
Abstract
Lipid accumulation and inflammation act together to induce, sustain, and further development of chronic liver disease. Hepatitis C virus (HCV) infection induces metabolic and immune changes in liver macrophages, promoting lipid accumulation and inflammation that synergize and culminate in the development of steatohepatitis and fibrogenesis. Chronic HCV patients have increased liver macrophages with disruptions in cholesterol metabolism and alterations in inflammatory mediators. While HCV-induced changes in inflammatory mediators are well documented, how HCV triggers metabolic change in macrophages is unknown. In this report, we examined the mechanism of macrophage sensing of HCV to cause metabolic impairment and subsequent immune dysfunction. We demonstrate that HCV protein and RNA kinetics in macrophages are distinct from hepatocytes. In macrophages, HCV RNAs and protein accumulate rapidly after exposure but internalized RNAs quickly decline to a low-level set point. Notably, exposure of macrophages to HCV resulted in increased lipids and cholesterol and activation of cholesterol-sensing, immunomodulatory liver X receptors (LXRs).
Furthermore, we provide evidence that HCV RNA accumulation in macrophages occurs through scavenging receptors. These results suggest that HCV released from infected hepatocytes stimulates accumulation of lipids and activation of LXR in macrophages contributing to metabolic changes involved in HCV-induced chronic liver disease. Our results provide novel insight into mechanisms through which impaired lipid metabolism in macrophages associated with HCV infection promotes development of liver steatohepatitis and fibrosis.
Introduction
Hepatitis C virus (HCV) is a significant public health concern despite antiviral agents that inhibit viral replication. HCV infection in humans is a major cause of mortality in the United States (42,43,47). Chronic HCV infection causes severe liver diseases, including hepatocellular carcinoma, a major cause of cancer-related deaths (40,42,66). Infection alters host lipid metabolism such that HCV increases hepatic lipids but decreases serum cholesterol through impaired secretion of very-low-density lipoprotein (VLDL) from hepatocytes (29). HCV does not increase de novo cholesterol biogenesis but instead induces accumulation of cholesterol and cholesterol esters through alterations in global lipid metabolism and activity of cholesterol esterifying enzyme sterol O-acyltransferase 2 (SOAT2, acyl-coenzyme A: cholesterol acyltransferase 2, ACACT2, or ACAT2) (29,55,70).
HCV depends on cellular lipid and lipoprotein metabolism as viral particles and the most infectious fraction of virus in plasma resemble VLDLs (4,5,50). Importantly, targeting cellular lipid metabolism has antiviral effects and could reduce the development of severe liver dysfunction (33,44,48,62). However, it is not well understood how HCV induces metabolic defects and liver inflammation.
HCV infection in hepatocytes regulates lipid metabolism and inflammation through the activation of transcription factors, such as sterol regulatory element-binding protein-1c (SREBP-1c). SREBP-1c is a master regulator of lipogenesis and is a downstream target of liver X receptors (LXRs) involved in the regulation of cholesterol flux (27,45,51,56). Accumulation of cholesterol in hepatocytes activates LXRs, in turn activating SREBP-1c and mechanisms to degrade β-hydroxy β-methylglutaryl coenzyme A reductase, the rate-limiting step of cholesterol biogenesis.
Notably, LXR activation in macrophages induces genes involved in the reverse cholesterol transport pathway of cholesterol efflux, including ATP-binding cassette transporter A1 (ABCA1). Activation of LXRs in macrophages has anti-inflammatory effects, principally through suppressing inflammatory gene expression (3,30,31,67,72). Thus, LXR activation limits cholesterol formation, activates cholesterol removal, and influences innate immunity.
Liver is a major site of lipid and cholesterol metabolism and maintains immune surveillance with a significant macrophage population (8,9,15). Two distinct populations of macrophages are present in liver, particularly in the context of pathology, such as long-term tissue-resident macrophages derived from the fetal yolk sac known as Kupffer cells and blood monocyte-derived macrophages (MDMs).
Kupffer cells are estimated to account for ∼15% of total liver cells (49). These cells are difficult to isolate and culture in vitro and several studies have used culturable monocyte-like cells to complement analyses of Kupffer cells to examine HCV effects on liver macrophages (36,49,61). These studies have shown that both macrophage populations respond to virus exposure by producing a similar array of cytokines, including IFNβ, interleukin (IL)-1β, and IL-18 (36,49,61). In this study, we sought to model interaction of MDMs with virus in the absence of a pre-existing immune microenvironment that could potentially predetermine immune and metabolic characteristics.
It is important to acknowledge the limitations of the cell culture THP-1 macrophage-like cell model and that there are likely differences in responses of Kupffer cells and MDMs to HCV exposure that our system may not reproduce. However, the THP-1 model has proven particularly useful for studying the effect of virus exposure in macrophages on tissue-level immune responses to acute and chronic HCV (36,49,61,73). In this context, these cells represent an appropriate model of metabolic changes likely to occur in blood MDMs acutely exposed to HCV released from hepatocytes. Of note, it is generally agreed that productive HCV infection in Kupffer cells and MDMs is unlikely (9) although differences in uptake mechanisms cannot be ruled out.
HCV affects functional activity, differentiation, and polarization of myeloid cells at multiple stages of differentiation without productive replication. HCV induces Galectin-9 expression and differentiation of monocytes (26). Macrophages take up HCV resulting in the activation of inflammasomes and induction of inflammatory cytokines (14,49,61,73). HCV-induced monocyte differentiation leads to anti-inflammatory and fibrotic M2-like macrophage polarization (57,58).
Finally, HCV can induce functional changes and expression of molecules involved in fibrosis, including procollagen, transforming growth factor-β (TGF-β), and C-C motif chemokine ligand 5 (CCL5) in macrophages, generating virus-induced fibrocytes (57 –59). Importantly, anti-inflammatory mediators secreted from HCV-exposed macrophages are able to induce the activation of hepatic stellate cells, promoting regulatory T cell expansion and liver fibrosis observed in chronic HCV patients after direct-acting antiviral therapies (57 –59).
