
Abstract
Transmissible gastroenteritis virus (TGEV) is a coronavirus, which causes fatal severe diarrhea and leads to high mortality in newborn piglets. Inflammasomes are hub molecules that induce proinflammatory cytokine production and maturation to initiate innate immune defenses upon cellular infection. To date, the potential role of inflammasome in TGEV infection in porcine intestinal epithelial cells has not been elucidated. The present study aims to investigate the function of the inflammasome in response to TGEV infection in porcine intestinal epithelial cells. Our results revealed that TGEV infection induced the production of pro-interleukin-1β (pro-IL-1β) and enhanced its processing and maturation in porcine intestinal epithelial cells through caspase-1 activation. In addition, TGEV infection in porcine intestinal epithelial cells induced pyroptosis, indicated by cell death and the production and cleavage of gasdermin D (GSDMD). Meanwhile, TGEV infection sufficiently activated the expression and assembly of the NOD-like receptor protein 3 (NLRP3) inflammasome in porcine intestinal epithelial cells, and inhibition of NLRP3 blocked TGEV-induced IL-1β release. We also found that inhibition of NLRP3 enhanced the replication of TGEV without inducing cell death. In conclusion, these data demonstrated that activation of IL-1β release and pyroptosis is dependent on NLRP3 inflammasome, thus NLRP3 inflammasome may play a central role in the innate immune response to TGEV infection.
Introduction
Porcine transmissible gastroenteritis, characterized by vomiting, rapid dehydration, severe watery diarrhea, and with high morbidity, is caused by infection of transmissible gastroenteritis virus (TGEV) (19). TGEV is a member of genus Coronavirus with a 28.5 kb, positive-sense, single-stranded RNA genome, and its diameter is about 100–150 nm (14). TGEV can infect both newborn and adult pigs, and although it often leads to just mild diarrhea in adult pigs, in suckling piglets younger than 2 weeks, the mortality often reaches 100%, thus bringing about huge losses in the breeding industry (14).
TGEV is mainly transmitted through the digestive and respiratory tracts and it infects epithelial cells from the apical side, infuses into cells and replicates, and then gets released into the lumen, initiating cell-to-cell spreading (30). The membrane infusion of TGEV and epithelial cells are mediated by cellular receptor such as CD13 epidermal growth factor receptor (1). Because intestinal epithelial cells are the major cells involved in the nutrition absorption, balance of water absorption, and secretion, their infections and thereafter impairment are underlying mechanisms of diarrhea (4).
Porcine IPEC-J2 intestinal epithelial cells are susceptible to TGEV, making them as practical model for pathological study of TGEV (3). Although previous studies have demonstrated that unbalanced inflammatory response, complete mitophagy, mitochondrial injury, and apoptosis, are important mechanisms underlying epithelial cell impairment (48). The precise molecular mechanisms of intestinal injury are poorly understood and limited information is known about the potential signaling pathways mediating inflammatory response caused by TGEV.
Previous studies on TGEV infection-induced intestinal epithelial cell death mainly focus on apoptosis (15,45,48). Pyroptosis, which is NOD-like receptor protein 3 (NLRP3) inflammasome, and caspase-1 dependent, is a novel modality of inflammatory cell death (24,43). However, to date, few studies have explored the role of pyroptosis in TGEV infection-induced intestinal epithelial cell death. In this study, we explored the role of inflammasome and pyroptosis in TGEV infection pathology in IPEC-J2 cells. We observed that TGEV infection significantly induced interleukin (IL)-1β production and cell pyroptosis in a NLRP3 inflammasome-dependent manner. In addition, NLRP3 inflammasome inhibition effectively blocked pyroptosis and promoted TGEV replication, indicating that the NLRP3 inflammasome may act as a therapeutic target in porcine TGEV injection.
Materials and Methods
All experimental procedures were approved by the Southwest University Animal Care Committee.
Cells and antibodies
IPEC-J2 cell lines were obtained from the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 μg/mL penicillin/streptomycin (Gibco), and 16 mM HEPES and were incubated at 37°C in an atmosphere with 5% CO2. Primary antibodies used in the study included rabbit polyclonal anti-IL-1β (AF-401-NA; R&D Systems), rabbit monoclonal anti-NLRP3 (19771; Proteintech), mouse monoclonal anti-GAPDH (10494; Proteintech), rabbit monoclonal anti-activating signal cointegrator (ASC) (10500; Proteintech), rabbit polyclonal anti-Gasdermin D (GSDMD, 39754S; Cell Signaling Technology), rabbit polyclonal anti-cleaved N-terminal GSDMD antibody (ab215203; Abcam), rabbit monoclonal anti-Caspase-1 antibody (sc-398715; Santa Cruz Biotechnology), and rabbit polyclonal anti-caspase-1 p10 (PA5-39882; Thermo Fisher Scientific). Corresponding secondary antibodies were horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG), HRP-conjugated goat anti-mouse IgG.
