
Abstract
The human cytomegalovirus (HCMV) UL24 and UL43 are tegument proteins that have recently been shown to interact with each other in a yeast two-hybrid system. By their overexpression in MRC5 cells, we demonstrate that these viral proteins interact with several important host proteins, especially Dicer and trans-activation response RNA binding protein. As these hots proteins are involved in regulating the production of cellular micro-RNAs, the cytomegalovirus (CMV) proteins could interfere with their actions to favor viral replication directly or through an immune escape mechanism. Double knockout of UL24 and UL43 does not show a remarkable effect on CMV entry or replication, but it significantly downregulates the expression of CMV-encoded miR-UL59, which is thought to regulate the expression of a downstream target UL16 binding protein 1 (ULBP1). Interestingly, the double knockout increases the expression of the ULBP1 recognized by the NKG2D activating receptor of natural killer cells. This study investigates the potential role of several proteins encoded by HCMV in regulating the host cellular environment to favor escape from immunity, and it also provides some basis for the future development of RNA-targeted small molecules to control HCMV infection.
Introduction
The human cytomegalovirus (HCMV) viral genome encodes ∼200 proteins. The US22 family is a member of its genome, including 12 viral genes, known as UL23, UL24, UL28, UL29, UL36, UL43, TRS1, IRS1, US22, US23, US24, and US26. The encoded product has at least one of the family's four conserved amino acid sequences (48). The viral proteins encoded by these viral genes are mainly used as interlayer proteins and are essential structural components of HCMV virus particles. Most of the US22 family genes are necessary for the growth of the virus, and their deletion will impair or reduce the virus growth.
This is, however, that some genes are not required for the growth development of the virus and their absence will not affect the growth of the virus or are nonessential genes for viral growth such as UL24 and UL43. Besides, some of the US22 family genes may work solely or in cooperation to perform some critical roles in viral infection. For example, UL23 can uniquely regulate the immune response induced by IFN-γ, thereby enhancing the resistance of the virus and helping the virus escape (16); UL29 and UL28 can cooperate to activate early gene expression (34,45); UL36 has antiapoptotic activity (31) TRS1 and IRS1 work together to regulate the transcription of some genes, to participate in regulating the protein kinase RNA-activated (PKR) signaling pathway, and to promote virus replication (30,44).
Our understanding of the UL24 and UL43 members of the US22 family is that they encode the interlayer protein, which is a structural tegument protein, but their functions are still poorly understood (2). A research report using yeast two-hybrid to explore the interaction of HCMV virus proteins proved that pUL24 could interact with pUL43 (47). Some recent studies have shown that the interlayer protein of HCMV plays various vital roles in different stages of the virus life cycle. For example, include pp65 (immune escape), pp71 (regulate gene expression), pp150, and pp28 (virus assembly and release) (3,42,48). Therefore, we speculate whether these two genes cooperate in the process of viral infection to participate in some intracellular reactions and thus play some essential functions.
We constructed two overexpression plasmids of UL24 and UL43 and found through immunoprecipitation and silver staining that compared with the control group, these two proteins can co-precipitate some specific interacting proteins (Supplementary Fig. S1), through mass spectrometry (MS) analysis, the interactions with cellular proteins TRBP and Dicer were found. Trans-activation response (TAR) RNA binding protein (TRBP) is a double-stranded RNA binding protein, which can cooperate with Dicer to promote the processing of pre-miRNAs and load the processed miRNAs onto Ago2 (Argonaute 2) to form RNA induction. The RNA-induced silencing complex (RISC) ultimately regulates the expression of some genes (9,20,26,50).
Currently, the most studied HCMV miRNAs mainly include miR-UL112, miR-UL148D, miR-US5-1, miR-US5-2, and miR-US25-1, which play an essential role in the process of viral infection, such as miR-UL112 regulates the latency and reactivation of CMV; miR-UL148D or miR-UL112 can regulate immune response and help virus escape; miR-US25 regulates the cell cycle; miR-US5-1 or miR-US5-2 can regulate inflammatory cytokines. These data indicate that HCMV-encoded miRNAs have essential roles in targeting host immune response, cell cycle, and vesicle transport (12,22,36).
In addition, there are many studies to investigate the functions of HCMV miRNAs to discover other novel miRNAs. The analysis of viral miRNAs became very important in therapeutic intervention, and recently several drug development strategies have been used to develop RNA-targeted small molecules. These strategies can be used to explore the role of HCMV miRNAs in pathogenesis and develop unique modes of action of small molecule drugs as a new treatment against HCMV infection (1).