In the present study, we investigated the status of lipid metabolism and functional characterization in macrophages exposed to HCV. Herein, we report that HCV exposure results in alteration of macrophage lipid content and metabolic pathways. Our data suggest that macrophages respond to HCV exposure in a mechanism involving scavenger receptors. Collectively, these results suggest that HCV induces metabolic changes in macrophages that may complement the profibrotic inflammatory response and contribute to the development of liver fibrosis and severe liver diseases.
Materials and Methods
Cells, virus, reagents
THP-1 human monocytic cell line was purchased from ATCC and maintained in RPMI-1640 Media (Gibco, cat. no. 11875093) supplemented with 0.05 mM 2-mercaptoethanol (Sigma, cat. no. M7522), 10% fetal bovine serum (FBS) (HyClone/Cytiva, cat. no. SH30071.03), 100 U/mL penicillin/streptomycin (Corning, cat. no. 30001CI), and 2 mM L-glutamine (Gibco, cat. no. 25030081). Cells were differentiated with 100 nM phorbol 12-myristate 13-acetate (PMA; LC Labs) for 72 h. Macrophages were rested 24 h without PMA before use in experiments. Unless otherwise noted, THP-1 macrophage-like cells were differentiated and rested as described above for all experiments, then exposed to 0.1 multiplicity of infection (MOI) HCV or mock supernatant for 48 h.
Human hepatoma cell line Huh7.5.1 was maintained in Dulbecco's modified Eagle's medium (Corning, cat. no. 10-017-CV) with 10% FBS (HyClone/Cytiva, cat. no. SH30071.03), 100 U/mL penicillin/streptomycin (Corning, cat. no. 30001CI), 2 mM L-glutamine (Gibco, cat. no. 25030081), and 1% nonessential amino acids (Gibco, cat. no. 11140050).
Primary human MDMs were generated from peripheral blood mononuclear cells (PBMCs). Deidentified donor blood was obtained from a commercial source (Virginia Blood Services). PBMCs were isolated using Ficoll in SepMate separation tubes (StemCell) followed by enrichment of monocytes using the EasySep Human Monocyte Enrichment Kit without CD16 depletion (StemCell). Monocytes were further enriched by plastic adherence for 2–4 h, followed by removal of nonadherent cells and culture in X-VIVO 15 (Invitrogen) supplemented with M-CSF (PeproTech) for a total of 14 days with half-media changes every 3–4 days to obtain mature resting macrophages.
All experimental protocols were approved by the Institutional Review Board of the University of Virginia (IRB no. 8233). All Methods were carried out in accordance with relevant guidelines and regulations required by the National Institutes of Health (NIH) and the University of Virginia.
JFH-1 HCV was kindly provided by Takaji Wakita (69). Virus stocks were prepared by infecting Huh7.5.1 cells. Virus was collected following 6–8 days of infection and tittered by Focus-Forming assay on Huh7.5.1 cells exposed to virus for 3 days. Cells were fixed and stained with anti-Core antigen (Thermo Fisher) followed by APC-labeled anti-mouse IgG (Life Technologies) (48). Virus stocks were cleared of debris by centrifugation, aliquoted, and stored at −80°C until use. Mock supernatants were prepared from uninfected cells maintained under identical conditions.
siRNA reagents, including control and scavenger receptor class B type 1 (SR-B1) siRNAs (sc-44752), transfection reagent, and media were from Santa Cruz.
Real-time reverse transcriptase quantitative polymerase chain reaction
Total RNA was extracted using RNA-Bee as per the manufacturer's instructions (Tel-Test) and converted to first-strand cDNA with the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). Real-time reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) was run on StepOnePlus System with SybrGreen (Life Technologies). Primers were HCV inNeg F1 ACTGTCTTCACGCAGAAAGCGCC and HCV inNeg R1 CAAGCGCCCTATCAGGCAGTACC (modified for Genotype 2A) for HCV (JFH-1) 5′ untranslated region (5′-UTR) and qNS3-2aF CTGCCACCCTGGGGTTTGGG and qNS3-2aR GCAGCCCCCATCGGCGAGAA for HCV nonstructural protein 3 transcript region (NS3) (19,52).
Target gene expression analysis with SybrGreen chemistry was normalized to housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT) with primers HPRT-F CATTATGCTGAGGATTTGGAAAGG and HPRT-R AGACGTTCAGTCCTGTCCATAA. TaqMan gene expression assays for ABCA1, CD81, Claudin-1 (CLDN1), dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) (CD209), HPRT, LXRα (NR1H3), SR-B1 (SCARB1), and SREBP-1c (SREBF1) were obtained from (Thermo Fisher).
Western blot
Cell lysates were prepared in lysis buffer (HEPES pH:7.4, NaCl, EDTA, DTT, Triton X-100). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with TGX precast gels (Bio-Rad) and were transferred to polyvinylidene difluoride membranes (Bio-Rad) using a semidry transfer cell (Bio-Rad). Membranes were blocked with milk in TBS-T, followed by incubation in primary antibodies and incubation with secondary antibodies for development. Blots were developed using enhanced chemiluminescent (ECL) detection.
Antibodies used for western blotting were: Hep C cAg (C7-50; Santa Cruz), actin (Santa Cruz), HCV E2 (Santa Cruz), HCV NS3 (Abcam), SR-B1 (GeneTex), DC-SIGN (Cell Signaling), CD81 (Systems Biosciences), CLDN1 (ProSci) LXRα (R&D Systems), LXRβ (R&D Systems), ABCA1 (Novus), Vinculin (Cell Signaling), and SREBP-1 (Santa Cruz).
Flow cytometry
For analysis of surface expression, THP-1 macrophage-like cells were differentiated as above. Adherent macrophages were washed and incubated in Cell Dissociation Buffer, enzyme free (Thermo Fisher/Gibco) for 20 min on ice with gentle disruption with a vacuum pipette every 5 min. Cells were collected, washed, and resuspended in TruStain FcX Fc Receptor Blocking Solution (BioLegend) in fluorescence activated cell sorting (FACS) buffer for 15 min. Fc Blocking reagent was removed and cells were incubated in primary antibodies for 20 min. Cells were washed twice in FACS buffer and analyzed on a CytoFlex Flow Cytometer (Beckman Coulter). Antibodies used were PerCP CD45 (Clone 2D1; Tonbo), PE CD36L1 (SCARB1, SR-B1, Clone m1B9; BioLegend), FITC CD81 (TAPA-1, Clone 5A6; BioLegend), and CD209 (DC-SIGN)-APC-Vio770 (Clone DCN47.5; Miltenyi).
