
Abstract
As a zoonotic disease, ovine contagious pustular dermatitis (Orf) is a serious threat to sheep as well as humans. Orf virus (ORFV) interferon resistance protein (VIR) is the principal virulence protein that encodes a dsRNA-binding protein to inhibit host antiviral response. p53 is one of the key proteins of the host antiviral innate immunity. It not only enhances type I interferon secretion but also induces apoptosis in infected cells, and plays a crucial role in the immune response against various viral infections. However, it remains to be elucidated what role p53 plays in ORFV replication and whether ORFV's own protein VIR regulates p53 expression to promote self-replication. In this study, we showed that p53 has an antiviral effect on ORFV and can inhibit ORFV replication. In addition, ORFV nonstructural protein VIR interacts with p53 and degrades p53, which inhibits p53-mediated positive regulation of downstream antiviral genes. This study provides new insight into the immune evasion mediated by ORFV and identifies VIR as an antagonistic factor for ORFV to evade the antiviral response.
Introduction
Ovine contagious pustular dermatitis (Orf) is a zoonotic infection caused by Orf virus (ORFV) and has been reported to affect livestock in several countries (7). ORFV can infect antelopes, cats, deer, and other animals, and occasionally humans (4). Goats and sheep infected with ORFV results in infectious pustules, also known as sheep's mouth sores (1). ORFV usually is an acute infection with a high infection and low mortality rate. However, lambs are susceptible to be infected with other pathogens after infection, resulting in increased mortality (8). ORFV is a member of the parapoxvirus genus of Poxviridae. It has a linear double-stranded (ds) DNA with a genome of 134 to 139 Kbs in length. The genome expresses 35 proteins with molecular masses between 10 and 220 KDa on the surface of virus particles (12). ORFV genome consists of a long central coding region with inverted repeats at both ends, and with the major virulence factors distributed at both ends (3). ORFV infection could result in a strong immune response and could repeatedly infect the same host. During infection, ORFV initiates a unique immune evasion mechanism that enables it to survive the host's defense mechanisms.
Major virulence genes in ITRs (inverted terminal repeat structure) included ORFV homologous ovine gene encoding cytokine IL-10 (vIL-10), ORFV interferon resistance gene (OVIFNR), dUTP pyrophosphatase (dUTPase), vascular endothelial growth factor (VEGF), the virus encoding chemokine-binding protein (vCBP), ankyrin (ANK), granulocyte/macrophage colony-stimulating factor (GM-CSF)-inhibiting factor (GIF), apoptosis inducing and inhibiting genes, and ORFV121gene that inhibits the host nuclear factor kappa B (NF-κB) pathway. interferon resistance protein (VIR) is a nonstructural protein expressed early in ORFV infection. It can antagonize the host immune response and create a favorable condition for virus replication and proliferation (6). The VIR is located about 20 kb from the 5′ region of the ORFV genome, and encodes a protein that is 190 amino acids (10,15). VIR encodes two proteins, that is, VIR (25 kDa) and shVIR (12 kDa) in host cells (17). VIR protein is a dsRNA-binding protein homologous to E3L of anti-interferon gene of vaccinia virus (VACV), and its nucleotide sequence identity is 44%. Studies have demonstrated that the VIR gene could functionally replace E3L in VACV (11,23). Consistent with this, previous studies have also demonstrated that VIR protein inhibited the activation (by autophosphorylation) of an interferon-inducible, ds RNA-dependent kinase (PKR), so as to win the opportunity for virus proliferation in host cells (5).
p53 is a multifunctional protein that can be activated by a variety of stress signals. p53 can regulate several cellular pathways that determines cell fate, such as cell cycle, differentiation, senescence, and apoptosis (2,17,24). p53 as a tumor suppressor in very early studies has been shown to play a role in various physiological cellular processes (21). Recent studies have shown that p53 is a key protein involved in innate antiviral immunity. It not only enhances type I interferon response to viral infection, but also induces apoptosis in infected cells (13,14,16,22). Both of these properties function to prevent replication of multiple viruses. p53 has been reported to inhibit coronavirus infection (27), and successive studies afterward have demonstrated its ability to inhibit influenza A and dengue virus (DV) replication (9,26). These studies demonstrated that p53 plays a crucial role in the immune response against viral infections.
In the present study, we investigated the role of p53 during ORFV infection and demonstrated that p53 had an antiviral effect on ORFV. We also found that the VIR protein encoded by ORFV itself can interact with p53 and reduce the expression of p53 at the transcription stage, thus inhibiting the positive regulation of downstream antiviral genes mediated by p53, leading to the increase of ORFV replication. So far, we have clarified the role of p53 in ORFV infection, and revealed the new mechanism of ORFV antagonizing the antiviral effect of the host through proteins encoded by itself, deepening the understanding of the natural immunity of the host and the pathogenesis of ORFV.
Materials and Methods
Virus and cell culture
Goat skin fibroblasts (GSF) and Hamster kidney cells (BHK-21) were purchased from the Kunming Cell Bank of the Chinese Academy of Sciences. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and grown at 37°C, 5.0% CO2. ORFV LY/2012/Chinese strain ORFV virus was isolated in our laboratory and propagated using GSF cells.
