
Abstract
Natural killer-like B (NKB) cells are a newly identified immune subset, which are separated from NK cells and B cells. NKB cells demonstrated immunoregulatory functions in elimination of microbial infection and inflammation through secretion of interleukin (IL)-12 and IL-18. However, the role of NKB cells in hepatitis B virus (HBV)-related diseases has not been reported. In this study, peripheral T cells, B cells, NK cells, and NKB cells in HBV-associated acute-on-chronic liver failure (ACLF), chronic hepatitis B (CHB), asymptomatic HBV carriers (AsC), and controls were investigated by flow cytometry. Plasma IL-12 and IL-18 levels were measured by enzyme-linked immunosorbent assay. Peripheral blood mononuclear cells from HBV-ACLF patients were stimulated with recombinant IL-12 or IL-18. Changes of immune cell percentage and nuclear factor-kappa B (NF-κB) phosphorylation were assessed. There were no statistical differences of T cell percentage, B cell percentage, or NK cell percentage among groups. NKB cell percentage within lymphocytes, and plasma IL-12 and IL-18 levels in HBV-ACLF patients were significantly elevated compared with in CHB, AsC, and controls. NKB cell percentage and IL-18, but not IL-12, had a better prognosis function for the 28-day survival status in HBV-ACLF patients. Recombinant IL-12 enhanced T cell and NK cell percentage, while only high concentrations (10 ng/mL) of IL-18 promoted NKB cell percentage in HBV-ACLF patients. High concentrations (10 ng/mL) of IL-18 induced NF-κB phosphorylation in NKB cells probably through suppression of IL-18 binding protein in HBV-ACLF patients. The current data indicated that elevated NKB cells and IL-18 might be important indicators for poor prognosis of HBV-ACLF patients. Increased IL-18 might play a positive feedback activity to NKB cells in HBV-ACLF patients.
Introduction
Acute-on-chronic liver failure (ACLF) is acute decompensation in patients with chronic liver diseases, which presented as both hepatic dysfunction and extrahepatic organ failures due to infection or acute liver injury-induced systemic inflammation (1, 2). In China, hepatitis B virus (HBV) infection is still the most common cause of chronic liver disease and a severe exacerbation risk factor for the development and progression of ACLF (14, 24). HBV-associated ACLF presents the shift from low-grade inflammation to intensive systemic inflammatory response, which is the hallmark of ACLF in the setting of immune dysfunction or exhaustion (10, 15, 20). However, the inflammatory response to the pathogenesis of HBV-ACLF was still not completely understood.
The natural killer-like B (NKB) cell subset is a newly identified lymphocyte subpopulation that exists in peripheral blood, spleen, and mesenteric lymph nodes (MLNs) of humans and mice (18, 23). NKB cells present the phenotype of both NK cells and B cells, and have the unique features that differ from T cells, B cells, and NK cells. Both interleukin (IL)-12 and IL-18 are signature cytokines produced by NKB cells in the early stage of microbial infection, and play vital roles in the eradication of infection and proinflammatory response (18, 23).
Importantly, toll-like receptor 3 preactivated bone marrow-derived mesenchymal stem cells protected alcohol-induced intestine and hepatic damage through downregulation of hypoxia-inducible factor-2α signaling pathway, reduction of NKB cell proportion, and IL-18 secretion (3), indicating the potential regulatory activity of NKB cells in liver injury. However, few studies focused on the modulatory function of NKB cells in HBV-associated liver disorder, including asymptomatic HBV carriers (AsC), chronic hepatitis B (CHB), and HBV-ACLF.
Due to the proinflammatory activity of NKB cells and secreted IL-18 in periodontitis (23) and alcohol-induced liver injury (3), we hypothesized that NKB cells and secreting cytokines also contribute to hepatic damage in HBV-induced liver diseases. To test this possibility, NKB cell proportion was investigated in patients with HBV infection. The regulatory function of IL-12 and IL-18 to immune cells in HBV infection was also assessed in vitro.
Patients, Materials, and Methods
Ethical statement
The study was approved in compliance with the Declaration of Helsinki. The study protocol was approved by the Ethics Committee at 964th Hospital of PLA and the Ethics Committee at Tangdu Hospital, Fourth Military Medical University. Written informed consent was obtained from all studied subjects.
Studied population
Thirty-seven HBV-ACLF patients were enrolled from 964th Hospital of PLA and Tangdu Hospital between July 2018 and December 2019. The diagnosis of HBV-ACLF was based on the underlying chronic HBV infection and clinical criteria of recent development of jaundice (total bilirubin >170 μmol/L) and a prothrombin activity <40%. ACLF patients could be divided into type A (chronic liver disease, n = 7), type B (compensated cirrhosis, n = 16), and type C (decompensated cirrhosis, n = 14) (5).