Virus and infection
TGEV (SHXB strain) was obtained from the Chinese Academy of Sciences (Shanghai, China), and propagated in swine testicular cells. The complete genome sequence for TGEV SHXB is available in GenBank (KP202848.1) (39). IPEC-J2 cells were infected with TGEV at the indicated multiplicity of infection (MOI) at 37°C. The medium and unattached virus were removed and replaced with fresh growth medium after infection. Infected cells were collected and analyzed after the indicated incubation period.
Caspase-1 activity assays
The Caspase-1 Activity Kit (Beyotime, Shanghai, China) was used to measure caspase-1 activity following the manufacturer's protocols. The basic principle is that caspase-1 catalyzes the substrate acetyl-Tyr-Val-Ala-Asp p-nitroanilide (Ac-YVAD-pNA) to produce yellow pNA. In brief, cells were collected and lysed on ice for 15 min, then the cell lysis was centrifuged at 16,000 g for 10 min and the supernatant (50 μg protein) was mixed with synthetic tetrapeptide Ac-YVAD-pNA and incubated at 37°C for 2 h. After incubation, the optical density at 405 nm was measured using a 96-well plate reader (Thermo Fisher Scientific).
Enzyme-linked immunosorbent assay
The concentrations of IL-1β were determined using a Porcine Quantikine Enzyme-Linked Immunosorbent Assay (ELISA) Kit (R&D Systems) following the manufacturer's protocols.
Detection of lactic acid dehydrogenase release
Lactic acid dehydrogenase (LDH) release was detected to measure cell lysis using the LDH Cytotoxicity Detection Kit (Roche Applied Sciences, Mannheim, Germany), following the manufacturer's protocols. Briefly, 100 μL of cell supernatant was collected and the amount of LDH released was immediately measured (sample LDH released), Then target cells were lysed in 1% Triton X-100 to measure maximal LDH release. The percentage of lysed cells was calculated as (sample LDH released/maximal LDH release) × 100%.
Western blot analysis
Cell lysis buffer (P0013; Beyotime) supplemented with 1 mM phenylmethylsulfonyl fluoride (ST506; Beyotime) was chilled on ice. Cell lysates from TGEV-stimulated or control samples were centrifuged at 13,000 g for 25 min at 4°C, and the supernatant was collected. Next, the protein concentration of the supernatant was determined by the Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology, China). The same amount of proteins for each sample was separated using 12% sodium dodecyl sulfate/polyacrylamide gel electrophoresis gels and then transferred onto polyvinylidene fluoride (PVDF) membranes. Next, the PVDF membranes were blocked with 5% skim milk and then incubated with primary antibodies, and after being washed, the membranes were incubated with corresponding HRP-conjugated secondary antibodies. The protein bands were detected and imaged by the ECL Plus Kit (P0018; Beyotime) and chemiluminescence imaging system (GS-800; Bio-Rad), and finally quantified by Image Lab 6.0 software according to the user's guide. Complete Western blot membranes and representative images of flow-cytometry were shown in Supplementary Data.
Cell death assay
The cell death was measured by Propidium Iodide (PI) staining as previously described (25). Briefly, cells were collected and stained with PI at a final concentration of 1 μg/mL. PI is membrane impermeant and usually excluded from live cells. Stained cells were analyzed using BD FACS Array software on a BD FACS Array flow cytometer (BD FACSVerse; BD Biosciences).
Statistical analyses
Data are presented as the mean ± standard deviation. Student's t-test was used to compare the mean between two groups and one-way analysis of variance and Bonferroni correction was used to compare the mean among three or more groups. SPSS 13.0 (SPSS, Inc.) was used to perform statistical analyses. Significance was defined as p < 0.05.
Results
TGEV stimulated the maturation of IL-1β in porcine intestinal epithelial cell
First, the effects of TGEV infection on expression and secretion of IL-1β was investigated. IPEC-J2 cells were cultured and infected with TGEV at MOIs of 0.1 or 1 for 12, 24, 36, and 48 h. Cell supernatant and lysate samples were harvested at indicated time points. Then secreted mature IL-1β in cell supernatant was assessed using the Porcine IL-1β Quantikine ELISA Kit. We observed that IL-1β secretion in TGEV infection groups was markedly increased compared with that in mock-infected cells, and increase in IL-1β secretion was dose dependent (Fig. 1A). Meanwhile, western blot analysis revealed that pro-IL-1β expression was enhanced by TGEV infection, and similar changes was observed in IL-1β p17 subunit (Fig. 1B).