Oil Red O staining
Cells in tissue culture plates were stained in 1% Oil Red O (Sigma) in 60% isopropanol for 1 h. Staining was differentiated in three changes of 60% isopropanol followed by a running water wash. Slides were counterstained in Hematoxylin, washed, and blued in running water and imaged in water before extraction. Neutral Lipids (Oil Red O) were extracted using 100% isopropanol. A separate aliquot was also used for determination of cell protein. Total cell protein was similar between mock- (1.031 mg/mL) and HCV-exposed (1.027 mg/mL) cell groups and results are presented non-normalized as Raw Absorbance values.
Cholesterol content analysis
Free and esterified cholesterol mass analysis was conducted according to published methods (38). Media were removed, and cells were washed twice. Lipids were extracted in hexane/isopropanol (3:2) for 1 h with mixing. Lipid extracts were collected, and cells were reextracted with hexane/isopropanol. Pooled extracts were dried under nitrogen and lipid films resuspended in reaction buffer (0.1 M potassium phosphate, pH 7.4, 50 mM NaCl, 5 mM cholic acid, and 0.1% Triton X-100) for 5 h with shaking.
Aliquots were assayed for free cholesterol and cholesterol ester using Amplex Red cholesterol assay (Molecular Probes) in the presence or absence of cholesterol esterase, respectively. Free cholesterol was calculated as difference of total cholesterol and esterified cholesterol for matched samples. Cells were dissolved in NaOH/SDS, and an aliquot used for protein determination by BCA protein assay. Cholesterol mass values were normalized to cellular protein.
Cholesterol staining
Cells were grown in a chamber slide (Lab-Tek II, Nunc). Media were removed and cells were washed twice before fixing and staining with the Cholesterol Cell-Based Detection Assay Kit (Cayman) according to the manufacturer's recommendations. Cholesterol (Filipin III)-stained cells were imaged with an LSM710 laser-scanning confocal microscope (Zeiss). Nuclei were counterstained with DRAQ5 (BioStatus).
Blocking antibody experiments
For blockade of E2 envelope interaction, HCV cell-free supernatants were incubated with anti-E2 (1:30) for 1 h before exposure to cells and antibody maintained throughout exposure. Cells were treated with blocking antibodies to target HCV host receptors for 1 h before exposure to HCV and maintained throughout exposure. Blocking antibodies used included: anti- E2 (GeneTex, cat. no. GTX103353), CD81 (Santa Cruz sc-23962, 1:50), SR-BI (GeneTex, EP1556Y GTX61567, 1:50), and DC-SIGN (R&DMAB161, 10 μg/mL).
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. Samples for qPCR and metabolic assessments were measured in triplicate (three biological replicates, n = 3, each sample assayed in technical duplicate) and results presented as mean + standard error. Experiments presented graphically were performed a minimum of two times with biological triplicate replicates. Measurements of biological triplicates from a single representative experiment are presented. Individual measures for all graphs in main Figures are included as Supplementary Fig. S9. t-Tests at 95% Confidence Interval (CI) were used to assess differences in the means between two grouped variables. One-way analysis of variance (ANOVA) with post hoc Tukey's multiple comparison analyses at 95% CI was used to assess differences in means between multiple grouped variables. *,# p < 0.05, **,## p < 0.01, ***,### p < 0.001.
Software use
All graphs were prepared using statistical software Prism (GraphPad). Photographic films were scanned with an Epson professional desktop scanner and Epson Scan software. Flow Cytometry data were collected on a CytoFlex flow cytometer (Beckman Coulter) and processed with FlowJo Software (FlowJo). Light microscopic images were collected using instrument-associated software (Leica Microsystems).
Spectrophotometric Absorbance readings were collected using a SpectraMax plate reader and Softmax software. Confocal microscopy images were processed with instrument-specific software (Zen, Zeiss Microscopy) and further processed for publication using ImageJ (NIH). Real-time quantitative PCR data were collected and analyzed with instrument-specific software (ABI) and statistical comparisons used in analysis of RT-qPCR were conducted with Prism software (GraphPad).
Results
HCV RNA accumulates and persists in macrophages without viral protein expression
Induction of innate immune mediators, including CCL5, interferons, IL-1β, IL-6, IL-8, IL-10 (TGF-β), and tumor necrosis factor alpha in macrophages upon exposure to HCV has been reported along with a gradual decline of viral RNA between 24 and 72 h (36,49,57 –59,61,73). To characterize viral RNA dynamics immediately upon virus exposure, we determined HCV RNA kinetics in macrophages differentiated from THP-1 promonocytic cells within 24 h of HCV exposure.
THP-1 macrophage-like cells were exposed to clarified supernatants from HCV-infected Huh7.5.1 hepatoma cells and viral RNA and protein were analyzed in macrophage lysates. Viral genomic RNA was detectable by reverse transcriptase quantitative PCR (RT-qPCR) of the 5′-UTR and NS3 in macrophages within 1 h of HCV (0.1 MOI–MOI) exposure and gradually decreased throughout 6 h (Fig. 1A).