Thirty CHB patients and 22 AsC were recruited from Tangdu Hospital between July 2018 and March 2019. The diagnosis of chronic HBV infection was made in accordance with the HBV DNA and hepatitis B surface antigen positive from more than 6 months. The liver function of all AsC was normal, while CHB patients exhibited symptoms or signs of abnormal hepatic function. The exclusion criteria were the following: patients with other chronic viral infections, malignancies, autoimmune diseases, diabetes, or with pregnancy. No patients received antiviral or immunomodulatory therapies at least 3 months before sampling. Twenty-five age- and sex ratio-matched healthy controls (HC) were also included. The characteristics of all the enrolled subjects are shown in Table 1.
Characteristics of All Enrolled Subjects
Data are shown as median and range.
AsC, asymptomatic HBV carrier; CHB, chronic hepatitis B; HBV-ACLF, hepatitis B virus-associated acute-on-chronic liver failure; HC, healthy control; MELD, model for end-stage liver disease.
Isolation of plasma and peripheral blood mononuclear cells
Total anticoagulant blood was obtained from all enrolled subjects. Plasma was obtained by centrifuging at 1,000 g at 4°C for 5 min. Peripheral blood mononuclear cells (PBMCs) were prepared with the Ficoll-Hypaque density centrifugation reagent Hisotopaque-1077 (Sigma, St Louis, MO) according to the manufacturer's instruction. PBMCs, 106, were stimulated with either recombinant human IL-12 (final concentration, 1 or 10 ng/mL, Cat# 219-IL; R&D Systems, Minneapolis, MN) or recombinant human IL-18 (final concentration, 1 or 10 ng/mL, Cat# B001-5; R&D Systems) for 48 h. Cells were harvested for further investigation.
Enzyme-linked immunosorbent assay
Plasma IL-12p70 and IL-18 levels were measured by commercial enzyme-linked immunosorbent assay (ELISA) kits (Cat# D1200 for IL-12p70 and Cat# DL180 for IL-18; R&D Systems) according to the manufacturer's instruction. IL-18 binding protein (IL-18BP) in the cultured supernatants was measured by the commercial ELISA kit (Cat# CSB-E13687h; Cusabio, Wuhan, Hubei Province, China) according to the manufacturer's instruction.
Flow cytometry
PBMCs were transferred into fluorescence-activated cell sorting (FACS) tubes, and washed twice. Cells were stained with the fluorescein isothiocyanate mouse anti-human CD3 (clone HIT3α, Cat# 555339; BD Pharmingen, San Jose, CA), peridinin-chlorophyll-protein complex-cyanine5.5 (PerCP-Cy5.5) mouse anti-human CD56 (clone B159, Cat# 560842; BD Pharmingen), allophycocyanin (APC) mouse anti-human CD16 (clone 3G8, Cat# 561248; BD Pharmingen), phycoerythrin cyanine 7 (PE-Cy7) mouse anti-human NKp46 (clone 9E2/NKp46, Cat# 562101; BD Pharmingen), and APC-Cy7 mouse anti-human CD19 (clone SJ25C1, Cat# 557791; BD Pharmingen) for 30 min in the dark.
In certain experiments, PBMCs were then treated with the Cytofix/Cytoperm solution and Perm/Wash buffer (BD Bioscience, San Jose, CA), and they were intracellularly stained with PE mouse anti-nuclear factor-kappa B (NF-κB) p65 (pS529) (clone K10-895.12.50, Cat# 558423; BD Phosflow; San Jose, CA). The labeled cells were examined on the FACS Aria II flow cytometer (BD Bioscience). The FACS results were analyzed with FlowJo Version 11.0 software (Tree Star, Ashland, OR).
Statistical analysis
Data are presented as mean ± standard deviation, and they were statistically analyzed using SPSS Version 23.0 software (Chicago, IL). One-way ANOVA, SNK-q test, or paired t-test was used for comparison. Pearson or Spearman correlation was used for correlation analysis. The prognosis for the 28-day survival status was analyzed by the receiver operating characteristic (ROC). A p-value <0.05 was considered statistically significant.
Results
NKB cell percentage was increased in HBV-ACLF patients
The gating strategies for T cells, B cells, NK cells, and NKB cells are shown in Figure 1. Lymphocytes within PBMCs were gated using forward scatter and side scatter. CD3+CD19− cells were defined as T cells, while CD3−CD19+ cells were defined as B cells. CD16+CD56+ cells within CD3−CD19− cells were defined as NK cells. The phenotype for NKB cells was CD3−CD16+CD56+NKp46+CD19+. There were no statistical differences of T cell percentage, B cell percentage, or NK cell percentage among groups (p > 0.05, one-way ANOVA, Fig. 2A–C).