Plasmids and antibodies
The plasmid expressing wild-type p53 was constructed from cDNA derived from BHK-21 cells. The polymerase chain reaction (PCR) product was digested with HindIII and BamHI and cloned into the pcDNA3.1 vector to generate pcDNA3.1-p53 eukaryotic expression vector. The VIR protein used in this study was expressed and purified in our laboratory. Commercial antibodies used in the study included anti-p53 monoclonal antibody (Santa Cruz Biotechnology), anti-GFP monoclonal antibody (ProteinTech), and anti-β-actin monoclonal antibody (Santa Cruz Biotechnology). The monoclonal antibody against the B2L protein of sheep mouth sore virus was prepared in our laboratory.
RNA extraction and quantitative polymerase chain reaction
Total RNA was extracted using TRIzol Reagent (Invitrogen). The isolated RNA was reverse transcribed into cDNA using M-MLV reverse transcriptase (Promega) and random hexamer primer (TaKaRa). mRNA levels were quantified by relative quantitative polymerase chain reaction (qPCR) using SYBR Premix ExTaq reagent (TaKaRa) run on the Mx3005P qPCR system (Agilent Technologies). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as the internal control. The relative expression of mRNA was calculated using the comparison cycle threshold (2−ΔΔCT) method. ORFV genome copies in GSF cells were assessed with absolute quantitative qPCR assays. All experiments were performed in triplicate.
Western blot analysis
Cells were cultured in culture dishes until semiconfluent, and then transfected the plasmid expressing the target gene. Collected and lysed cells after transfection. Cell lysates expressing the target protein was separated using sodium dodecyl sulfate/polyacrylamide gel electrophoresis. Proteins were then transferred to Immobilon-P membranes (EMD Millipore, Billerica, MA). The membranes were blocked and then the appropriate primary and secondary antibodies were used to detect the protein of interest. Antibody/antigen complexes were visualized using enhanced chemiluminescence detection reagents (Thermo Fisher Scientific, Waltham, MA).
p53 inhibitor experiments
The p53 inhibitor used in this study was purchased from SIGMA. The p53 inhibitor dissolves in dimethyl sulfoxide (DMSO) and the inhibitor was added to the cell culture medium with Lipofectamine 2000 transfection reagent (final concentration 100 μM).
Transient transfection and virus infection
Transient transfections were performed in BHK-21 cells seeded in cell culture dishes by using Lipofectamine 2000 transfection reagent. The ratio of plasmid to Lipofectamine 2000 transfection reagent is 1 μg: 2 μL. At 24 h posttransfection, cells were infected with ORFV at a multiplicity of infection (MOI) of 0.5. After 1 h, the viral inoculum was removed and refed with DMEM containing 2% FBS.
Proteasome, lysosome, autophagy, and caspase inhibitor assay
The proteasome inhibitor MG132 was purchased from Merck & Co.; the lysosome inhibitor chloroquine diphosphate (CQ), the autophagy inhibitor NH4Cl, and the caspase inhibitor benzyloxycarbony (Cbz)-l-Val-Ala-Asp (OMe)-fluoromethylketone (Z-VAD-FMK) were purchased from Sigma. BHK-21 cells were cultured to a confluence of 80% to 90% and then transfected with pEGFP-VIR plasmid or empty vector using Lipofectamine 2000. The cells were maintained in the presence or absence of MG132, CQ, NH4Cl, or Z-VAD-FMK for 36 h. The collected cells were then subjected to Western blotting.
Coimmunoprecipitation assay
BHK-21 cells were cultured in 10-cm dishes, and the monolayer cells were transfected with various plasmids. The collected cells were then lysed and immunoprecipitated with the indicated antibodies as described previously. The immunoprecipitates and whole cell lysates were then analyzed by Western blotting with specific antibodies.
Statistical analysis
Statistical analysis was performed using SPSS Statistics for Windows. The Student's t-test was used to compare three independent experiments. The data represent results from one of the triplicate experiments. *, p < 0.05 was considered significant; **, while p < 0.01 was considered highly significant.
Results
p53 inhibits ORFV replication during viral infection
The intracellular multifunctional protein p53 mediates the antiviral effects of the immune system. To determine if p53 was involved in the replication of ORFV, a loss-of-function assay was performed on p53. The effect of the p53 inhibitor was evaluated by measuring the p53 mRNA and protein levels in GSF cells. GSF cells in the experimental and control group were harvested after treatment with the p53 inhibitor (Pifithrin-a [PFT]) to measure p53 mRNA and protein levels. As shown in Figure 1A and B, PFT reduced p53 mRNA and protein levels by >80% at 48 h posttreatment. The copy number of ORFV in PFT-treated cells was then analyzed and compared with control treated cells at the indicated time points after viral infection. We found that ORFV replication levels in GSF cells were increased in the absence of p53 expression (Fig. 1C). The results indicated that p53 had an inhibitory effect on ORFV replication during ORFV infection.