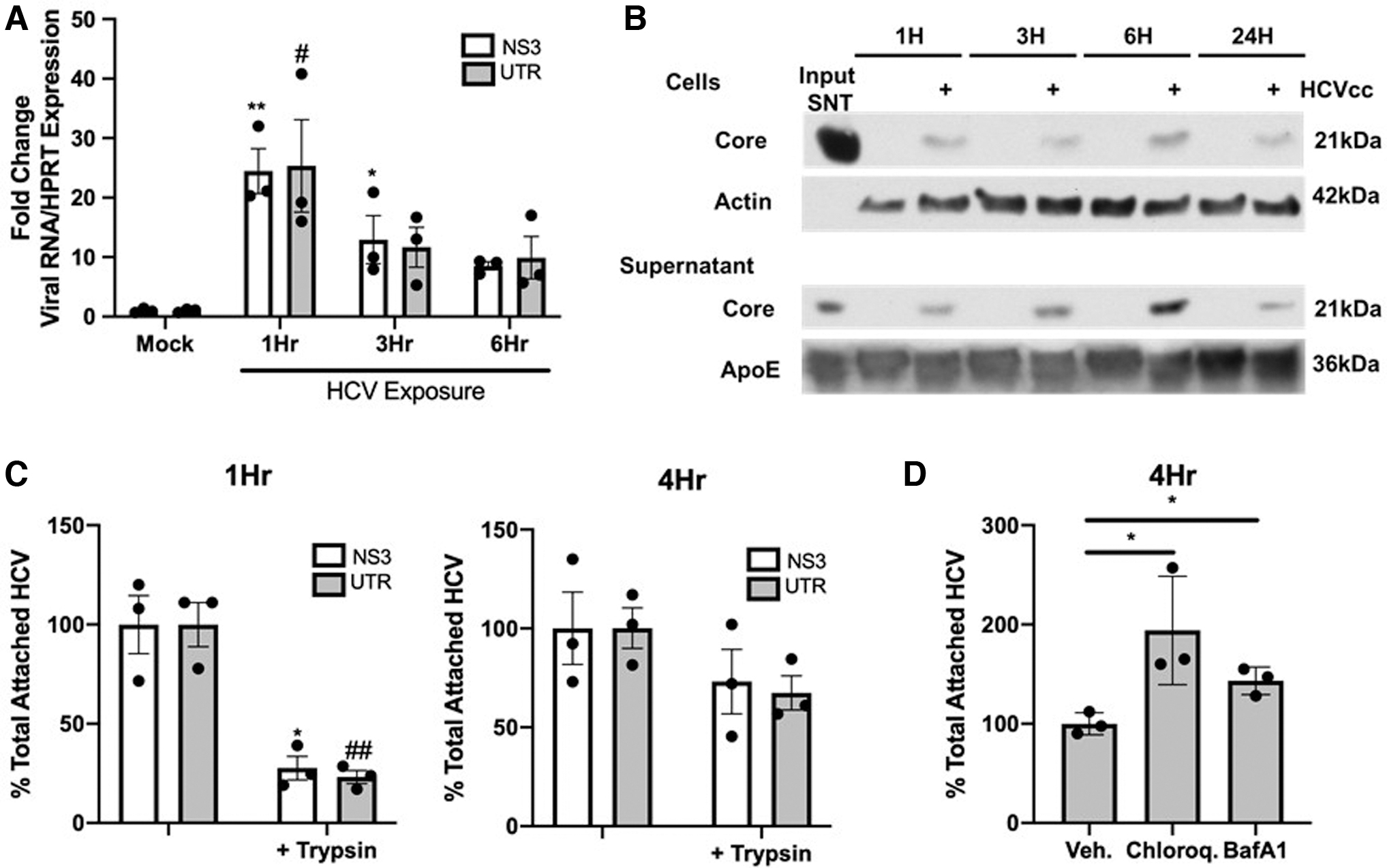
When we extended our studies to examine the presence of viral genomic RNA during a 3-day exposure to 0.1 MOI HCV in THP-1 macrophage-like cells, viral RNA above background was detectable throughout time course, gradually declined throughout 24 h, and stabilized at a low level by 72 h (Supplementary Fig. S1A). In contrast, viral protein (NS3 or core) was not detectable at any time with this dose of HCV (Supplementary Fig. S1B). Increasing viral inocula (1 MOI) allowed detection of low levels of viral protein in macrophages, potentially derived from inocula, with a transient increase in viral protein at 6 h postexposure (Fig. 1B) (36). These results suggest that virus exposure in macrophages leads to accumulation and maintenance of viral genomic RNA in these cells that persists at least for 72 h.
To compare the level of HCV RNA detected in macrophages upon HCV exposure with HCV infection in hepatocytes, we examined viral genomic RNA and protein during HCV infection in hepatoma cells. In Huh7.5.1 cells exposed to HCV, viral genome was detectable within 24 h, increased rapidly between 24 and 72 h, and peaked between 96 and 120 h postinfection before a precipitous drop with marked cytopathic effect at 144 h (Supplementary Fig. S1C).
Viral life cycle is intact in this system as negative-strand RNA replication intermediates were detectable from 72 h until peaking at 120 h postinfection (Supplementary Fig. S1D). Viral polyprotein (high mw reactivity to NS3 antibody) was detectable within 24 h and polyprotein processing (multiple bands of lower mw reactivity to NS3 antibody) was evident by 48 h, peaking with the presence of viral genomes between 72 and 120 h postinfection (Supplementary Fig. S1E).
Structural (Core), nonstructural (NS3), and Envelope (E2) proteins were all detectable demonstrating intact and complete nature of the viral replication cycle within Huh7.5.1 cells (Supplementary Fig. S1E). This contrasts with the viral replication cycle in THP-1 macrophage-like cells, which is either abortive or significantly delayed under the conditions of these experiments.
Based on common features of the expression of surface receptor for HCV entry in macrophages and hepatocytes, it is possible that macrophages may become productively infected. However, viral genomic RNA did not increase significantly throughout 3 days of exposure indicating THP-1 macrophage-like cells do not become productively infected.
Envelope-independent internalization of viral RNA in macrophages and degradation in endo/lysosomes
Viral RNAs can nonspecifically attach to the surface of macrophages. Thus, the presence of viral RNA in cell homogenates does not directly address whether these RNAs are entering cells. In addition, a recent study demonstrated that HCV can attach to the surface of both primary monocytes and macrophages but only macrophages internalize the virus (73). To measure viral binding and uptake, macrophages (Fig. 1C) or monocytes (Supplementary Fig. S2A) were incubated with HCV followed by treatment with trypsin to remove viral particles that remained attached to the cell surface as in Zhang et al. (73). Bound or internalized virus was quantified by qPCR for viral genomic sequences in the absence or presence of trypsin treatment, respectively.