The gating strategies for T cells, B cells, NK cells, and NKB cells were shown in HBV-ACLF patients, CHB patients, AsC, and HC. Lymphocytes within PBMCs were gated using FSC and SSC. CD3+CD19− cells were T cells. CD3−CD19+ cells were B cells. CD3−CD19−CD16+CD56+ cells were NK cells. CD3−CD16+CD56+NKp46+CD19+ cells were NKB cells. ACLF, acute-on-chronic liver failure; AsC, asymptomatic HBV carrier; CHB, chronic hepatitis B; FSC, forward scatter; HBV, hepatitis B virus; HC, healthy control; NKB, natural killer-like B; PBMCs, peripheral blood mononuclear cells; SSC, side scatter.
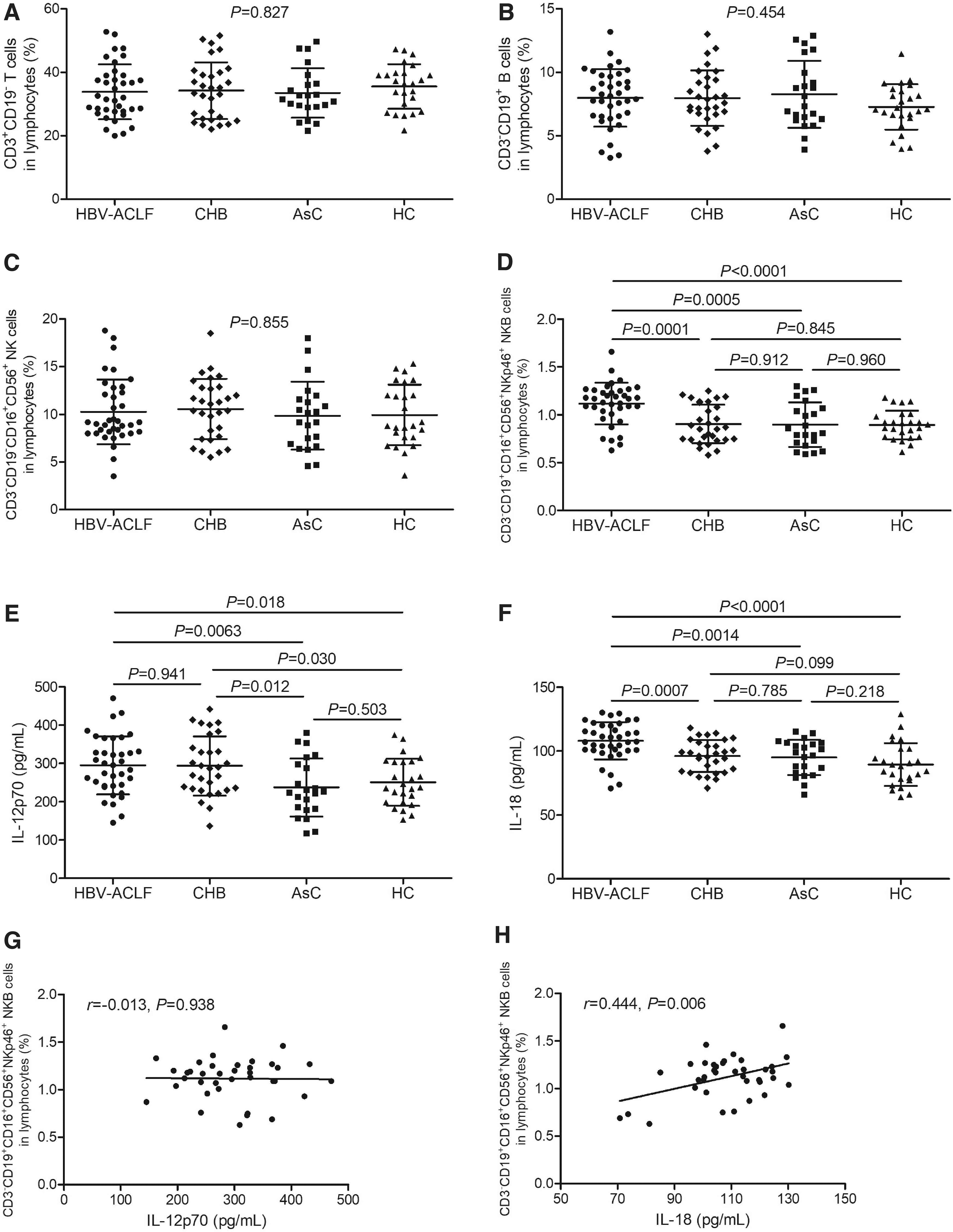
Comparison of T cell, B cell, NK cell, and NKB cell percentages and plasma IL-12 and IL-18 levels among HBV-ACLF patients, CHB patients, AsC, and HC.