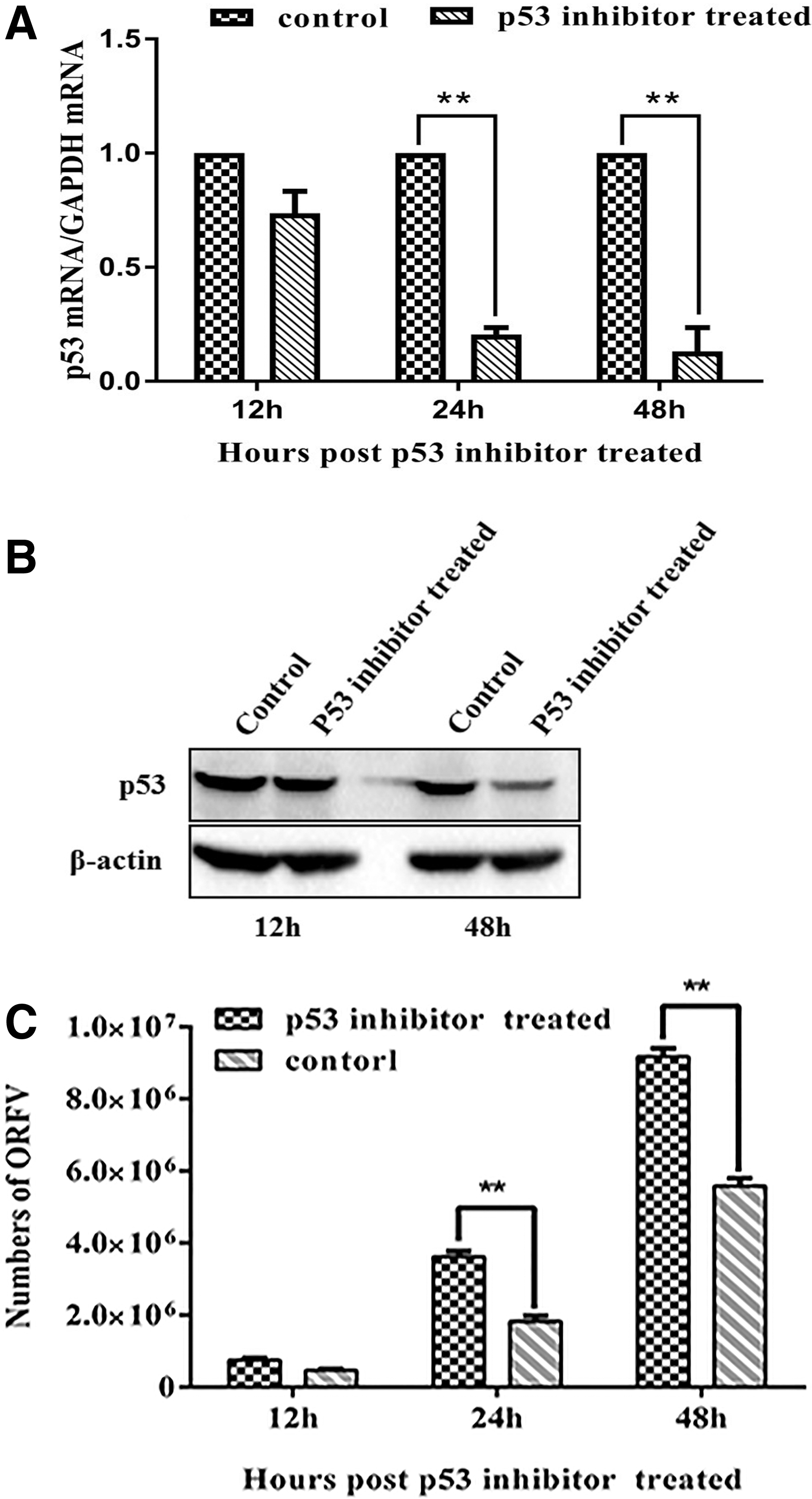
p53 inhibition enhances ORFV replication.
p53 is a positive regulator of downstream antiviral genes
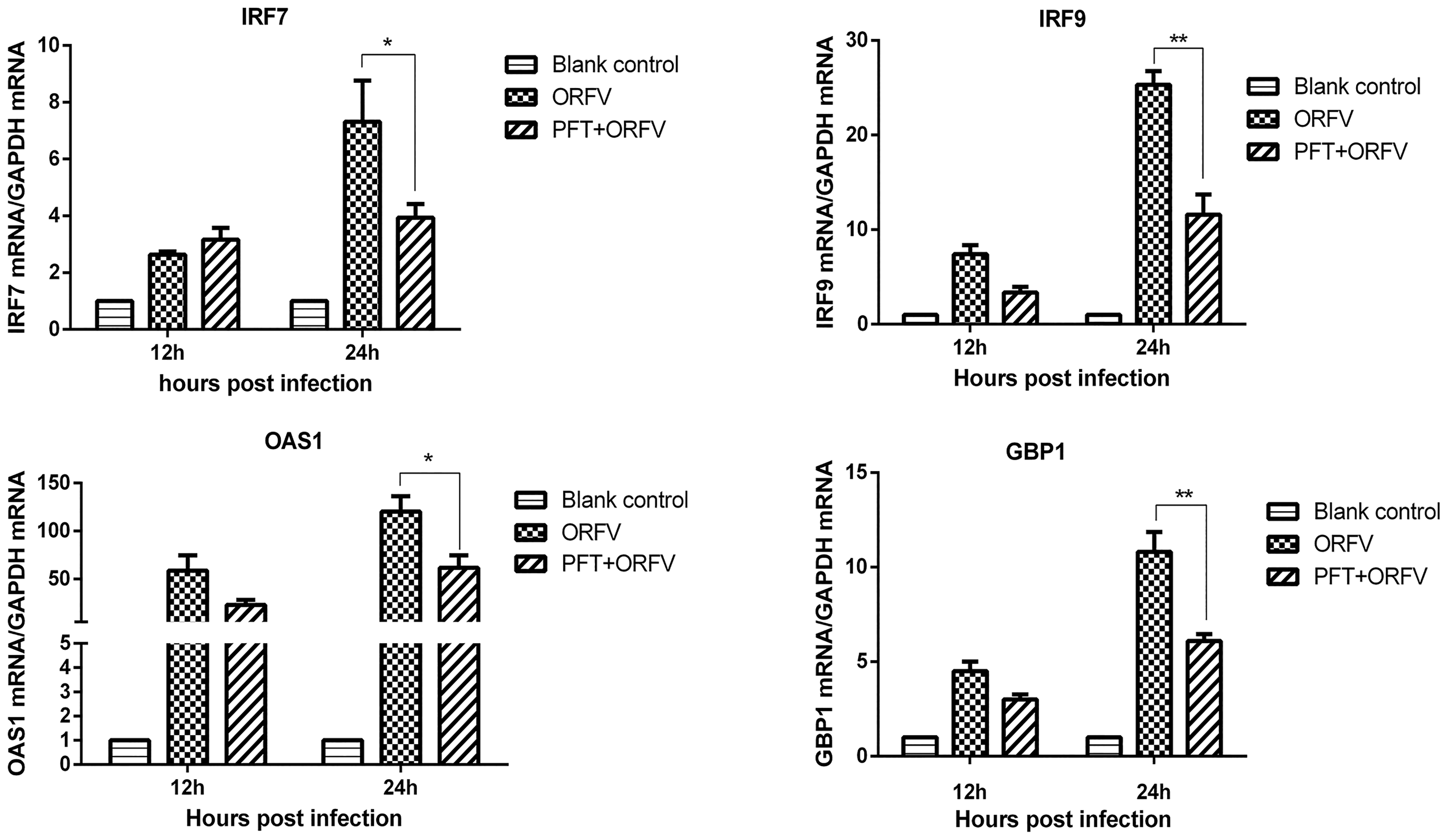
Previous studies had demonstrated that enhanced IAV replication in p53 knockout cells was associated with decreased interferon-stimulated gene (ISG) expression (27). We investigated whether p53 inhibited ORFV infection by increasing the expression of ISGs. We transfected pcDNA3.1-p53 into GSF cells and measured ISGs mRNA levels 12 and 24 h posttransfection by quantitative real-time PCR. As shown in Figure 2A, p53 overexpression increased the transcriptional levels of ISG15, ISG20, IRF7, IRF9, OAS1, and GBP1. After GSF cells were treated with p53 inhibitor, they were infected with ORFV at an MOI of 