The quantity of virus bound (-trypsin) was normalized to 1 and uptake (+ trypsin) presented as a percentage of total attached HCV. Based on our studies indicating detection of highest viral RNA at 1 h postexposure, we examined trypsin sensitivity at both 1 and 4 h as in Zhang et al. (73). At 1 h postexposure ∼80% of viral RNA in macrophages was sensitive to trypsinization, indicating that virus is surface associated, reflecting binding (Fig. 1C). By 4 h postexposure ∼70% of viral RNA in macrophages was insensitive to trypsinization, indicating virus uptake and internalization (Fig. 1C) (73).
To test the possibility that a difference in virus uptake between monocytes and macrophages (73) might reflect upregulation of a factor required for virus recognition upon differentiation, we analyzed virus uptake in monocytes exposed to virus. At 4 h postexposure monocytes internalized considerably less virus than macrophages as a percentage of the amount of trypsin-insensitive RNA bound to cells as well as the ratio of trypsin-insensitive viral RNA to housekeeping gene HPRT (Supplementary Fig. S2A). Blockade of cell surface entry factor CD81 is incapable of preventing HCV protein or RNA uptake in macrophages but can prevent infection and uptake in other cell types (17,35,36,49,64). Studies from other groups have been inconsistent on the requirement for viral E2 protein in HCV uptake and cytokine induction.
Thus, we examined whether preincubating virus with an E2-blocking antibody previously demonstrated to block HCV infection in hepatocytes (confirmed in Supplementary Fig. S2B) could prevent virus binding or uptake in macrophages (12). Preincubating virus with E2-blocking antibody failed to prevent HCV binding or uptake in macrophages (Supplementary Fig. S2C). These results suggest that unlike results in DCs, receptor-mediated entry of HCV is not responsible for uptake in macrophages. It also implicates an alternative process distinct from the envelope-mediated process that leads to productive infection in hepatocytes (35).
Envelope–receptor interactions lead to fusion to the cellular membrane and release of viral genomes into the cytoplasm where they are degraded by cellular factors or used as templates for viral replication (28). We next examined whether inhibition of endosomal acidification with chloroquine or lysosomal acidification with the v-ATPase inhibitor Bafilomycin A1 (BafA1) could prevent loss of viral RNAs during 4 h exposure in macrophages (7,12,24,36,46,49,53,68,71,73).
Viral RNA in chloroquine- or BafA1-treated cells at 4 h postexposure was increased ∼1.5–2-fold relative to vehicle-treated macrophages (Fig. 1D and Supplementary Fig. S2D). Thus, chloroquine or BafA1 treatment, disrupting degradation in the endo/lysosomal network, increases recoverable viral RNA at 4 h, implicating these disposal pathways in the overall reduction of viral RNA between 1 and 24 h (Fig. 1A and Supplementary Fig. S1A). Combined, these results suggest that internalization of HCV RNA in macrophages does not require viral E2 interaction and RNA is subsequently degraded by acidification in the endo/lysosomal network.
Macrophages express receptors for HCV
As macrophages specifically internalize HCV independent of viral E2 entry factor, differentiation may upregulate surface factor(s) that enable cells to recognize HCV. Expression of SCARB1 (SR-B1), CD209 (DC-SIGN), CD81, and CLDN1 have been reported to mediate interaction between HCV particles and hepatocytes (20,37). We analyzed the expression of these receptors in THP-1 macrophage-like cells, monocytes, and Huh7.5.1 hepatocytes. Notably, SR-B1/SCARB1 was expressed in all three cell types, DC-SIGN/CD209 and CD81 were expressed at higher levels in myeloid cells, and CLDN1/CLDN1 expression was restricted to hepatocytes (Fig. 2A, B). We hypothesized that surface expression differences of SR-B1, DC-SIGN, and CD81 in macrophages relative to monocytes may explain the increase in virus uptake in these cells.
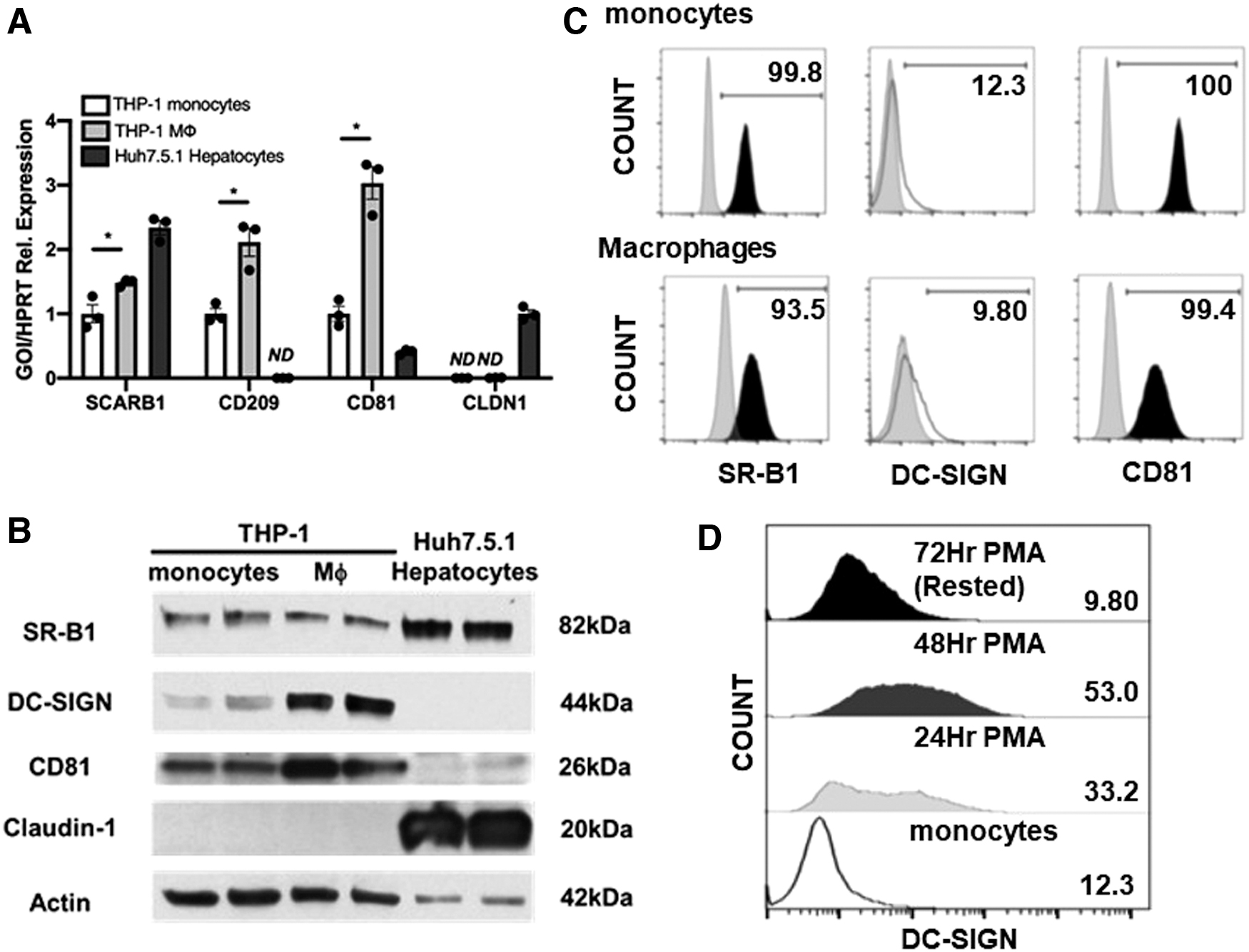
Despite lower protein expression compared with hepatocytes, cell surface expression of SR-B1 was detectable in THP-1 monocytes and macrophage-like cells by Flow Cytometry (Fig. 2C). CD81 was also detectable on the surface of both cell types (Fig. 2C). Surprisingly, THP-1 macrophage-like cells displayed minimal surface expression of DC-SIGN similar to that observed in monocytes (Fig. 2C). DC-SIGN surface expression showed an initial upregulation for THP-1 macrophage-like cells during PMA treatment but was rapidly lost upon removal of PMA to achieve a “resting” macrophage phenotype (Fig. 2D). These results suggest that macrophages express receptors involved in recognition of HCV.