Peripheral NKB cell percentage within lymphocytes in HBV-ACLF patients (1.12% ± 0.22%) was significantly elevated compared with in CHB patients (0.90% ± 0.20%, p = 0.0001), AsC (0.90% ± 0.23%, p = 0.0005), and HC (0.89% ± 0.15%, p < 0.0001) (SNK-q test, Fig. 2D). The plasma IL-12p70 level was remarkably increased in HBV-ACLF patients (294.8 ± 75.29 pg/mL) and CHB patients (293.4 ± 77.33 pg/mL) compared with AsC (237.2 ± 75.59 pg/mL) and HC (250.7 ± 61.38 pg/mL) (p < 0.05, SNK-q test, Fig. 2E). The plasma IL-18 level was also notably increased in HBV-ACLF patients (108.0 ± 14.52 pg/mL) compared with in CHB patients (96.07 ± 12.56 pg/mL, p = 0.0007), AsC (95.08 ± 12.90 pg/mL, p = 0.0014), and HC (89.42 ± 16.79 pg/mL, p < 0.0001) (SNK-q test, Fig. 2F).
There were no significant differences of either immune cell subpopulation percentage or cytokine level among type A, type B, and type C ACLF patients (p > 0.05). NKB cell percentage did not correlate with IL-12p70 level (r = −0.013, p = 0.938, Pearson correlation analysis, Fig. 2G), but positively correlated with IL-18 level (r = 0.444, p = 0.006, Pearson correlation analysis, Fig. 2H) in ACLF patients.
HBV-ACLF patients were divided into the survival group (n = 26) and death group (n = 11) according to the survival outcome on 28 days after admission. NKB cell percentage yielded an area under curve (AUC) of 0.836 (95% confidence interval 0.697–0.975) in HBV-ACLF patients (p = 0.0014, Fig. 3A). However, IL-12p70 level in plasma was analyzed by ROC curve with an AUC of 0.605 (95% confidence interval 0.420–0.790) in HBV-ACLF patients (p = 0.319, Fig. 3B). Moreover, plasma IL-18 level yielded an AUC of 0.724 (95% confidence interval 0.523–0.925) in HBV-ACLF patients (p = 0.034, Fig. 3C).
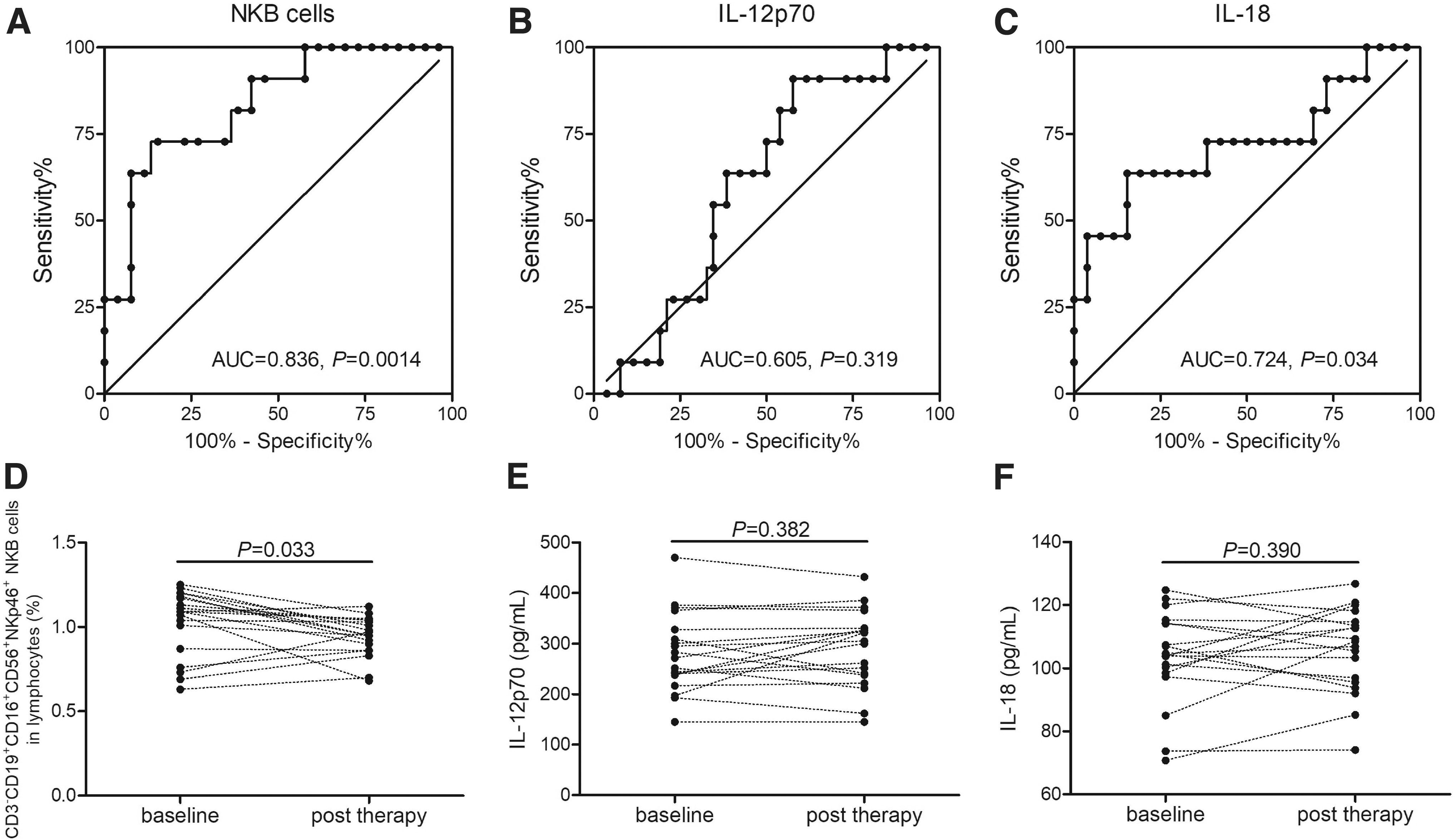
ROC curve analysis as well as comparison between baseline and post-therapy of NKB cell percentage and plasma IL-12 and IL-18 levels in HBV-ACLF patients.