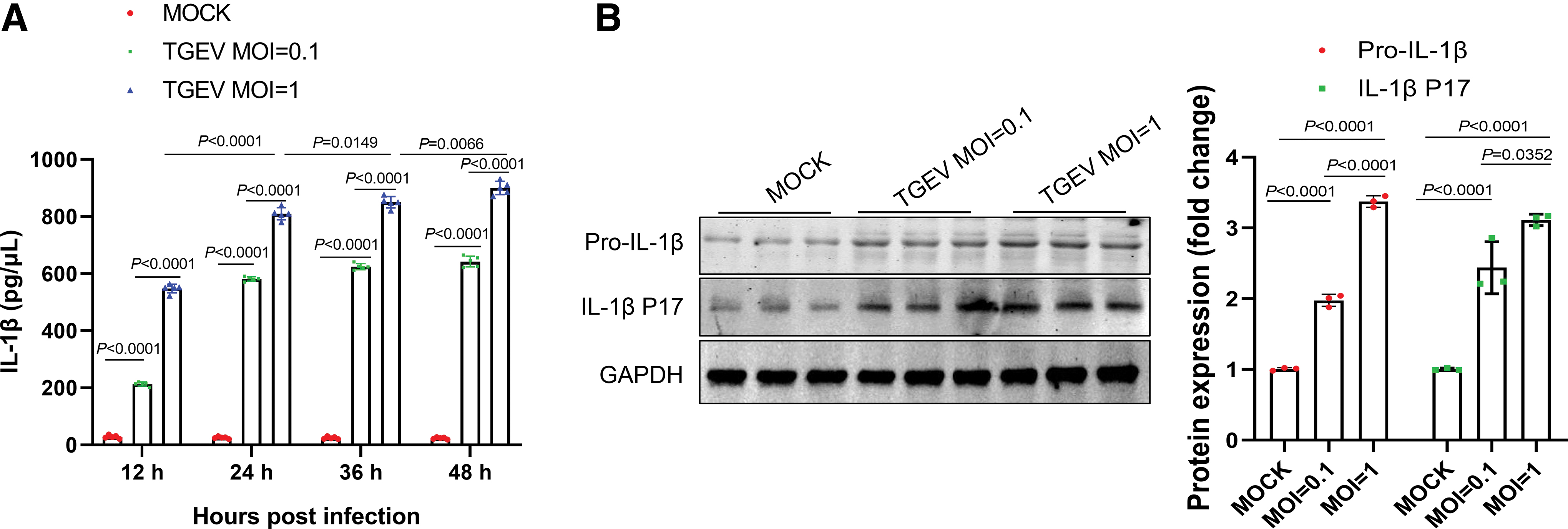
TGEV infection induced the maturation and secretion of IL-1β.
Caspase-1 activation was required for TGEV infection-induced IL-1β induction in porcine intestinal epithelial cell
The immature pro-IL-1β will be cleaved into mature IL-1β upon inflammatory stimuli, during which activation of caspase-1 is necessary (42). Caspase-1, one of the proinflammatory subfamily of caspases, has been reported to mediate pyroptosis, in which cells swell, lyse, and then release intracellular contents, including proinflammatory cytokines (9). Therefore, we asked whether TGEV infection in IPEC-J2 cells causes activation of caspase-1.
As shown in Figure 2A, TGEV infection of IPEC-J2 cells at MOI of 1 and 0.1 significantly increased the activity of caspase-1 compared with that in mock-infected cells. Even at an MOI of 0.1, the activity of caspase-1 in IPEC-J2 cells infected with TGEV was ∼1.7 times higher than that in the mock group (Fig. 2A). Furthermore, protein levels of the cleaved caspase protein and the pro-caspase-1 were further evaluated by western blot analysis. As shown in Figure 2B, TGEV infection resulted in increased protein level of both cleaved caspase protein and the pro-caspase-1.