Macrophage exposure to HCV alters lipid metabolism causing cholesterol accumulation
HCV particles, replicons, and proteins induce accumulation of multiple lipids, including fatty acids and cholesterol in hepatocytes (1,10,32,39,44,62,63). We hypothesized that virus exposure in macrophages may also elicit metabolic disturbance. To test this possibility, we extracted and quantified Oil Red O as a measure of total neutral lipids in macrophages exposed to virus. Oil Red O extraction and quantification as a measure of neutral lipids indicated increased lipids in macrophages exposed to HCV for 48 h (Fig. 3A). Microscopic observation demonstrated that HCV-exposed macrophages were larger with increased cytoplasm and prominent lipid inclusions (Supplementary Fig. S3). We determined no difference in triglyceride levels (Supplementary Fig. S5A), however, total cholesterol demonstrated a 20% increase between mock- and HCV-exposed macrophages (Fig. 3A).

We next determined cholesterol species accumulated in macrophages exposed to HCV by analyzing free versus esterified cholesterols. Cholesterol esters accumulated in macrophages exposed to HCV such that increase in total cholesterol included 40% increase in cholesterol esters coupled with 15% increase in free cholesterol (Fig. 3A).
We performed filipin staining in macrophages to determine if virus exposure induced discreet intracellular cholesterol accumulations in these cells. Confocal microscopy demonstrated diffuse intracellular accumulations of cholesterol in a perinuclear region in HCV-exposed macrophages, in contrast to weaker staining localized to membranes, in mock-exposed macrophages (Fig. 3B and Supplementary Fig. S4, arrows in insets).
Fluorescence traces through mock-exposed cells demonstrated two polar peaks of Filipin staining corresponding to cell membrane with a nuclear peak in the center of cells. In contrast, HCV-exposed cells demonstrated a dramatic skewing of Filipin staining preferentially toward one side of cells indicating perinuclear accumulation (Supplementary Fig. S4). Increased cholesterol biogenesis, reduced cholesterol efflux, or increased lipid uptake can increase cellular cholesterol.
To determine whether cholesterol accumulation in macrophages occurred from internal or exogenous sources, we assayed uptake of DiI-labeled LDL and C12-NBD-labeled cholesterol. No significant increase in the capacity of virus-exposed cells to take up lipoproteins or cholesterol was detected when these features were assayed with commercially available kits utilizing fluorescently labeled lipoprotein and cholesterol species (Supplementary Fig. S5B). These data indicate that HCV exposure causes macrophages to accumulate cholesterol in diffuse pools derived from an intracellular process. These data prompted us to examine pathways of cholesterol flux in macrophages to determine if HCV-induced cholesterol accumulation could activate these pathways.
HCV exposure in macrophages increases proteins in the LXRs—dependent metabolic pathway
LXRs regulate macrophage cholesterol efflux and are involved in immune responses (65). LXR activation can induce or repress gene expression. Considering HCV-induced accumulation of cholesterol in macrophages (Fig. 3), we wanted to examine effects on LXR protein expression and activation. HCV exposure had no effect on LXRβ, but resulted in a slight increase in LXRα in macrophages (Supplementary Fig. S6A). Next, we examined the effect of HCV exposure on LXR targets in macrophages. HCV induced upregulation of LXR target ABCA1 beginning at 24 h and increasing throughout 48 h postexposure in THP-1 macrophage-like cells (Supplementary Fig. S6B). Cell culture virus preparations contain factors released from producer cells. Thus, we validated our findings using sucrose-gradient purified HCV isolates from Genotypes 1A, 1B, and 3A.
Within 24 h of exposure to primary isolates, THP-1 macrophage-like cells demonstrated a roughly two- to three-fold increase in ABCA1 protein expression compared with mock-exposed cells indicating that LXR activation is a specific effect of virus exposure (Fig. 4A and Supplementary Fig. S6C). THP-1 macrophage-like cells exposed to HCV also demonstrated increased SREBP-1c (Fig. 4B). We examined LXR activation in primary MDMs exposed to HCV. Forty-eight hours exposure to HCV caused upregulation of multiple LXR target proteins in MDMs, ABCA1, SREBP-1c, and LXRα itself (Fig. 4C).