Blood samples were also collected from 19 survival HBV-ACLF patients after effective therapy before discharged. NKB cell percentage was significantly downregulated after effective treatment in HBV-ACLF patients (0.94% ± 0.12% vs. 1.03% ± 0.19%, p = 0.033, paired t-test, Fig. 3D). However, there were no statistical differences of either plasma IL-12p70 or IL-18 level between baseline and post-therapy in HBV-ACLF patients (p > 0.05, paired t-tests, Fig. 3E, F). There were no significant correlations between NKB cell percentage and liver injury index (including alanine aminotransferase, bilirubin, and prothrombin activity) in HBV-ACLF patients (p > 0.05, Spearman correlation analysis).
High concentration of IL-18 promotes NKB cell percentage in HBV-ACLF patients
PBMCs, 106, from 18 HBV-ACLF patients were stimulated with either recombinant human IL-12 (1 or 10 ng/mL) or recombinant human IL-18 (1 or 10 ng/mL). Cells were harvested 48 h poststimulation for T cell, B cell, NK cell, and NKB cell analysis by flow cytometry. T cell and NK cell percentage was significantly increased in response to both 1 and 10 ng/mL of IL-12 stimulation (p < 0.05, SNK-q tests, Fig. 4A, C). However, there were no remarkable differences of either B cell or NKB cell percentage among no stimulation, 1 ng/mL of IL-12 stimulation, and 10 ng/mL of IL-12 stimulation (p > 0.05, one-way ANOVA, Fig. 4B, D).
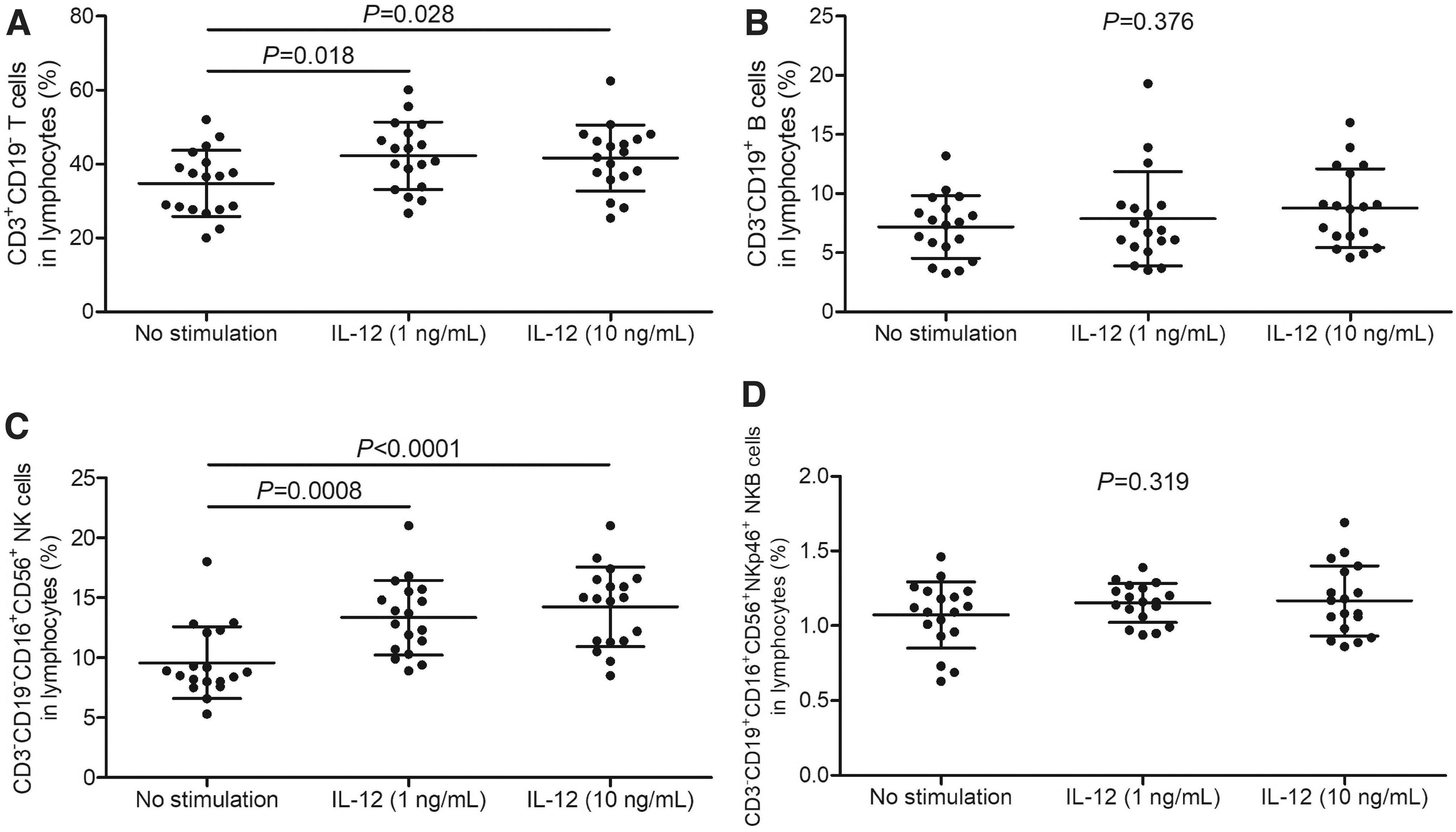
Comparison of T cell, B cell, NK cell, and NKB cell percentage in response to recombinant human IL-12 stimulation in HBV-ACLF patients. PBMCs, 106, from 18 HBV-ACLF patients were stimulated with recombinant human IL-12 (1 or 10 ng/mL) for 48 h.
