exacerbate airway disease when NETs are involved in pathogen elimination. Neutrophils can have additional adverse effects on the airways, including airway stenosis caused by airway remodeling, neutrophil elastase-mediated mucus secretion, and increased airway smooth muscle reactivity; this causes a rapid decline in lung function (2). Thus, although inflammation and remodeling may not always occur simultaneously, it is clear that neutrophils produce chemokines and proteases and, thus, actively participate in both processes. In turn, the formation of NETS may be regulated by lung epithelial cells and other recruited neutrophils (57).
Dendritic cells
Dendritic cells (DCs) are macrophage-like cells with antigen-presenting functions in airway epithelial cells (13,50,67). DCs process allergens into peptides and migrate to the local lymph nodes, where they provide the allergic peptides to unformed T lymphocytes to program the production of allergen-specific T cells; DCs that reach the site of infection in the lungs are susceptible to infection, which may improve their ability to present antigens to CD8 T cells (5,11,20,54). Immature DCs in the respiratory tract promote the differentiation of helper T (Th2) cells (18). DCs have a distinctive ability to activate naive T cells and to elicit a primary immune response in the lymph nodes. However, DCs are not just carriers for delivering antigens to lymph node T cells. The DC has an active, fine-tuning function that integrates information from the lungs and delivers it to the regional lymph nodes. Subsequent immune responses depend on the type of pathogen and whether the tissue is damaged; however, this transmission is often disrupted in a variety of lung diseases, particularly in serious diseases, such as influenza A infection (28).
NK cells
NK cells are large granular lymphocytes that are armed with cytotoxic and cytokine-producing capabilities. Killing occurs through three main mechanisms: release of perforin and granzymes, Fas ligand-mediated induction of apoptosis, and antibody-dependent cellular cytotoxicity. A large number of NK cells are present in steady-state lung tissue and are recruited in even greater numbers within the first few days following influenza infection in mice. NK cells in the lung are activated to secrete IFNγ, release perforin, and granzymes to kill target cells and strengthen the subsequent CD8+ T cell response. In human influenza infection, peripheral blood NK cells were decreased in