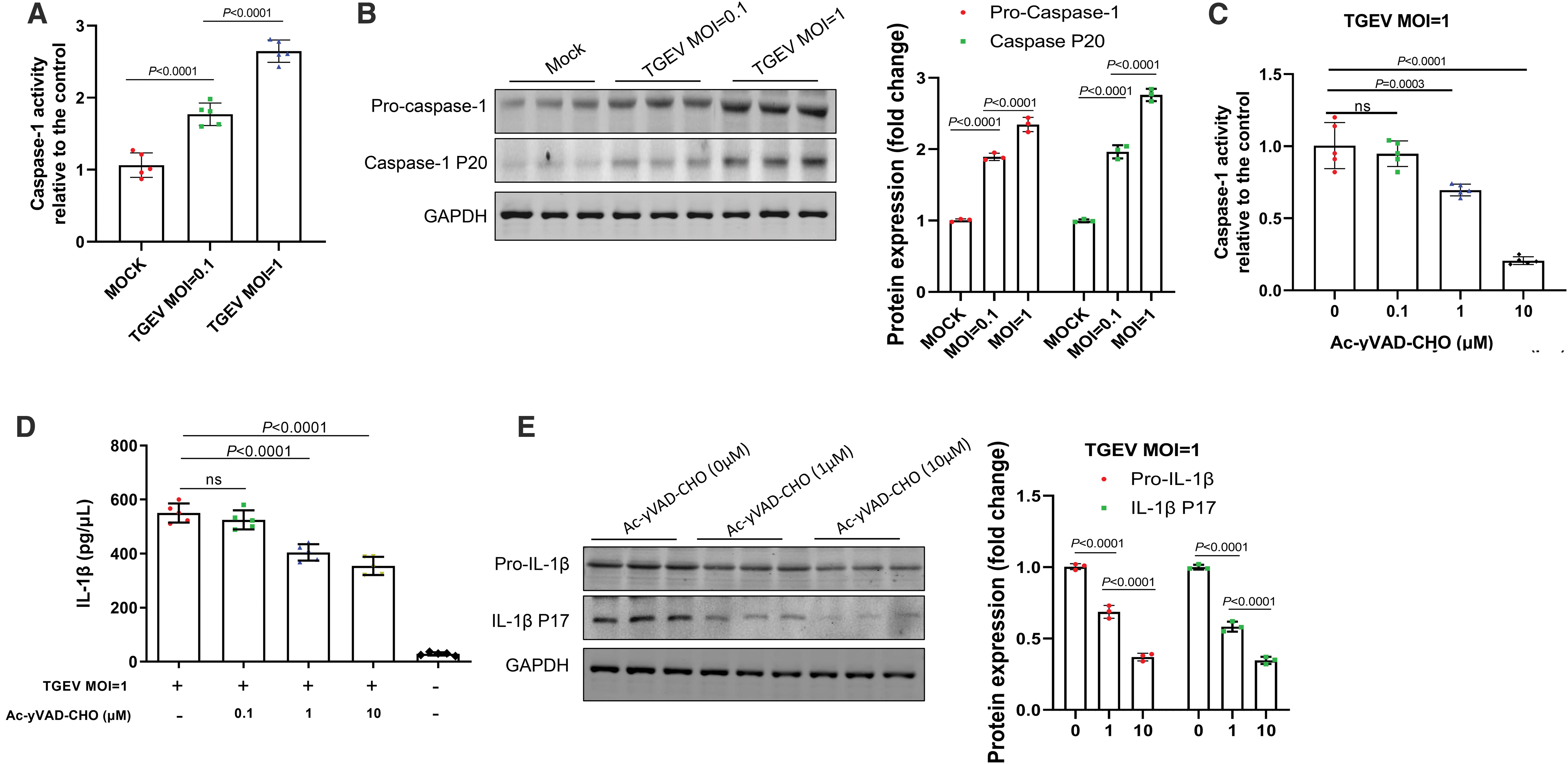
Activation of IL-1β induced by TGEV infection was dependent on caspase-1.
In addition, Ac-yVAD-CHO, which is a caspase-1 inhibitor, was used to investigate whether interfere in activation of caspase-1 could block TGEV infection induced IL-1β generation in IPEC-J2 cells. We found that caspase-1 activity was effectively inhibited by Ac-yVAD-CHO in a dose-dependent manner (Fig. 2C). Notably, inhibition of caspase-1 activity significantly blunted TGEV infection-induced IL-1β levels in an inhibitor dose-dependent manner in IPEC-J2 cells (Fig. 2D, E). Taken together, these data indicated that caspase-1 is a key mediator during TGEV infection-induced maturation of IL-1β in IPEC-J2 cells.
TGEV infection-induced pyroptosis of porcine intestinal epithelial cell was dependent on activation of caspase-1
Large amount of evidence has suggested that caspase-1 is a key factor in cell death (31). We, therefore, asked whether TGEV infection induces pyroptosis in porcine intestinal epithelial cell and investigated the role of caspase-1 in this process. IPEC-J2 cells were subjected to infection of TGEV (MOI = 1) with or without Ac-yVAD-CHO treatment and at 24 h postinfection, the cell death and membrane damage was evaluated with the Calcein-AM/EthD-III Cell Activity Assay Kit. We found that TGEV infection significantly decreased the proportion of live cells and Ac-yVAD-CHO treatment blunted these changes in a dose-dependent manner in IPEC-J2 cells (Fig. 3A). Meanwhile, Ac-yVAD-CHO treatment also dose dependently decreased the proportion of dead cells with membrane damage (Fig. 3B). In addition, we also observed that TGEV infection significantly increased LDH release of IPEC-J2 cells and administration of either Ac-YVAD-CHO or NLRP3 inhibitor glibenclamide blunted this change in a dose-dependent manner (Fig. 3C). These data indicate that TGEV infection induced caspase-1-dependent pyroptosis in IPEC-J2 cells.