Furthermore, there were no robust differences of T cell, B cell, or NK cell percentage among no stimulation, 1 ng/mL of IL-18 stimulation, and 10 ng/mL of IL-18 stimulation (p > 0.05, one-way ANOVA, Fig. 5A–C). One nanogram per milliliter of IL-18 stimulation did not significantly increase NKB cell percentage (1.19% ± 0.15% vs. 1.07% ± 0.22%, p = 0.080, SNK-q test, Fig. 5D). In contrast, 10 ng/mL of IL-18 stimulation notably elevated NKB cell percentage (1.30% ± 0.29%) in HBV-ACLF patients (p = 0.012, SNK-q test, Fig. 5D).
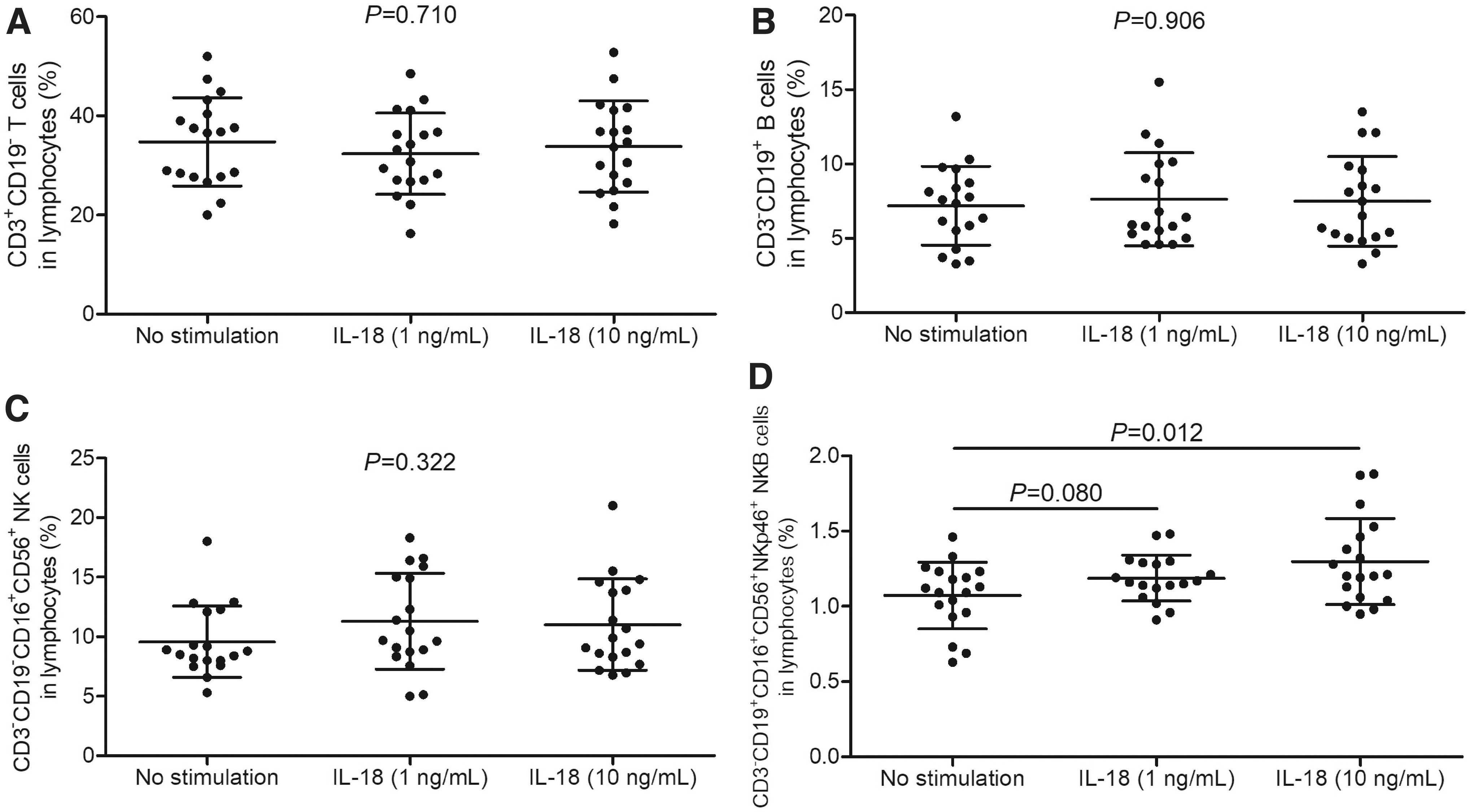
Comparison of T cell, B cell, NK cell, and NKB cell percentage in response to recombinant human IL-18 stimulation in HBV-ACLF patients. PBMCs, 106, from 18 HBV-ACLF patients were stimulated with recombinant human IL-18 (1 or 10 ng/mL) for 48 h.
High concentration of IL-18 enhanced NF-κB phosphorylation in NKB cells via suppression of IL-18BP in HBV-ACLF patients
PBMCs, 106, from 15 HBV-ACLF patients were stimulated with recombinant human IL-18 (1 or 10 ng/mL) for 48 h. Phosphorylated NF-κB p65 in NKB cells was assessed by flow cytometry, while IL-18BP level in the supernatants was measured by ELISA. The representative flow histograms for phosphorylated NF-κB p65 within NKB cells in response to IL-18 stimulation are shown in Figure 6A. The percentage of phosphorylated NF-κB p65+ cells in NKB cells was significantly elevated in response to 10 ng/mL of IL-18 stimulation (5.26% ± 1.16%) compared with no stimulation (1.76% ± 0.36%, p < 0.0001, SNK-q test, Fig. 6B) and 1 ng/mL of IL-18 stimulation (1.70% ± 0.20%, p < 0.0001, SNK-q test, Fig. 6B).

Recombinant IL-18 stimulation enhanced NF-κB phosphorylation and reduced IL-18BP level in HBV-ACLF patients. PBMCs, 106, from 15 HBV-ACLF patients were stimulated with recombinant human IL-18 (1 or 10 ng/mL) for 48 h.