multiple studies. Unlike in mouse studies, NK cells were not found in lung tissues but these studies were limited to examining tissue from fatal cases that may not have allowed them to capture the time point of NK cell infiltration. In terms of immune pathology, NK cells are relatively clean innate cells in part because of their control by a multitude of inhibiting receptors. However, compared with other innate cells, they exhibit far greater host genetic variations and susceptibility to change in function. These polymorphisms often result in loss of function and therefore increased viral escape from control contributing to worse outcome (Fig. 1).
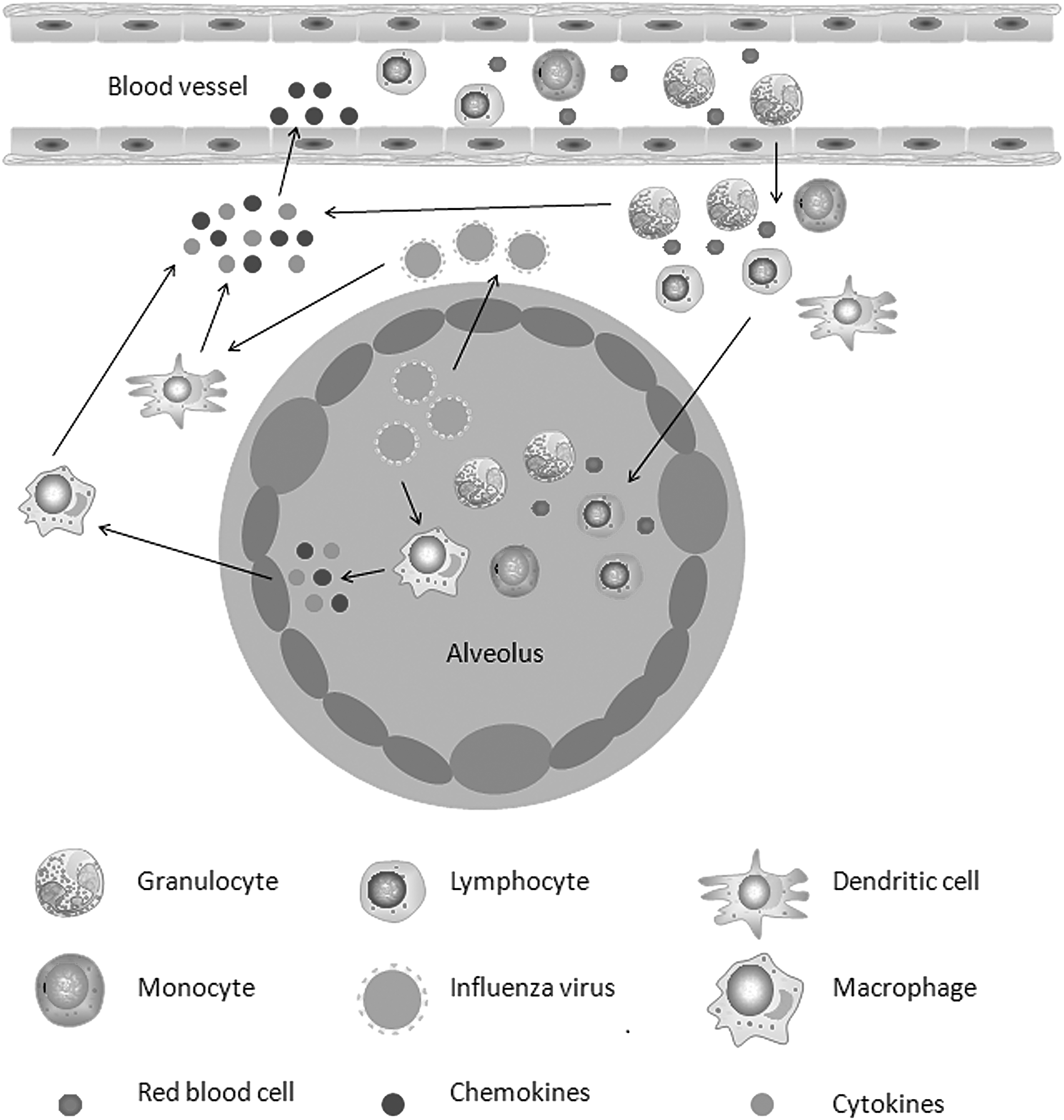
Cytokine storms in the lungs following an influenza virus infection. A viral infection of the lung epithelial cells and alveolar macrophages produces progeny viruses and leads to the production and release of cytokines and chemokines. Cytokine/chemokine-activated macrophages and virus-infected dendritic cells result in a broader immune response and in the production of more cytokines and chemokines. The released chemokines draw more lymphocytes, monocytes, and granulocytes from the blood vessels to the site of inflammation, releasing additional chemokine and cytokine formation cycles to amplify cytokine storms.
Innate Immune Responses and Inflammatory Responses After an Influenza A Infection
An influenza A infection stimulates the innate immune system, the first line of defense against viral infections, to produce type I IFNs and proinflammatory cytokines. Cytokines act through receptors, and are especially important in the immune system; cytokines modulate the balance between humoral and cell-based immune responses, and they regulate the maturation, growth, and responsiveness of particular cell populations. Some cytokines enhance or inhibit the action of other cytokines in complex ways. IAVs and their infected cells can be recognized by pattern recognition receptors (PRRs) on innate immune cells, such as airway epithelial cells, inflammatory DCs, macrophages, monocytes, and neutrophils. There are several types of cellular PRRs, as follows: membrane-associated Toll-like receptors (TLRs), C-type lectin receptors, cytoplasmic nucleotide-binding oligomerization domain-like receptors, and retinoic acid-induced gene-like receptors (49). The viral RNA in the endosome and cytoplasm is predominantly recognized by membrane-associated TLRs and RIG-I-like receptors, respectively.