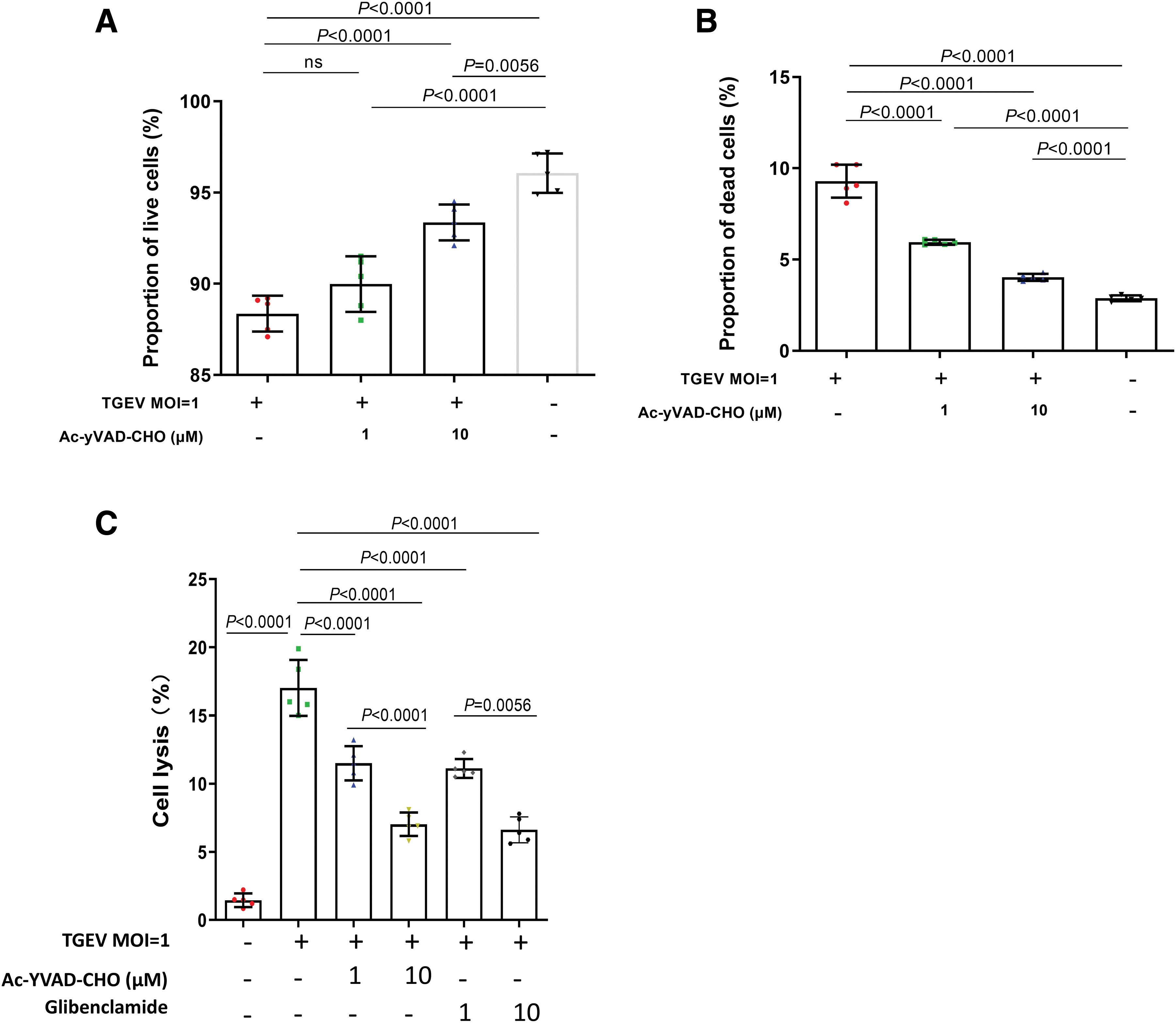
Induction of cell death and membrane damage by TGEV infection was depended on the activity of caspase-1. IPEC-J2 cells were treated with 1 or 10 μM AC-yVAD-CHO. Subsequently, IPEC-J2 cells were mock treated or infected with TGEV at MOIs of 1 for 24 h. The proportions of live IPEC-J2 cells
TGEV infection induced GSDMD production and cleaving in porcine intestinal epithelial cells
Several researches have demonstrated that GSDMD cleavage by inflammatory caspases is a key execution event in pyroptosis (12,34,46). In the process of pyroptosis, the gasdermin-N domain in GSDMD, which is intrinsically capable to bind to membrane lipids, forms plasma membrane pores and thereafter initiates cell lysis (34,42). To confirm whether TGEV infection induces pyroptosis of porcine intestinal epithelial cells, IPEC-J2 cells were subjected to infection of various doses of TGEV (MOIs of 0.1 and 1), then protein level of GSDMD and its cleavages were detected. We found that TGEV infection induced drastic increase in protein level of GSDMD, GSDMD-C, and its active form GSDMD-N (Fig. 4). Notably, these effects were positively correlated with infection level by TGEV. Collectively, these data suggest that TGEV infection induced pyroptosis in IPEC-J2 cells.