HCMV miRNAs play an essential role in the regulation of the viral genes during infection of the host and impact on the gene regulation of the host as well. They participate in all aspects of virus infection through different mechanisms and regulate some immune responses to provide a suitable environment for growth and replication. Based on research reports, the viral UL24 and UL43 genes can encode viral interlayer proteins, which are not necessary for the development of the virus, and the proteins encoded by these two viral genes can interact.
Therefore, we speculate whether these two genes are similar to UL29/28 or TRS1/IRS1 in the US22 family, which are cooperating to help the HCMV growth and replication. According to MS analysis, we demonstrate that the interacting proteins TRBP and Dicer of pUL24 and pUL43, these two host proteins are essential components for processing mature miRNAs, and can load mature miRNAs onto RISC to regulate the expression of some genes. Therefore, we speculate whether these two viral genes can regulate some miRNAs, thereby affecting some immune responses in the host, helping the virus to escape the host's immune system, and promoting the immune escape of the virus.
A lot of recent research has focused on developing RNA-targeted small molecule drugs to prevent the immune escape of the virus. This project aims to investigate the potential role of several proteins encoded by cytomegalovirus (CMV) in regulating the host cellular environment to favor escape from immunity as well as to explore the mechanism of miRNAs and provide some theoretical basis for the future development of RNA-targeted small molecule drugs to help HCMV disease control and treatment.
Materials and Methods
Plasmid
The overexpression plasmids constructed in this study are all based on the empty plasmid pLKO.DCMV.TetO (15). pLKO-3 × Flag-sf-GFP plasmid overexpresses GFP, pLKO-3 × Flag-sf-UL43 qualitatively overexpresses pUL43, pLKO-3 × Flag-sf-UL24 qualitatively overexpresses pUL24, pLKO-3 × Flag-sf- US31 qualitatively overexpressed pUS31, pLKO-HA-UL43 qualitatively overexpressed pUL43, pLKO-TRBP-HA qualitatively overexpressed TRBP, cloning was constructed by enzyme digestion and ligation or in-fusion method, the primers required to construct the aforementioned plasmids are shown in Table 1.
Primers for Plasmids Construction
F, forward; R, reverse.
Antibodies and reagents
The antibodies used in this topic are as follows: rabbit polyclonal anti-Flag (Proteintech), Polyclonal anti-TRBP (Proteintech), Polyclonal anti-HA (Proteintech), Polyclonal anti-Dicer (Santa Cruz), Monoclonal anti-IE1 (a great gift from Jay Nelson, Oregon Health & Science University), Monoclonal anti-UL44 (Virusys), anti-pp28, anti-pp150 (a great gift from Thomas Shenk, Princeton University), Polyclonal anti-ULBP1 (Invitrogen), and antitubulin (Proteintech; 66031-1-Ig).
The reagents used in this subject are as follows: DNA ligase (TOYOBO), proteinase K (Beyotime Biotechnology), Anti-FLAG M2 Magnetic Beads (Sigma-Aldrich), Dynabeads™ Protein A (Invitrogen), Complete Protease Inhibitor Cocktail (l Roche), Phosphate-buffer saline (Hyclone); high-glycemic cell culture medium Dulbecco modified Eagle's medium (DMEM) and fetal bovine serum from Gibco; DNA polymerase PrimeStar, In-fusion ligase, and restriction enzymes EcoRI, SalI, XbaI, the I comes from Takara company; plasmid small-scale extraction kit, gel recovery kit, PCR product purification kit are all from Axygen company; high-purity plasmid small-scale medium-scale kit (Tiangen), MN large extraction kit, silver staining kit (Beyotime Biotechnology).
Cells and viruses
The cells used in this study are as follows: human embryonic lung fibroblasts (MRC5) (were obtained from the American Type Culture Collection (ATCC), available at (
Wild-type HCMV carries the whole genome of HCMV laboratory strain AD169; HCMV-GFP is a recombinant virus derived from it, but the GFP replaces the viral US4-US6 region HCMV under the control of a simian virus 40 early promoters (14,17). We used HCMV-GFP to perform our experiments throughout this research; GFP indicates infected cells.
BAC mutagenesis and recombinant viruses
In this project, two wild-type HCMV bacterial artificial chromosomes (BAC) are used and changed based on requirements. The two wild-type virus BACs are pBAC-AD/Cre-GFP and pBAC-TB40E-Mcherry. The pBAC-AD/Cre-GFP is changed from pBAC-AD/Cre, which carries the complete viral genome of the wild-type HCMV experimental virus strain. pBAC-AD/Cre-GFP is generated by replacing the pBAC-AD/Cre virus gene US4-6 with the green fluorescent protein gene (GFP), which is expressed under the control of the simian virus (SV40) early promoter. The clinical strain pBAC-TB40E-Mcherry is produced the same way as the experimental strain. We used pBAC-AD/Cre-GFP and pBAC-TB40E-Mcherry to create wild-type viruses as experimental controls.