PRRs recognize pathogen-associated molecular patterns (PAMPs) on different types of microorganisms (4,14). The engagement of PRRs by PAMPs on IAVs activates PPR-related signaling and induces the expression of various proinflammatory cytokines and chemokines to recruit innate immune cells (such as macrophages, neutrophils, and DCs) into the infected airway. These inflammatory cells control the replication and spread of the virus in the early process of infection by secreting inflammatory cytokines and inducing phagocytosis. However, aberrant inflammatory responses may lead to the development of cytokine storms and severe IAV-induced immunopathy (34). Therefore, a therapeutic strategy that targets PRRs should control an appropriate innate response to limit IAV replication, inflammation, and the potential for the development of immunopathy in the lung.
The TLRs on the cellular membrane can interact with their specific extracellular and endosomal PAMPs through the toll-interleukin receptor (TIR)-containing adaptor molecule-1 (TICAM-1)/TIR domain-containing adaptor-induced IFN β (TRIF) and myeloid differentiation primary response gene 88 (MyD88) adaptor. Subsequently, the activated TLRs trigger downstream signaling, such as NF-κB and/or IRF, to induce the expression and production of type I IFN, proinflammatory cytokines, and costimulators (25,62). Different TLRs selectively activate relative signaling. For example, TLR4 and its associated MyD88 selectively activate NF-κB and IRF signaling, whereas TLR3 and its related TRIF activate TRIF-dependent signaling. In addition, NF-κB activation is regulated by many factors. TANK-binding kinase 1 (TBK1) and IκB kinase (IKK) can phosphorylate IκB proteins, an inhibitor of NF-κB, to promote the nuclear translocation of NF-κB and the transcription factor IRF3 that induce proinflammatory cytokine expression. Other factors, including tumor necrosis factor (TNF) receptor type 1-associated death domain, fast-associated protein and death domain, receptor interacting protein 1 (RIP1), caspase-8, and caspase-10, can activate the IKKα, IKKβ, and IKKγ proteins to form the IKK complex; this regulates the NF-κB cascade (26,59,75). The expressed IFNs can, in turn, induce the expression of many IFN-stimulated genes (ISGs) to amplify antiviral activity and regulate cellular metabolism, proliferation, and immunomodulation (58,59). During the process of an IAV infection, activated transcription factors, including NF-κB and IRF3/7, can act synergistically to promote antiviral proinflammatory responses and to produce high levels of cytokines and chemokines that coordinate inflammation in the lungs (Fig. 2) (1,3,8).