To confirm the widely reported role of LXR activation on expression of these metabolic targets in THP-1 macrophage-like cells, cells were treated with LXR agonists GW3965 (GW) or TO-901317 (TO) and protein expression was examined. As expected, LXR agonists increased expression of proteins involved in cholesterol efflux (ABCA1) and lipogenesis (SREBP-1c) (Supplementary Fig. S6D), confirming that LXR activation regulates these proteins involved in cholesterol and fatty acid metabolism paralleling the observed effects of HCV in macrophages. These results confirm that LXR regulates metabolic signaling in macrophages and suggest that HCV activates expression of proteins in this pathway in macrophage models and primary macrophages.
Macrophages recognize HCV through SR-B1 resulting in LXR activation
Blockade of viral envelope protein E2-CD81 interaction did not inhibit viral RNA uptake in macrophages (Supplementary Fig. S2C), although CD81 is expressed in this system (Fig. 2C). Resting macrophages also displayed minimal surface DC-SIGN (Fig. 2C). Therefore, we examined the role of SR-B1 in HCV uptake in macrophages. HCV produced in hepatocytes through the apolipoprotein pathway is most infectious as lipoviral particles containing lipoprotein constituents (6,11,13,18,21,25). In the liver, SR-B1 functions to uptake high-density lipoproteins from the circulation (2,60). SR-B1 also plays a role in HCV infection in hepatocytes. We hypothesized that macrophages sense HCV similar to a lipoprotein through SR-B1.
Therefore, we sought to determine if targeting SR-B1 could reduce or delay accumulation of viral RNA observed upon exposure of macrophages to HCV. Macrophages and hepatocytes express SR-B1, although the role of this receptor in recognition of HCV in immune cells has not been described. Inhibitors of SR-B1, termed blockers of lipid transport (BLT) uncovered the role of SR-B1 in HCV uptake in hepatocytes. Blocker of lipid transport 4 (BLT-4) reduces HDL enhancement of pseudo-particle infectivity and blocks soluble E2 binding to SR-B1-expressing cells. Among CD81, DC-SIGN, and SR-B1, SR-B1 has been described to have a role in lipid binding at the cell surface of macrophages and hepatocytes.
We examined whether BLT-4 could reduce or delay HCV RNA binding upon exposure in macrophages. As we observed the greatest amount of viral RNA binding to macrophages at 1 h postexposure (Fig. 1A, C) and blockade of lipid binding by SR-B1 would be expected to effect viral binding, we examined the effect of BLT-4 treatment at this time point. For comparison, we utilized an inhibitor of actin polymerization, cytochalasin D (CytoD), which has been demonstrated to inhibit HCV genome uptake in macrophages (36,49).
One hour pretreatment with BLT-4 (Sigma, cat. no. SML0512) reduced bound total viral RNA in association with macrophages to ∼75% of levels in vehicle-treated cells similar to the effect of CytoD (Fig. 5A and Supplementary Fig. S7A). We also tested the effect of blocking antibodies to DC-SIGN and CD81 on HCV RNA uptake in macrophages. In agreement with minimal surface expression, we observed no effect of DC-SIGN blockade on virus uptake (Supplementary Fig. S7B). Blocking CD81 also had no effect on virus uptake, consistent with data showing that blocking E2 did not affect this process (Supplementary Figs. S2C and S7B).

These results suggest that macrophages recognize HCV through scavenger receptors involved in lipoprotein uptake. Data with CytoD confirm the requirement for phagocytosis in HCV uptake in macrophages (36,49). Taken together, this suggests a mechanism whereby lipid-associated virus binds to cells and is internalized through a mechanism involving SR-B1 and phagocytosis.
LXR activation by HCV in macrophages has not been previously described. This finding was unexpected as HCV exposure increases cholesterol in macrophages and LXR activation should reduce cellular cholesterol, suggesting virus may act directly on LXR. To demonstrate if the effect of HCV exposure on LXR activation was specific to virus, we assessed the ability of antibody blockade of SR-B1 to prevent HCV-induced ABCA1 protein induction alongside reduction in viral RNA accumulation by BLT-4. We used anti-SR-B1 antibody demonstrated to prevent HCV infection of Huh7.5 hepatocytes (12).
In the presence of nonblocking isotype, HCV exposure induced the expected increase in ABCA1 protein compared with mock supernatants (Fig. 5B). The addition of SR-B1 blocking antibody from 1 h before infection throughout 48 h of virus exposure prevented the HCV-induced increase in ABCA1. SR-B1 blocking antibody reduced ABCA1 levels independently of virus, perhaps indicating that this antibody interferes with cellular recognition of apolipoproteins, which could independently reduce ABCA1 expression.
We also assessed the involvement of SR-B1 in recognition of virus and induction of LXR activation in macrophages by utilizing SR-B1 knockdown THP-1 macrophage-like cells. Notably, knockdown of SR-B1 similarly reduced ABCA1 independent of HCV (Supplementary Fig. S8). These data suggest that blocking SR-B1 can generally reduce ABCA1 protein and also prevent the HCV-specific induction of ABCA1.
Discussion
HCV causes inflammation in the liver. Long-term inflammation associated with chronic HCV infection plays a role in the development of liver fibrosis and cirrhosis. Macrophages are central regulators of liver inflammation and fibrosis. In this report, we characterized viral RNA and protein along with metabolic activity in macrophages following exposure to HCV. Uptake of HCV and persistence of viral RNAs were detectable in macrophages for several days.
Our data demonstrate that the level of genomic RNA is sufficient to support a window of potentially de novo protein synthesis (˜6 h), but insufficient to support viral genomic RNA replication or high-level protein expression. The uptake of HCV by macrophages involves scavenger receptor SR-B1, endocytosis, and degradation in endo/lysosomes. Notably, viral exposure in macrophages caused accumulation of cholesterol esters.