For the construction of the recombinant mutant virus, the two-step Red Recombination System (Red Recombination System) is used as described previously (46), which can perform point mutation, deletion, and fragment insertion modifications to the BAC carrying the viral genome, and recombination of the modified BAC carrying the Kanna gene. The BAC is electroporated to the competent Escherichia coli GS1783. After induction with
We constructed four viruses based on the experimental virus strain pBAC-AD/Cre-GFP, named pBAC-AD-UL24-HA, pBAC-AD-UL43-Flag, and pBAC-AD-dd24-dd43 double deleted (knocked-out) virus, the other virus is in the clinical strain pBAC-TB40E-Mcherry. The recombinant BAC primers used in this experiment are shown in Table 2.
Primers for Bacterial Artificial Chromosomes Recombination
F, forward; R, reverse; BAC, bacterial artificial chromosomes.
Virus growth analysis
MRC5 cells were seeded in a 12-well plate. After 48 h, the cells were incubated with HCMV at a multiplicity of infection (MOI) of 0.1 or 1 in 300 μL of inoculum. The inoculum was removed and replaced with a fresh medium 2 h later. At different times postinfection, cell-free media from infected cultures were collected. The virus titers in the media were determined by a 50% tissue culture infective dose (TCID50) assay in human fibroblast cells. Reed–Muench method was used to calculate TCID50 (the 50% endpoint) data analysis.
Protein analysis
Protein interactions were analyzed by coimmunoprecipitation assay as previously described (39). HEK293T cells were transfected with the indicated plasmids and collected after 48 h. Collected cells were lysed in 1 mL lysis buffer (40 mM HEPES [pH 7.4], 1 mM EDTA, 300 mM NaCl, and 0.5% NP-40) supplemented with 250 U of Benzonase nuclease and PIC, incubated at 4°C for 1 h, and centrifuged at 13,200 g at 4°C for 15 min. The supernatant was collected and boiled in sodium dodecyl sulfate (SDS)-containing sample buffer. 40 μL of the supernatant saved as the input control, while the remaining part of the supernatant was incubated with either:
Then, the beads were washed five times with 1 mL lysis buffer. The immunoprecipitants were eluted by 150 ng/μL FLAG peptide (Sigma-Aldrich) or with Protein A beads at 4°C for 1 h. Then, the beads were washed five times with 1 mL lysis buffer. The immunoprecipitants were eluted by boiling in a heat block for 10 min/100°C.
Finally, the input and elution were analyzed by immunoblotting with the indicated antibody.
Proteins were analyzed by immunoblotting as described in the previous study (48). Cells were infected with HCMV at the MOI of 0.1 or 1, and cell lysates were collected in the SDS-containing protein sample buffer at different time points. Protein samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to polyvinylidene fluoride membrane, and incubated with primary and secondary antibodies. After incubation with antibodies, the proteins were visualized using Clarity Western ECL substrate (Bio-Rad).
Silver stain
Silver stain is performed as described previously (8,27,33). After the electrophoresis, the gel was put into about 100 mL fixative and shaken at room temperature on a shaker for 20 min/60–70 rpm. After washing processes and adding the silver-stained chromogenic solution, the silver staining solution was discarded. Silver staining stop solution (1 × ) was added and shaken at room temperature on a shaker for 10 min/60–70 rpm, Finally, discard the silver dye stop solution and wash with double-distilled water, and shake at room temperature on a shaker for 2–5 min/60–70 rpm (Fig. 1A, B).