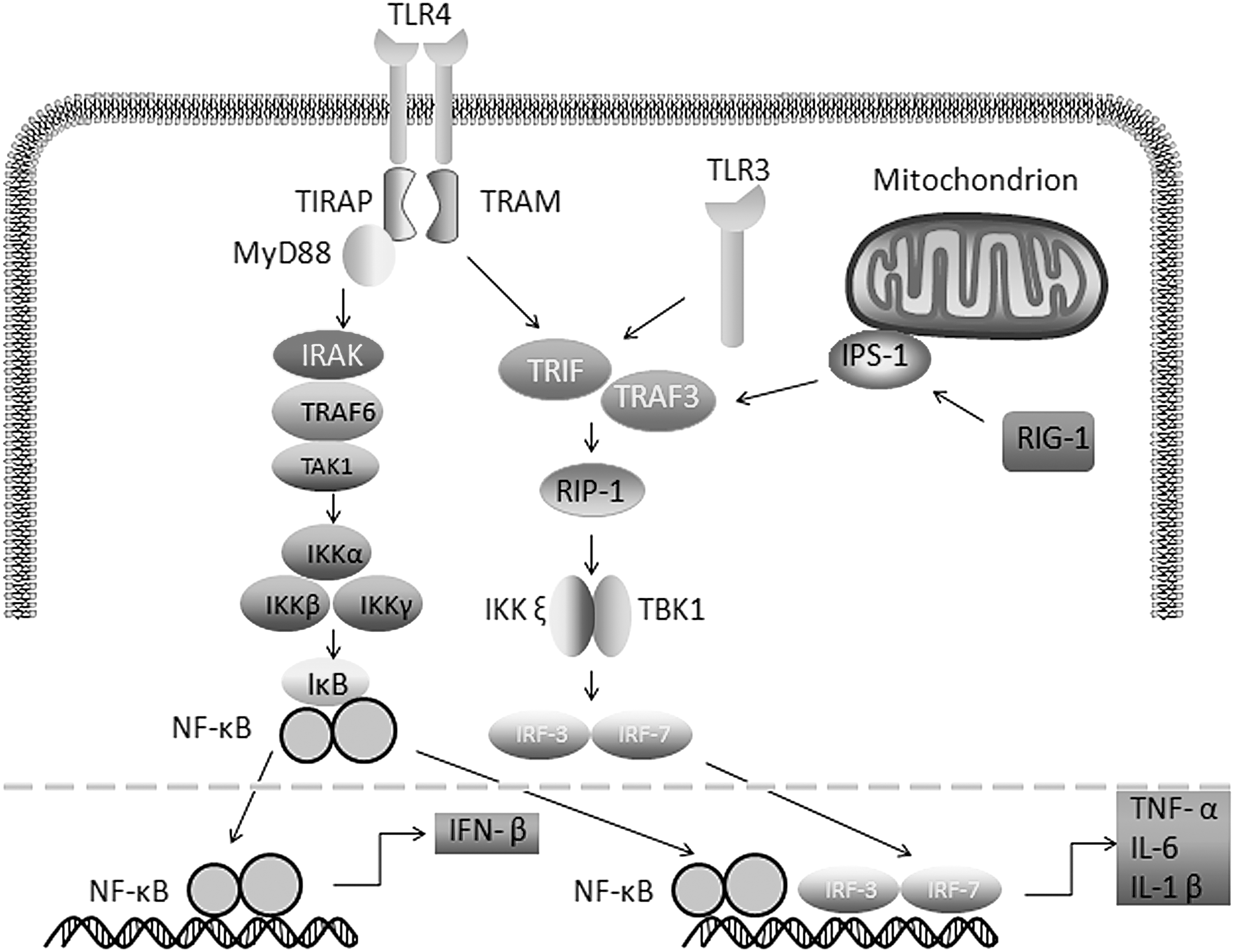
Cytokine signaling pathway following an influenza virus infection. TIRAP recruits MyD88 to TLR 4, and MyD88 signaling involves the activation of IRAKs. TRAF6 induces the activation of TAK1, which leads to the activation of IKK. The IKK signaling pathway leads to the induction of the transcription factor NF-κB. TLR3 recruits adaptors of the aptamer TRIF, which further activates NF-κB. After recognition of ssRNA by RIG-I in the cytoplasm, the complex further activates NF-κB and IRF-3. IFN, interferon; IL, interleukin; IKK, IκB kinase; IRAK, interleukin-1 receptor-associated kinase; IRF, IFN regulatory factor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; TIRAP, toll-interleukin receptor adaptor protein; TRIF, Toll/IL-1R domain to induce IFN-β.
While activated NF-κB signaling induces proinflammatory cytokine production, it also triggers the expression of IκB proteins and anti-inflammatory cytokines to attenuate NF-κB signaling and inflammation. Thus, the outcome of an IAV infection is determined by the balance of proinflammatory and anti-inflammatory responses in the lungs. Proinflammatory cytokines, such as IFNs, interleukins (ILs), TNFα, and chemokines, including RANTES, MCP-1, and IL-8, are major players in antiviral activity. They can communicate between immune or nonimmune cells, recruit leukocytes to activate epithelial cells by enhancing major histocompatibility complex expression, promote cell proliferation and differentiation, and mediate antigen-specific immune responses to drive several important reactions against IAV infection. On the other hand, these proinflammatory cytokines can also damage the airway epithelium. Thus, virus-related inflammation is associated with not only viral clearance but also pathological injury in the lungs. During the process of an IAV infection, there are different cytokine waves from the initial early responses of RANTES, MCP-1, IL-8, IFN-α, IFN-β, and IFN-κ (24,32,74) to the later responses of IL-1α/β, IL-6, TNF-α, IL-18, IFN type I, MCP-1, MIP-1α, MIP-1b, RANTES, MCP-3, MIP-3α, and IP-10, which are mainly secreted by infected macrophages in the lower respiratory tract (23). Uncontrolled inflammatory responses, together with viral virulence, may lead to severe lung injury (30,73). Therefore, the levels of cytokine storms will determine the severity of the disease during an influenza infection (29,38).