However, 1 ng/mL of IL-18 did not affect NF-κB p65 phosphorylation in NKB cells compared with no stimulation (p = 0.567, SNK-q test, Fig. 6B). Similarly, 10 ng/mL of IL-18 stimulation downregulated IL-18BP level in the supernatant (155.4 ± 61.39 pg/mL) compared with no stimulation (257.2 ± 87.75 pg/mL, p = 0.001, SNK-q test, Fig. 6C). One nanogram per milliliter of IL-18 stimulation slightly reduced IL-18BP level (201.7 ± 70.42 pg/mL), but this difference failed to achieve statistical significance (p = 0.067, SNK-q test, Fig. 6C).
Discussion
NKB cells had the phenotype of both NK cells and B cells. NK1.1+CD19+CD3− NKB cells existed in the mouse spleen and MLNs, while NKp46+CD19+CD3− NKB cells could be found in the human spleen and MLNs (18). Peripheral and periodontal-infiltrating NKp46+CD19+CD3− NKB cells were also found in humans (23). For the first time, we investigated NKB cells in HBV infection-induced liver diseases.
We used the classical B cell phenotype (CD19+), NK cell phenotype (CD3−CD16+CD56+), and the natural cytotoxicity receptor NKp46 for NKB cell identification. There were an approximate 1% of CD3−CD16+CD56+NKp46+CD19+ NKB cells within the total lymphocytes in all studied subjects. It should be pointed out that several studies reported that the putative phenotype of NKB cells existed in blood, tissues, and lymphoid organs of humans, rhesus macaques, and mice, but the proportion of the detected events was consistently lower, which accounted for a median of less than 0.1% in multiple organs (8, 12). They even speculated that NKB cells are a subpopulation of conventional B cells, which only shared some properties overlapping with NK cells (8, 12).