0.5. The effect of p53 inhibition on downstream ISGs and IRF7/9 expression during ORFV infection, and the changes in ISGs and IRF7/9 transcriptional levels before and after the addition of inhibitors were compared. As shown in Figure 2B, inhibition of p53 expression resulted in a decrease in the expression levels of ISG15, ISG20, IRF7, IRF9, OAS1, and GBP1 genes. This indicated that p53 had a significant positive regulatory effect on the expression of downstream antiviral genes during ORFV infection.
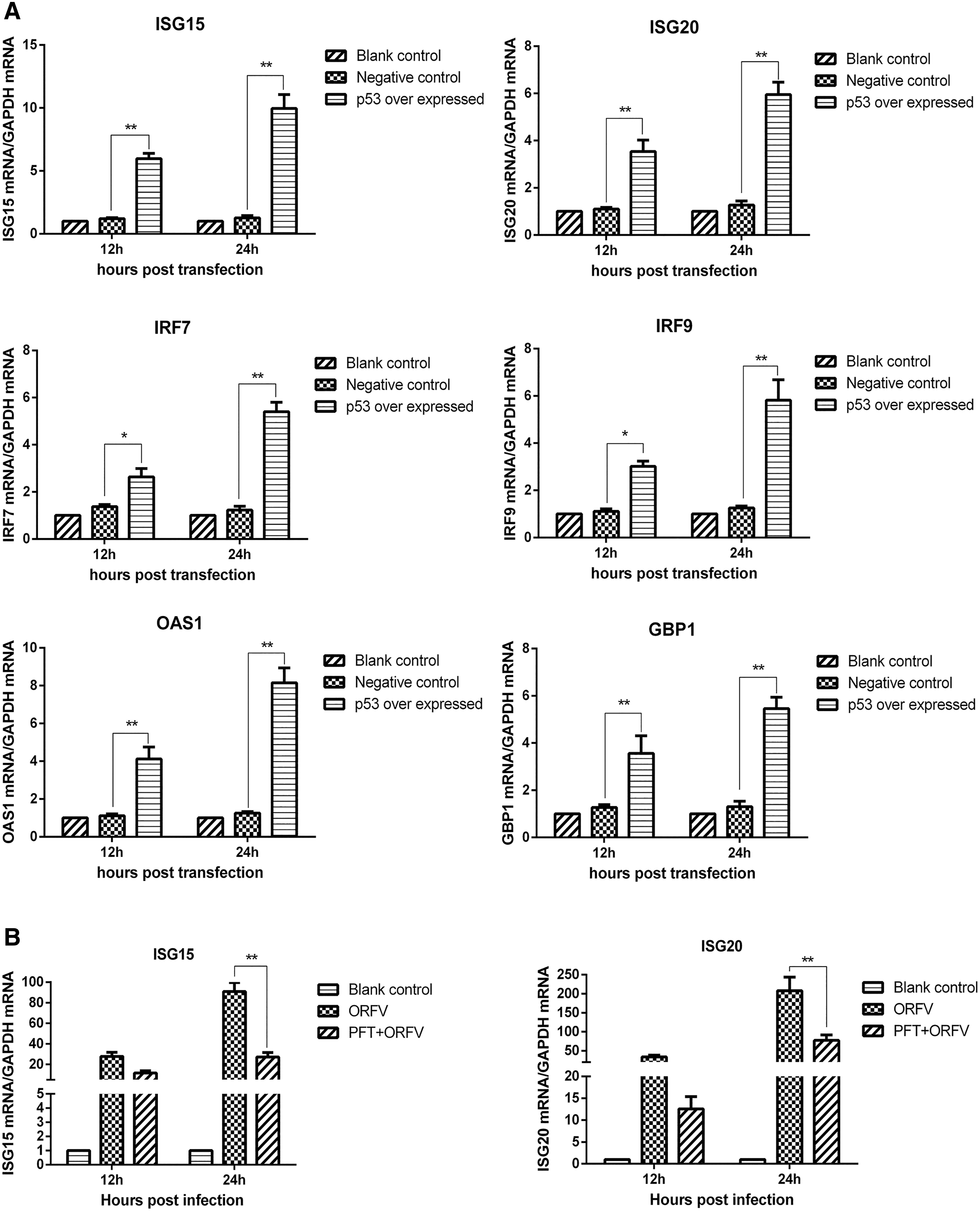
p53 increases downstream antiviral genes transcriptional levels.
ORFV infection inhibits the expression of endogenous p53
p53 had been shown to have an antiviral effect on ORFV, hence we investigated p53 mRNA and protein levels during ORFV