It is well known that a serious IAV infection may increase the risk of strong lung inflammation. Although a strong inflammatory response is also necessary for the early control of an acute infection and for inducing antigen-specific immune responses to a viral infection, it may also cause tissue damage, and even death, of the patients (8). Accordingly, controlling inflammatory responses to viral replication, inducing antigen-specific immunity, and avoiding tissue damage will be optimal for patients with severe influenza A infections.
Since the H1N1 and H5N1 subtypes are widely prevalent and have been studied more, the following two subtypes are taken as an example to focus on the role of the innate cytokine storm induced by the IAV. IAV is an orthomyxovirus that contains the glycoproteins HA and NA. For this reason, they are described as H1N1, H1N2 etc. depending on the type of H or N antigens they express with metabolic synergy. HA causes red blood cells to clump together and binds the virus to the infected cell. NA is a type of glycoside hydrolase enzyme which helps to move the virus particles through the infected cell and assist in budding from the host cells. H5 stands for the fifth of several known types of the protein HA. H1 stands for the first of several known types of the protein HA. N1 stands for the first of several known types of the protein NA. Although the two viruses have similar pathological features, virulence levels, and LD50 values, and they both cause diffuse alveolar damage, they have significant differences in the course and pathological features of ARDS. The difference between the two makes it easy to distinguish between the two subtypes of infection through different pathological features. In the H1N1 subtype infection, when the pulmonary edema intensity is at a low level and results in a histopathological image characterized by dense inflammatory cell infiltration, the disease becomes fatal and forms cuffs around the bronchioles and blood vessels. Second, the H1N1 subtype colonizes the epithelial cells in the upper and lower respiratory tract without any significant preference, whereas the H5N1 subtype is essentially associated with alveolar and terminal bronchioles. In the alveoli, unlike the H1N1 subtype, the H5N1 subtype has a preference for type II lung cells and alveolar macrophages. Finally, although the H1N1 subtype is still strictly restricted to the respiratory system, theH5N1 subtype is transmitted to other organs (51).
The cytokine/chemokine response in H1N1 is weaker than that in H5N1 but is stronger than that in the seasonal H3 flu. IL-8 may be a marker of serious disease because it is significantly higher in both the severe H1N1 and severe seasonal H3 cases. IP-10, MCP-1, MIG, and MIP-1α are proinflammatory chemokines and are significantly higher in H1N1 cases than they are in healthy controls; MCP-1 and MIG are more than twice as high in severe cases (15). TNF-α and IL-6 during H7N9 infection in DCs were similar to those observed in H5N1 or H3N2 virus-infected cells. The levels of other cytokines in DCs infected with H7N9 virus remained moderate and significantly lower than those seen with H5N1 infection or even seasonal H3N2 influenza infection. However, the proinflammatory cytokines, TNF-α and IL-1β, had no decreasing effects on virus replication, suggesting that these cytokines contribute to innate immunity by other mechanisms, like inducing inflammation on the site of infection and recruiting other inflammatory cells to infected tissues (46).