In contrast, Wang et al. argued that NKB cells were not only a separate subpopulation constitutively expressing mRNA encoding NK1.1 antigen and NKp46 molecule, but also triggered an innate immune response against microbial challenge (19). Chronic simian immunodeficiency virus infection induced circulating and colorectal-resident NKB cell expansion, which did not correlate with viral loads. In contrast, no expansion was found in the peripheral blood of human immunodeficiency virus-infected humans, partly due to the species differences (12). Similarly, elevated periodontal-resident NKB cells were positively correlated with the degree of periodontal supporting tissue destruction (23).
We found that chronic HBV infection did not significantly induce NKB cell elevation in AsC and CHB patients, who presented little or low-degrade liver inflammation. Moreover, NKB cell proportion was robustly increased in HBV-ACLF patients, which was even higher than AsC and CHB patients. This might be partly due to the severe liver injury and systemic inflammation in ACLF patients, although we did not find a statistical correlation between NKB cells and hepatic damage index. There were no significant differences in T cell, B cell, and NK cell frequency within total lymphocytes among groups. Thus, elevated peripheral NKB cells might contribute to the pathogenesis of HBV-ACLF.
Wang et al. suggested that CD19+ cell could also bind NK1.1 or NKp46 antibody in the mouse spleen in marginal zone B cells. Some mature B cell populations might fall into the NKB cell gate in cells from organs, lacking marginal-zone structures (18). Thus, further studies will focus on the proportion and function of liver-infiltrating NKB cells in HBV-infected patients.
NKB cells presented their own signature features, which were characterized by rapidly producing large amounts of IL-12 and IL-18 to prime NK cells and type 1 innate lymphoid cells (18, 19). IL-18 was constitutively expressed by NKB cells and robustly secreted upon infection, while IL-12 was not constitutively produced by NKB cells and only moderately increased in response to microbial infection (18, 23). HBV X protein induced IL-18 expression in the liver, and strongly associated with hepatic damage during HBV infection (9). Decreased IL-18 secretion in peripheral blood enhanced intrauterine HBV infection in pregnant rats (26). Importantly, IL-18 was one of the components of NLRP3 inflammasome.
Differential expression patterns of peripheral and liver-resident NLRP3 inflammasome (caspase-1, IL-1β, and IL-18) were related to CD14+ monocyte recruitment into the liver during HBV-ACLF progression (6). IL-12 promoted central memory CD8+ T cell response and functionally rescued the exhausted viral-specific CD8+ T cells in chronic HBV infection (13, 21).
We found that both IL-12 and IL-18 were elevated in HBV-ACLF patients. However, effective comprehensive therapies for HBV-ACLF (including antiviral, hepatoprotective, and plasma exchange) did not affect circulating IL-12 or IL-18 levels. This might partly be due to the fact that NKB cells were not the only source of these cytokines (23). Elevated NKB cell proportion and peripheral IL-18 level had better prognosis value for the survival status of HBV-ACLF, indicating that IL-18 might be the signature cytokine secreted by NKB cells for mediation of inflammation in HBV-ACLF.
Costimulation with IL-12 and IL-18 induced the secretion of interferon-γ by CD4+ T cells, which contributed to the anti-HBV function of Th1 cells and viral clearance in children with chronic HBV infection (16). Our in vitro experiments showed that IL-12 stimulation enhanced T cell and NK cell frequency, which was consistent with previous findings in cancers and infection (11, 17). Moreover, higher concentrations of IL-18 stimulation promoted NKB cell percentage in HBV-ACLF patients, indicating a potential positive feedback activity of IL-18 in the regulation of NKB cells during HBV-ACLF progression. The probable mechanism for IL-18 modulation to NKB cells was then assessed. IL-18BP was a high-affinity IL-18 decoy receptor, and was also a secreted immune checkpoint and a major therapeutic barrier to IL-18 immunotherapy in cancers (25).
We found that low concentrations of IL-18 slightly reduced IL-18BP level in the cultured supernatants, while high concentrations of IL-18 strongly inhibited IL-18BP secretion. Moreover, IL-18 signaling induced the activation of two major pathways, including the myeloid differentiation factor 88/IL-1 receptor-associated kinase/tumor necrosis factor receptor-associated factor 6 and signal transducer and activator of transcription/mitogen-activated protein kinase (4, 7, 22). Both pathways finally mediated the phosphorylation of NF-κB.
We found that low concentrations of IL-18 did not induce elevated NF-κB phosphorylation, which might be due to the insufficient neutralization of IL-18 to IL-18BP. In contrast, high concentrations of IL-18 might completely block IL-18BP activity, and further increased phosphorylated NF-κB, which finally induced NKB cell activity in HBV-ACLF. In vivo animal experiments are still needed for confirmation of the in vitro results.
Conclusion
Elevated NKB cells and IL-18 might be important indicators for poor prognosis of HBV-ACLF patients. Increased IL-18 might play a positive feedback activity to NKB cells in HBV-ACLF patients, which might contribute to disease progression for HBV-ACLF.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by a grant from the Natural Science Foundation of China (81671555).