infection. GSF cells were infected with ORFV at an MOI of 0.5 and harvested at specified time points. Using quantitative real-time PCR and WB methods, it was found that p53 mRNA and protein levels were decreased after infection (Fig. 3A, B).
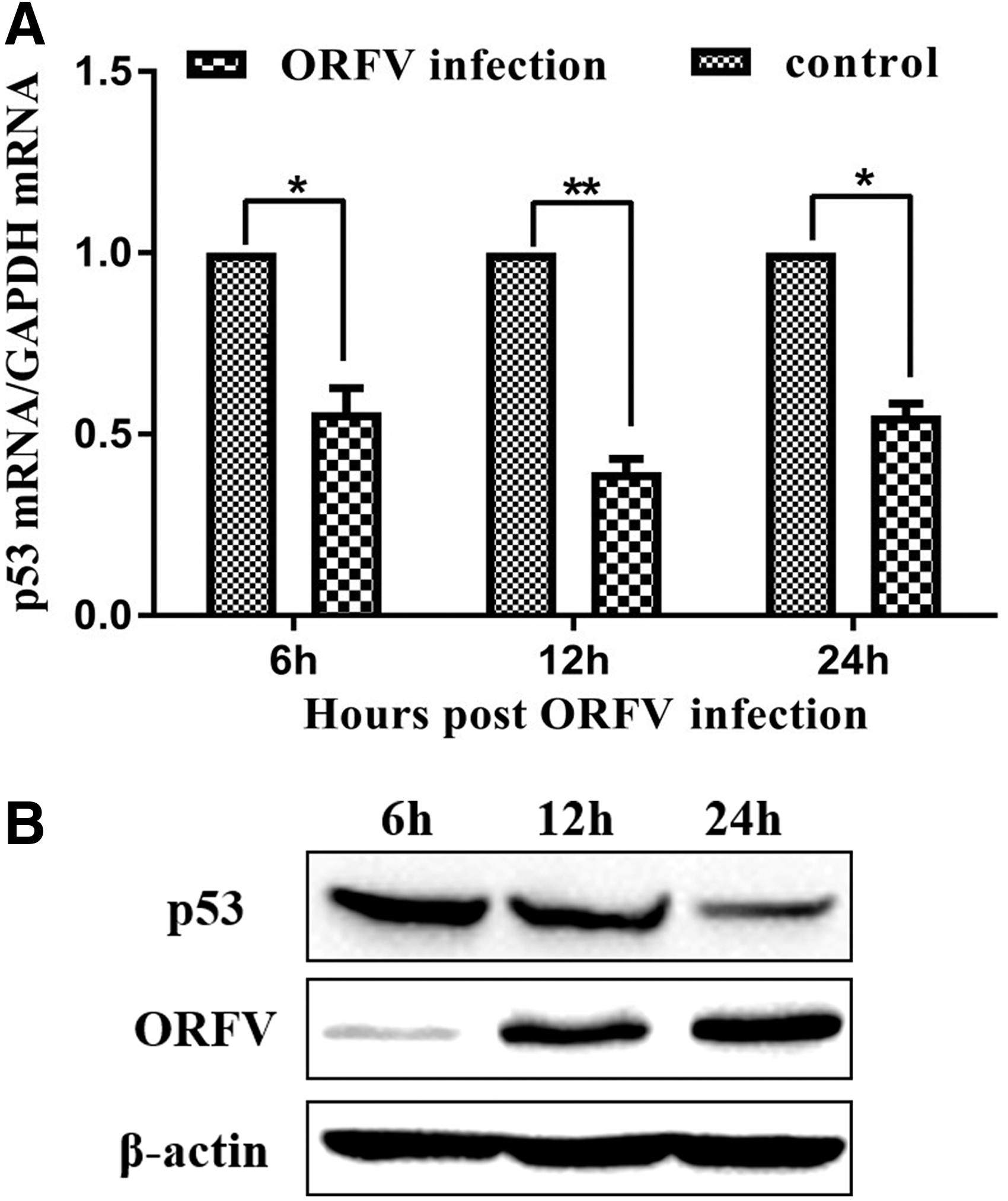
ORFV infection inhibits p53 expression. GSF cells were seeded in 3.5-cm cell culture dishes, and then infected with ORFV at an MOI of 0.5. Cells were harvested at the indicated time points (6, 12, 24 h) and total cellular RNA and proteins were extracted. p53 mRNA levels were quantified relative to the control group. Experiments were performed in triplicate. The results represent data from triplicate experiments. *, p < 0.05 was considered significant; **, while p < 0.01 was considered highly significant.