Influenza-related studies often use ferrets as model animals for the following reasons: First, the clinical symptoms of ferrets are similar to those of the human flu. Second, human influenza A can directly infect ferrets. The physiological characteristics of ferrets, including the respiratory tract and sneezing reactions, are consistent with the specific behavior of human influenza, making it the most suitable model for disease research. Early infection with H1N1 or H5N1 causes the TNF-α, IL-6, and IL-8 levels to be upregulated in the extrarespiratory tissues of ferrets. Both the H1N1 and H5N1 infections trigger cytokine responses in the central nervous system (CNS). When the H1N1 or H5N1 virus was inoculated in the CNS, the H5N1 virus infection caused a significant upregulation of cytokines in the spleen, liver, heart, pancreas, and jejunum. In contrast, an H1N1 infection caused a significant downregulation of proinflammatory cytokines in the spleen, liver, and kidneys (61). The upregulation of cytokines in the brain is mild, suggesting that the disruption of the blood–brain barrier is the result of the systemic effects of the cytokine storms produced by the IAV infecting the lungs. Anti-cytokine antibodies and trypsin inhibitors are effective in inhibiting boundary permeability.
The ferrets infected with H1N1 have lower amounts of external cytokine production than the amounts produced by the ferrets infected with H5N1 (53). Although extrarespiratory cytokine responses usually do not cause histological damage, both the TNF-α and IL-6 production by H1N1 and H5N1 infections induce neuronal apoptosis (68). Despite IAVs can enter the CNS and cause local damage (71), these data suggest that CNS diseases may also be mediated by cytokine storms in some Influenza A-infected cases (31,70).
TLR3-dependent inflammatory responses and RIG-I-dependent antiviral responses were triggered during H5N1 and H1N1 infections. The expression of TRIF (TLR3 adaptor molecule) was higher in cells infected with H5N1, whereas the expression of the RIG-I adaptor molecule IPS-1 was not significantly different. The H5N1 virus preferentially activates the TLR3 signaling pathway in lung epithelial cells and induces the release of cytokines (21,33). A significant increase in the levels of cytokines, such as TNF-α, IL6, and IL-1β (i.e., cytokine storms), exacerbates the inflammatory response that leads to apoptosis and decreases the levels of mitochondrial reactive oxygen species, which may lead to vascular dysfunction and multiple organ failure, leading to host death (7,27,56).
The IAV–cytokine–protease cycle of endothelial dysfunction that is induced by severe influenza early in infection may also affect a variety of innate immune cells, such as neutrophils, macrophages, and DCs, leading to elevated cytokine levels. Multiple organ failure is the end result of mitochondrial reactive oxygen species reduction, immunosuppressive agents, and tissue damage. The disruption to the molecular mechanisms of endothelial cell tight junctions and vascular permeability after a cytokine storm may be due to the upregulation of TNF-α, which alters the redox state of the cell, and a decrease in adenosine triphosphate (ATP) synthesis, which leads to mitochondrial damage (44,56,76). The depletion of ATP dissociates the polyprotein-linked complex in the actin cytoskeleton, increasing connectivity. Excessive inflammatory processes caused by IAV infection may become detrimental by the intracellular activation of the NF-κB signaling pathway activator (16,22,37,40,41,72). In addition, interactions between the cytokines and mitochondria lead to the production and activation of vasodilators, resulting in endothelial dysfunction and edema in various organs (76).
Conclusion
In this review, we highlight the innate immune cytokine storms caused by the infection of two common subtypes of IAV. The innate immune cells have a specific role in combating an IAV infection; however, these protective effects may be affected by the relevant immune pathology, which may result in even worse results. As we fully understand the multifaceted effects of innate immune effectors during influenza infections, traditional cytokine storms should be extended to include cytokines that promote pathogenic and protective immunity, including the induction of local inflammation and the elimination of infected cells. This will allow for the regulation of the cellular and molecular immune responses and for the promotion of tissue repair and homeostasis.
Footnotes
Acknowledgments
The project was funded by the Key Laboratory of Health and Family Planning Commission of Jilin Province (Grant no. 3D5172303426), and by the Provincial School Co-construction Industrialization Demonstration Project of Jilin Province [Grant no. SXGJSF2017-1-1(01)].
Author Disclosure Statement
No competing financial interests exist.