Our studies on the cholesterol efflux pathway in HCV-exposed macrophages indicate that HCV activated the LXR pathway and increased ABCA1 protein, responsible for cholesterol efflux, in macrophages. These results suggest that LXR activation and increased ABCA1 expression may result from HCV-induced cholesterol accumulation. Moreover, we demonstrated modulation of metabolic mediators by pharmacological activation of LXRs and HCV exposure in macrophages. Importantly, blockade of SR-B1 reduced virus surface binding and induction of ABCA1.
It is not clear how viral RNA is released in macrophages after HCV exposure. Inhibitors of endosomal acidification block HCV entry and infection in hepatocytes suggesting viral genomes are liberated into the cytoplasm following acidification at these sites (7,12,46,68). Endosomal acidification is important for HCV-induced interferon and cytokine induction in DCs (24,53,71). We observed significant increase in viral RNA during the 4-h incubation of macrophages in the presence of chloroquine and observed a similar effect through BafA1. These results suggest that viral RNAs are degraded in acidic compartments in macrophages. Gradual delivery to endo/lysosomes could allow maintenance of infectious virus upon fusion with other cells (16). Delivery to acidic compartments could facilitate recognition by RNA sensors to induce TGFβ, which is prevented by BafA1 (49).
Our data indicate that virus uptake in THP-1 macrophage-like cells occurs through SR-B1 rather than DC-SIGN. Notably, DC-SIGN expression, which is restricted to specific subsets of tissue macrophages, has been shown to mediate interaction of liver sections and Kupffer cells with HCV E2 and blockade of DC-SIGN can prevent HCV-mediated induction of certain cytokines in macrophages (34,73). Highlighting the subtle differences between Kupffer cells and MDMs, blood-derived monocytes and THP-1 macrophage-like cells, express only low levels of DC-SIGN basally that increase with cytokine stimulation (54).
Lack of DC-SIGN effect on virus uptake in our system coincided with the loss of this receptor from the surface of THP-1 macrophage-like cells following 72 h differentiation with phorbol ester (PMA) and PMA removal for 24 h to induce “resting” macrophages. However, other studies have shown that DC-SIGN expression is restricted to primary macrophages compared with monocytes, monocytoid dendritic cells (mDCs), and plasmacytoid dendritic cells (pDCs) (73). This same study demonstrated that blocking DC-SIGN could prevent HCV-induced cytokine induction in human macrophages, although this study did not assess viral RNA uptake (73).
While the reasons for this difference are unclear, it is important to note that our use of “resting” macrophages was intended to simulate a scenario where macrophage metabolic and inflammatory characteristics could be examined independent of interferences from differentiating agents (phorbol esters, PMA) and polarizing cytokines (IL-4, etc.). It is worthwhile to conduct further studies on HCV uptake upon recognition by DC-SIGN in different subsets of myeloid cells, particularly primary cells endogenously expressing the receptor. Likewise, it cannot be ruled out that DC-SIGN may play a role in HCV recognition by blood MDMs in other contexts.
In HCV-infected hepatocytes, triglycerides and cholesterol are drastically increased with particular cholesterol accumulation (55). Lipid and cholesterol accumulate close to HCV proteins in presumed replication complexes suggesting a role for lipid accumulation in viral life cycle. Our data indicate an absence of replication and low levels of viral protein in macrophages, suggesting that cholesterol accumulation in these cells is a consequence of viral activation of a secondary pathway rather than activation of cholesterol biogenesis itself. Similarity of cholesterol ester accumulation identified in our studies in macrophages and that in hepatocytes suggests future studies on cholesterol biogenesis and processing in virus-exposed macrophages and a potential role for ACAT2 in this process (55).
HCV activates the LXR pathway of cholesterol metabolism in macrophages. It is intriguing that virus exposure activates cholesterol efflux but affects an overall increase in lipids and cholesterol. This can be understood in the context of specific accumulation of cholesterol esters. Cholesterol is stored in macrophages as cholesterol ester but effluxed in the free or unesterified form. Hydrolysis of cholesterol esters by cholesterol ester hydrolase (CEH) has been suggested to be the rate-limiting step in cholesterol efflux (22,23,74).
These considerations suggest a scenario where virus exposure may cause an increase in total cholesterol through activating effects on acyl-CoA:cholesterol acyltransferase (ACAT), increasing cholesterol esterification, or inactivating effects on CEH, inhibiting hydrolysis of cholesterol esters, or both. Future studies will be needed to address which of these two pathways contributes to virus-induced cholesterol accumulation.
Our results suggest ABCA1 induction upon virus exposure is an adaptive response to cholesterol accumulation. It is intriguing to speculate that pharmacological manipulation of cholesterol ester balance in virus-exposed macrophages through ACAT or CEH may alleviate lipid accumulation by liberating esterified cholesterol to free cholesterol as substrate for efflux by the virus-induced abundance of efflux transporter.
While our data implicate SR-B1 in HCV recognition in macrophages and HCV-induced activation of LXR-dependent metabolic targets, the effects of SR-B1 blockade in our experiments were more general than specific. While SR-B1 antibody blockade prevented the HCV-specific induction of ABCA1, both SR-B1-blocking antibody and siRNA reduced basal ABCA1 levels.
Thus, it is important to acknowledge that while SR-B1 is likely one specific mediator between HCV recognition and increased metabolic targets in macrophages, we cannot conclude that this mechanism is entirely exclusive. In this regard, it is noteworthy that recent studies indicate a role for phagocytosis in HCV entry in macrophages (41). Thus, the ultimate effect of HCV on macrophages is likely a mosaic of effects mediated by specific receptors and effects that result from less-specific bulk uptake mechanisms.
Finally, our data raise an interesting specter that the manipulation of lipid metabolism may be an avenue for virus to alter the immune response of macrophages. LXR activation is anti-inflammatory, although the exact mechanism has remained elusive. Our data show that HCV can activate metabolic mediators downstream of LXR. In future studies, it will be important to address whether LXR activation may impact TGF-β or any of the other mediators, including procollagen and CCL5 that are induced by HCV, and impact hepatic stellate cell activation. Ultimately, this understanding will allow therapeutic strategies to impact metabolic and immune imbalance in infected patients and augment antiviral therapy to preserve and perhaps reverse liver pathology that persists in the absence of ongoing viral replication.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Institute of Health R01 grant (DK122737 to YSH).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.