VIR inhibits p53 expression
The VIR protein was expressed by ORFV during the early stages of infection, however, its role in p53 expression remains to be deciphered. To assess the role of VIR in p53 expression, we transfected the VIR eukaryotic expression vector, pEGFP-VIR and empty vector, into BHK-21 cells to determine its effect on p53 expression levels. As shown in Figure 4A and B, overexpression of VIR resulted in a significant decrease in intracellular p53 mRNA and protein levels.

VIR inhibits p53 expression.
To determine whether the proteasomes, lysosomes, autophagosomes, or caspase-dependent pathways play roles in VIR-induced reduction of p53, the proteasome inhibitor MG132, the lysosome inhibitor CQ, the autophagy inhibitor NH4Cl, and the general caspase inhibitor Z-VAD-FMK were used to evaluate the inhibitive effects. The pEGFP-VIR or empty vector plasmid was transfected into BHK-21 cells, and the cells were cultured with or without the inhibitors. The expression of p53 was detected at 36 h by western blotting. No inhibitory effect of MG132, NH4Cl, CQ, or Z-VAD-FMK on the reduction of p53 were observed (Fig. 4C).
VIR interact with p53
To investigate the interaction between VIR and p53, BHK-21 cells were transfected with Flag-p53 plasmid and pEGFP-VIR plasmid or its empty vector. The cells were lysed, and the lysates were immunoprecipitated (IP) with anti-GFP antibody and analyzed by western blotting. As shown in Figure 5A, GPF pulled down FLAG-p53, which indicates that VIR interacts with p53. Reverse immunoprecipitation experiments were also performed with anti-Flag antibodies, which showed that FLAG-p53 can also immunoprecipitate VIR (Fig. 5B).
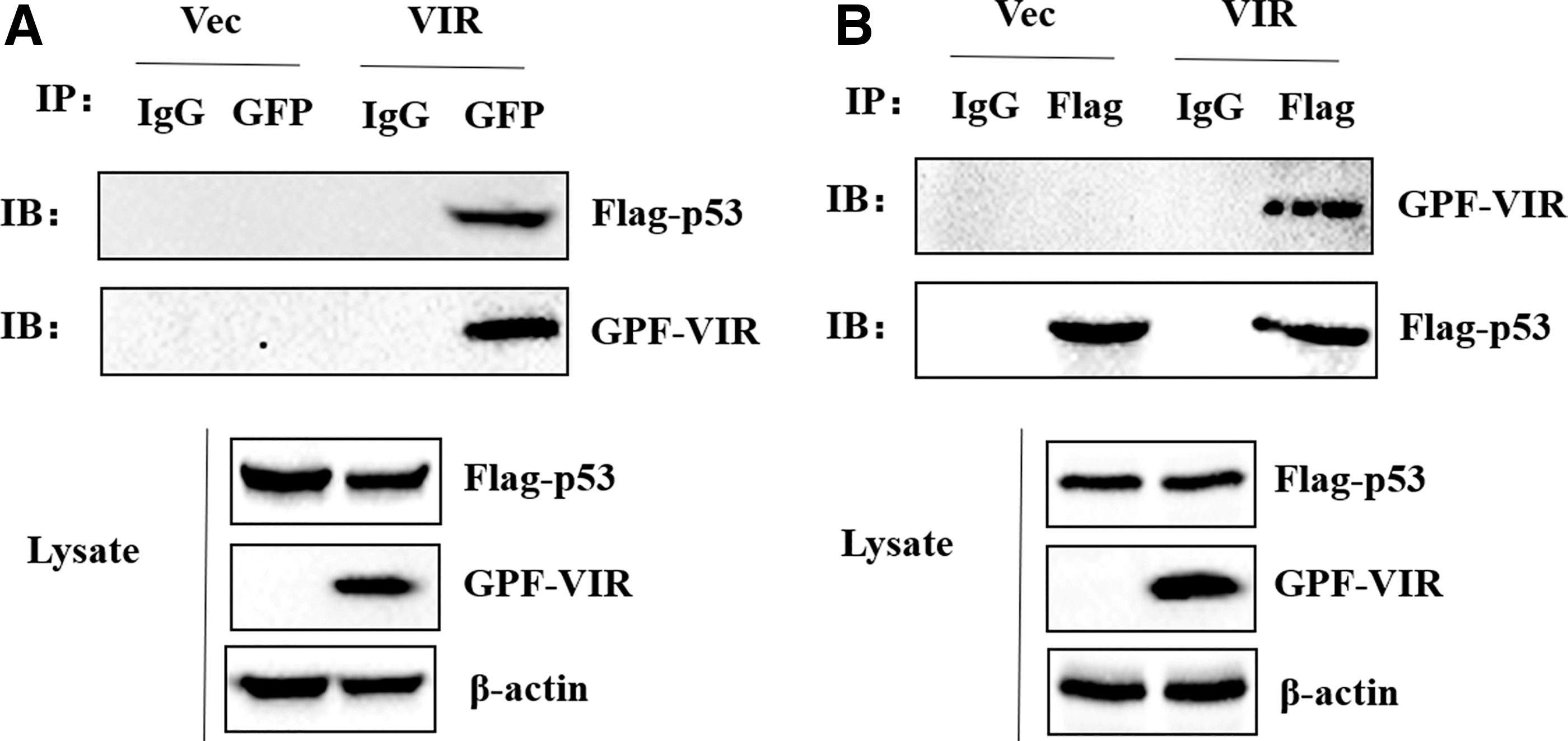
VIR interact with p53.