TGEV infection induced the production and cleavage of GSDMD in porcine intestinal epithelial cells. IPEC-J2 cells were mock treated or infected with TGEV at MOIs of 0.1 or 1 for 48 h. The expression levels of GSDMD, GSDMD-N, and GSDMD-C were determined by western blotting (N = 3). GAPDH was used as an internal loading control. GSDMD, gasdermin D. Color images are available online.
NLRP3 inflammasome was activated by TGEV infection in porcine intestinal epithelial cells
NLRP3 inflammasome, which can be activated by threatening endogenous signals of various pathogens, is the upstream hub activator of caspase-1 and its activation results in IL-1β production and pyroptosis (13). Therefore, the induction and activation of IL-1β are important hallmarks of NLRP3 inflammasome activation (20). The adapter protein ASC, which binds to the pyrin domain of the NLRP3 N-terminus, is important for recruitment and activation of pro-caspase-1 (36). We, therefore, investigated whether TGEV infection has any effect on activity of the NLRP3 inflammasome. As shown in Figure 5A, TGEV promoted the expression of the NLRP3 and ASC proteins. Notably, IL-1β release was dose dependently inhibited by NLRP3-specific inhibitor, glibenclamide (Fig. 5B). These results suggest that NLRP3 inflammasome is activated and its activation is essential for IL-1β production during TGEV infection in IPEC-J2 cells.
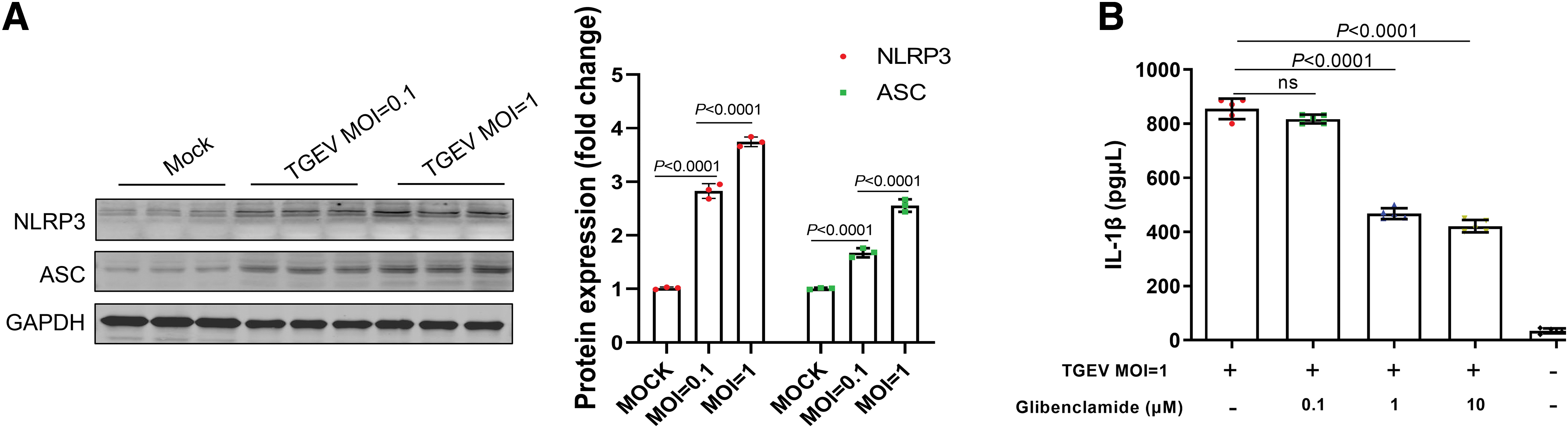
TGEV infection induced the formation of the NLRP3 inflammasome in porcine intestinal epithelial cells.
Activated NLRP3 inflammasome had an inhibitive effect on TGEV replication
NLRP3, as a pattern recognition receptor, can recognize a variety of viral and initiate antiviral response through IL-1β signaling activation. We, therefore, investigated whether NLRP3 inflammation have any effects on TGEV replication in IPEC-J2 cells. The NLRP3-specific inhibitor glibenclamide was used and then the virus copy number and virus titers were detected in infected IPEC-J2 cells. As shown in Figure 6A and B, NLRP3-specific inhibitor glibenclamide treatment increased the virus copy numbers and virus titers compared with that of the control group. To elucidate that the glibenclamide-induced increase in viral replication was not caused by cell death, EthD-III/calcein AM staining assays were used to explore effect of glibenclamide on IPEC-J2 cell activity. As shown in Figure 6C, glibenclamide exhibited no significant effect on the activity of IPEC-J2 cells. These results demonstrated that activation of NLRP3 has important antivirus effects on TGEV replication.
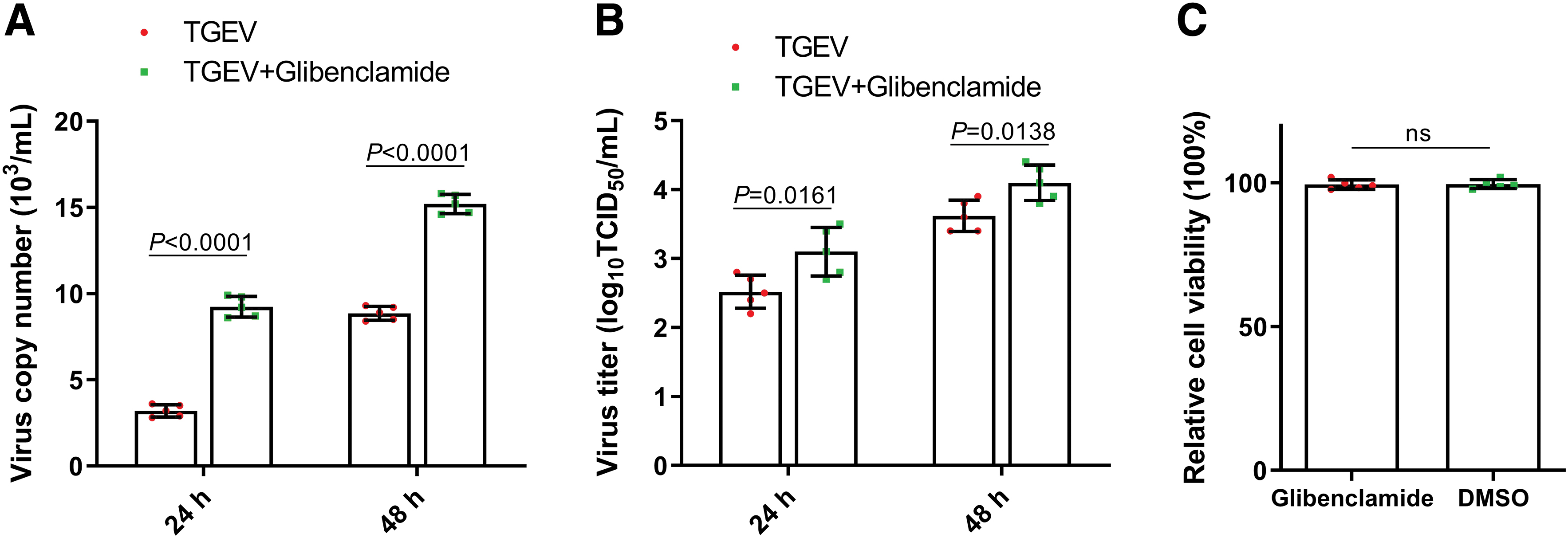
Inhibition of NLRP3 promoted the replication of TGEV. The effect of the NLRP3-specific inhibitor glibenclamide on the virus copy number
Discussion
Mechanisms mediating pathogen-induced cell death is still not well understood (8). Apoptosis and necrosis have long been regarded as the main mechanism for cell death, but the a large amount of evidence recently indicated pyroptosis as a novel form of cell death (8). Pyroptosis is a highly inflammatory form of programmed cell death that often happens upon infection with pathogens; it involves in the innate immune response and plays important roles in antiviral response (16,33). In the process of pyroptosis, cells release proinflammatory cytokines such as IL-1β after recognizing foreign danger signals, and then undergo swell, burst, and die (8). The cytokines can recruit other immune cells to fight against pathogen, but also lead to inflammation. Therefore, pyroptosis is important for rapid clearance of various pathogen infections (6,41). In this study, we observed that TGEV infection could induce porcine intestinal epithelial cell pyroptosis; Apoptosis and necrosis, no doubt, also existed in this infection process. Furthermore, TGEV infection reduced the cell survival, which can be blocked by inhibitor of caspase-1, which mediates pyroptosis (7,17). However, further detailed studies are needed to confirm the definite contribution of pyroptosis in TGEV infection-induced death of porcine intestinal epithelial cell.