VIR inhibits p53-mediated positive regulation of downstream antiviral genes
To further investigate the role of VIR in p53-mediated transcription regulation of downstream antiviral genes, pEGFP-VIR and pcDNA3.1-p53 plasmids were cotransfected into BHK-21 cells. Cells transfected with pcDNA3.1-p53 and empty vector were used as the control. After 12 and 24 h, p53 mRNA levels were measured by quantitative real-time PCR. The transcription levels of ISG15, ISG20, IRF7, IRF9, OAS1, and GBP1 genes in the cotransfected cells were reduced to different degrees compared with control cells (Fig. 6). This indicates that VIR could inhibit p53-mediated positive regulation of antiviral genes' transcriptional levels.
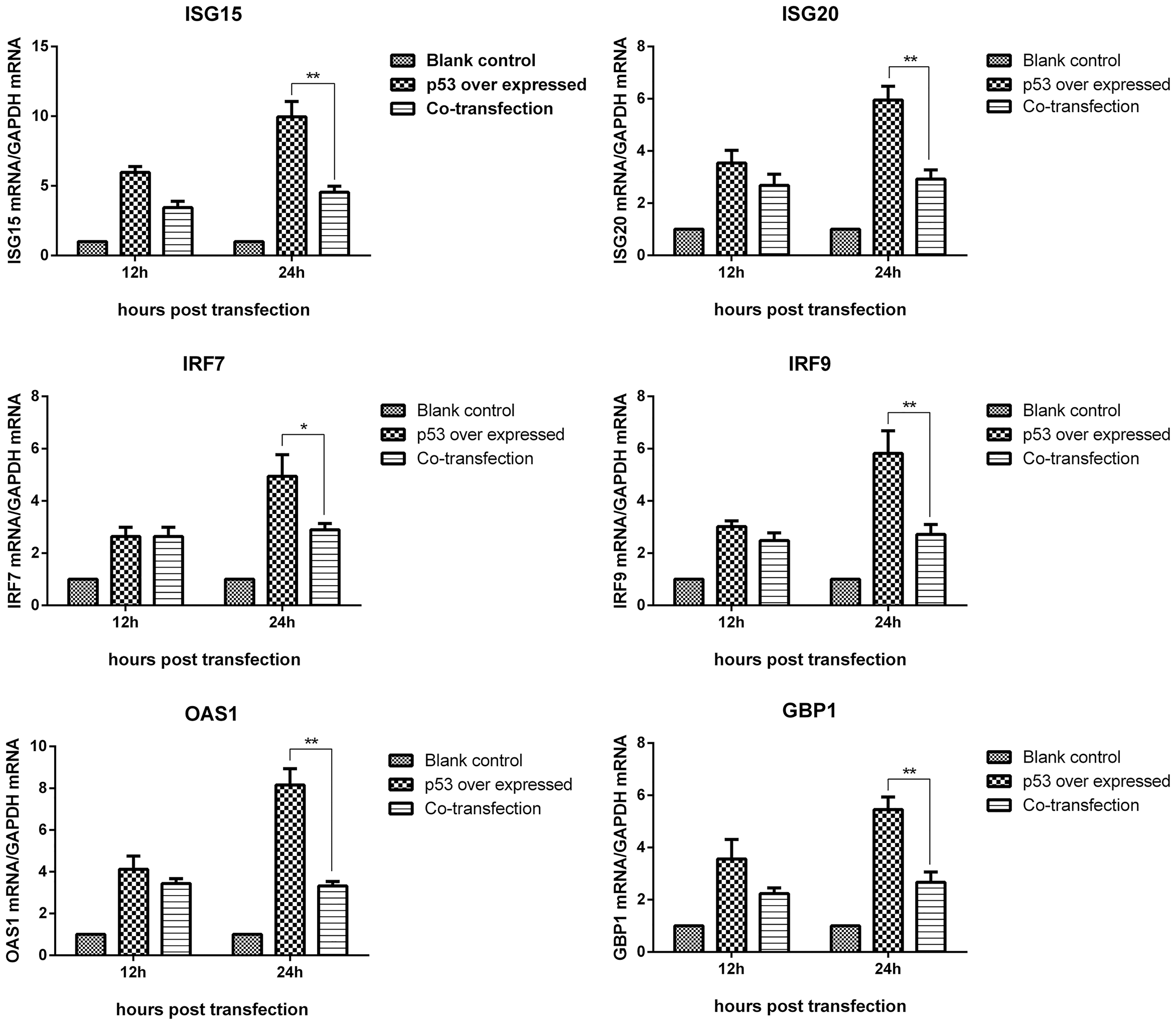
VIR inhibiting the positive regulation of downstream antiviral genes mediated by p53. BHK-21 cells were cotransfected with 2 μg pEGFP-VIR plasmid and 2 μg pcDNA3.1-p53 plasmid. BHK-21 cells transfected with 2 μg pcDNA3.1-p53 and 2 μg pEGFP-VIR EV were used as the positive control and BHK-21 cell transfected with 2 μg pEGFP-VIR EV and 2 μg pcDNA3.1-p53 EV were used as the blank controls. Cells were harvested after 12 and 24 h posttransfection. Expression of IRF7, IRF9, ISG15, ISG20, GBP1, and OAS1 was measured by quantitative real-time PCR. *, p < 0.05 was considered significant, while **, p < 0.01 was considered highly significant.