During infection of pathogens, IL-1β is secreted and act as an essential pyrogen, which is important for host immunity response against pathogens (28). Induction of IL-1β can be divided into two steps, namely expression of pro-IL-1β initiated by cellular signaling such as nuclear factor kappa B, which is activated by sensing of pathogen-associated molecular patterns, and activation of caspase-1 to promote cleavage pro-IL-1β into biologically active cytokine (10,38). In this study, we observed that TGEV stimulated the maturation of IL-1β in porcine intestinal epithelial cell, which was a critical signature of inflammasome activation. Meanwhile, TGEV could promote the cleavage and enzyme activity of caspase-1 in porcine intestinal epithelial cells and induce pyroptosis. Caspase-1 can cleave GSDMD to produce an N-terminal cleaved product (GSDMD-N), which is an essential component of pyroptosis (5). After GSDMD-N production, cells start to undergo pyroptosis, a process which contains many steps, such as chromatin condensation, membrane pore formation, cell lysis, and the release of intracellular proinflammatory cytokines (40).
We also observed induction of pyroptosis in this study, which indicated that pyroptosis is a vital form of cell death in TGEV infection of porcine intestinal epithelial cell. However, it should be noticed that pyroptosis is not the only one form of cell death induced by virus infection, other forms of cell death such apoptosis may also play an essential role (15), and this may help explain the result that caspase-1 inhibitor Ac-YVAD-CHO and NLRP3 inhibitor glibenclamide cannot completely block cell lysis. As caspase-1 spontaneously activates in cell lysates and the activity observed could result from increased pro-caspase, the result of caspase-1 activity assay does not necessarily reflect intracellular activation (2,26,37). We, therefore, analyzed caspase-1 P10 in cell lysates by Western blot and confirmed intracellular activation of caspase-1. In addition, treatment with the caspase-1-specific inhibitor inhibited the TGEV-induced activation of IL-1β, supporting the notion that TGEV infection enhances the release of IL-1β through caspase-1 activation. However, there still exists limitation that non-pyroptotic lysis may induce release of pro-IL-1β; this may to some extent interfere with the detection of mature IL-1β in cell supernatant.
The inflammasome pathway acts as an essential early response mechanism that enables the detection of pathogens and secretion of inflammatory cytokines to recruit immune cells to the site of infection (23). Sensor protein NLRP and adapter protein ASC are two components of inflammasome complex (29). Among these inflammasomes, NLRP3 is mostly focused because of its critical role in the activation of caspase-1 induced by multiple pathogens, including ssRNA and double-stranded DNA viruses such as Sendai virus, Influenza virus, as well as bacteria such as Staphylococcus aureus and Listeria monocytogenes (21,22). NLRP3 inflammasome promotes host immune defense during microbial infections (11), whereas dysregulated activation of the NLRP3 inflammasome also leads to pathogenesis of inflammatory diseases (18). Therefore, appropriate activation of NLRP3 inflammasome are important in the responses to the virus infection. In support of this notion, NLRP3 inflammasome has been demonstrated to be activated by many coronaviruses/coronavirus proteins, including SARS-coronavirus open-reading frame-8b, acute respiratory syndrome coronavirus ORF3a protein, and even COVID-19 (32,35,47).
It has been reported that the viruses cause a series of changes in cellular status host cells, including lysosomal maturation, mitochondria damage, aberrant ion concentrations, and the accumulation of misfolded protein aggregates. The maturation and acidification of lysosomes lead to the leaking of catalytically active cathepsin B, and the subsequent generation of reactive oxygen species, all of which are recognized as danger signals by the host and lead to the activation of the NLRP3 inflammasome (44). Further studies are needed to understand the contribution of these changes in NLRP3 inflammasome activation upon TGEV infection. We found that TGEV infection can promote porcine intestinal epithelial cell pyroptosis and the release of IL-1β, and this process is depended on NLRP3 inflammasome activation. Meanwhile, inhibition of NLRP3 inflammasome increased TGEV replication. Therefore, the anti-infectious effect of NLRP3 inflammasome may represent a restriction for TGEV. Pro-caspase-1, full-length GSDMD, and ASC are usually constitutively expressed and do not usually increase in abundance with activation. A recent study indicated that expression of the three proteins can be regulated upon infection in porcine cells, and activation of stimulator of interferon genes (STING) plays essential roles in this process and thus involves in NLRP3 inflammasome activation and innate immunity (27). Whether STING is also the mechanism for the increased expression of pro-caspase-1, full-length GSDMD, and ASC in the present study still needs to be explored in the future.
In conclusion, our data provided evidence for the first time that TGEV depends on NLRP3 and caspase-1 to enhance IL-1β production and activation, and thereafter pyroptosis in porcine intestinal epithelial cell. Overall, the aberrant IL-1β release and pyroptosis observed in this study offer a distinct perspective on the pathogenesis of TGEV infection and development of antiviral strategy.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Science and Technology Innovation Project of Social undertakings and People's Livelihood Guarantee of Chongqing Municipal Science and Technology Commission (cstc2017shms-xsny80061) and Youth Fund Project of Southwest University (132030/20700420).
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.