Discussion
Previous studies have demonstrated that p53 plays a crucial role in antiviral innate immunity. p53 blocks the transmission of DV by inhibiting the type I interferon pathway (9). During infection with pathogens such as influenza A and coronavirus, p53 plays a similar role (26,27). However, whether p53 has an antiviral effect on ORFV is yet to be deciphered. ORFV mainly invades the skin of goats and sheep after infection. Skin cells of sheep have been used as an in vitro model to study the replication and proliferation of ORFV (19). In this study, we compared the replication of ORFV in GSF cells before and after p53 inhibition. The inhibition of p53 can increase the replication of ORFV in GSF cells. Our study further extends the antiviral function of p53 and provides a mechanistic insight into ORFV pathogenesis.
p53 could induce the production of type I IFN, however, type I IFN itself has no antiviral activity. Its antiviral function is mainly through the expression of various ISGs on adjacent cells (25). These ISGs inhibit viral replication in host cells by inhibiting viral protein translation, degrading viral RNAs or inducing apoptosis. By performing gain and loss of function experiments, we demonstrated that p53 positively regulated ISGs and IRF7/9 genes. Overexpression of p53 resulted in the increase of ISGs and IRF7/9 to different degrees. Some of the genes that were positively regulated by p53 included IRF7, IRF9, ISG15, ISG20, GBP1, and OAS1. To confirm our results, we infected cells with ORFV in the presence of a p53 inhibitor. We found that inhibition of p53 expression resulted in a decrease in ISGs and IRF7/9 production. This indicates that p53 had a significant positive regulatory effect on the expression of downstream antiviral genes.
Since p53 can inhibit the replication of ORFV in vivo, we suspect that ORFV may have a series of effects on the expression of p53 to replicate better in vivo. Therefore, we detected the expression level of p53 in GSF cells infected with ORFV, and found that the expression of p53 was significantly inhibited by ORFV infection, thus proving the mutual antagonistic effect of ORFV and p53.
We were interested in how ORFV escaped the host's immune system. We functionally investigated VIR, as it was one of the major virulence factors of ORFV and was expressed early during infection (11). In addition, VIR was demonstrated to antagonize type I IFN (18) to inhibit the host immune response. During the early stages of viral infection, VIR was found to create favorable conditions for virus replication and proliferation, as well as to promote in vivo virus replication (20). However, the role of VIR in ORFV-induced innate immune response is unknown. In the present study, our data showed that ORFV VIR significantly induces the reduction of p53, however, VIR-induced reduction of p53 occurred in a proteasome-, lysosome-, autophagy-, and caspase-independent manner. Therefore, we speculate that VIR may reduce the expression of p53 in the transcription stage. Interactions between viral and host proteins play important roles in modulating viral replication. In this study, the interaction between VIR and p53 was also observed, which indicated that the interaction was involved in suppressing p53 expression. In addition, we found that VIR could inhibit the production of ISGs IRF7, IRF9, ISG15, ISG20, GBP1, OAS1 induced by p53 (Fig. 6). VIR counteracts IFN signaling through multiple mechanisms, such as suppression of PKR activation. We also clarify that the impact on ISG expression is directly due to p53, but not due to the inhibition of PKR activation by VIR (data not shown). Thus, our results indicated that in the early stage of ORFV infection, the inhibition of p53 expression is mediated by VIR, which inhibits p53-mediated positive regulation of downstream antiviral genes and promotes the replication of ORFV.
Conclusions
In summary, our results demonstrated the antiviral effects of p53 during ORFV infection. In addition, VIR protein encoded by ORFV itself can interact with p53 and reduce the expression of p53 at the transcription stage, thus inhibiting the positive regulation of downstream antiviral genes mediated by p53, leading to the increase of ORFV replication. The results of this study not only elucidates the pathogenic mechanism of ORFV, but also provides how ORFV virulence factors evade the host immune system. We hope that our findings help with the development of new vaccines and therapeutics to eliminate viral outbreaks in livestock.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Key R&D Program of China (2018YFD0502100), and the China Agriculture Research System (CARS-39).