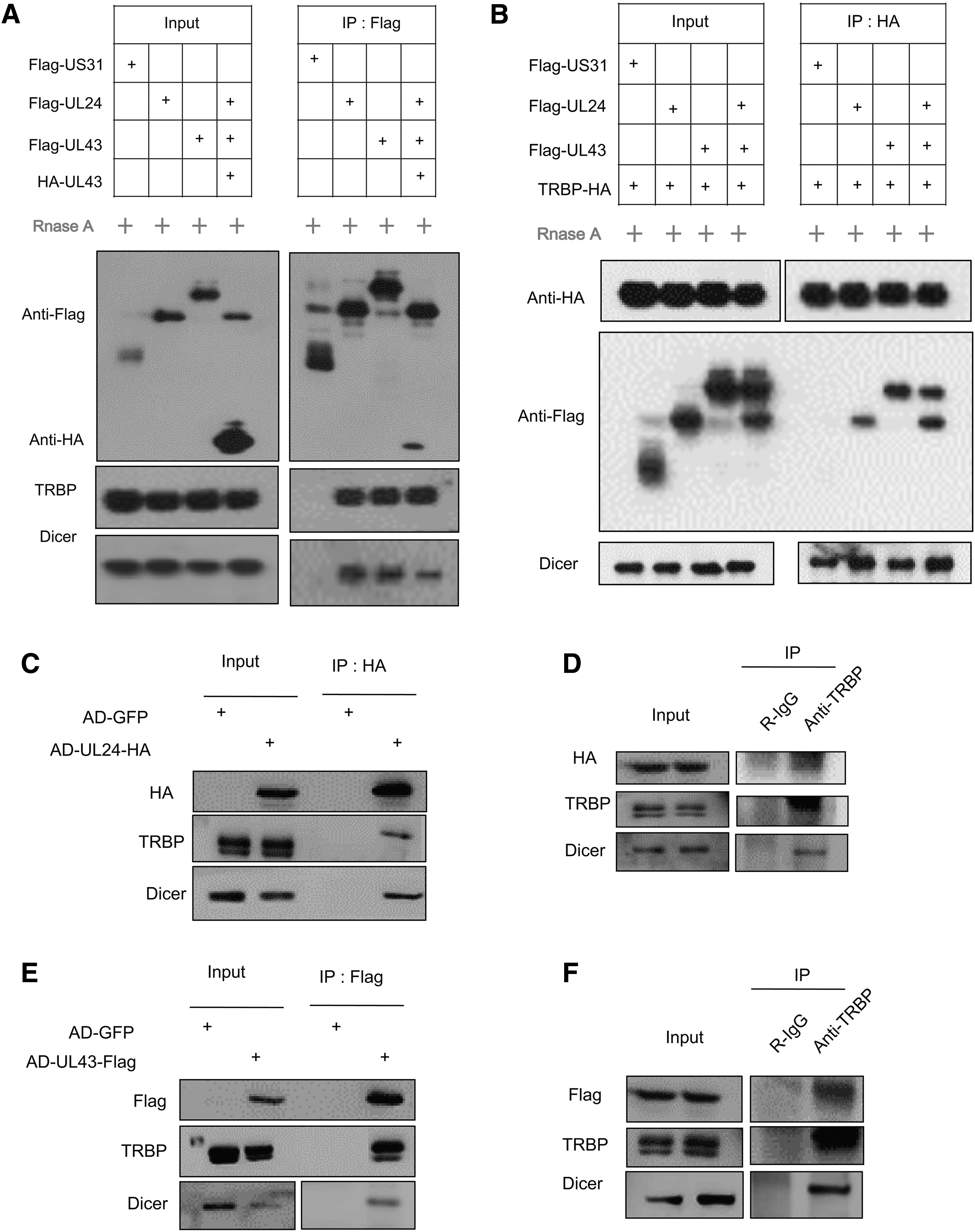
pUL24 and pUL43 interact with Dicer and TRBP specifically.
RNA and DNA analysis
As previously described, intracellular relative mRNA levels were determined by reverse transcription-quantitative PCR (RT-qPCR) (38). MRC5 cells were grown in six-well plates for 48 h and then infected with HCMV at an MOI of 1. Total RNA was extracted using the TRIzol reagent (Invitrogen), and the cDNA was synthesized with a PrimeScript real-time (RT) reagent kit (TaKaRa) and quantified using SYBR Premix Ex Taq (TaKaRa) by quantitative PCR (RT-qPCR) with specific primer pairs (Table 3) according to previously described protocol (21). All reactions were performed in two biological and two technical replicates. The amounts of viral transcripts were normalized to the Gapdh gene. The extracted total RNAs were sent to the company for RNA-seq according to the (SOLiD® Total RNA-Seq Kit) protocol. We used the Stem-loop method to design the reverse transcription primers of miR-UL59 and quantitative qPCR Primers.
Stem-Loop Primers for Reverse Transcription-Polymerase Chain Reaction
Intracellular DNA was measured by quantitative PCR as previously described (21). For cellular and viral DNA analysis, cells in 12-well plates were infected with HCMV at an MOI of 0.1 and collected in 200 μL 2 × digestion buffer (200 mM NaCl, 20 mM Tris-HCl [pH8.0], 50 mM EDTA [pH8.0], 1% SDS) at indicated time points. And then, 200 μL TE with 100 μg/mL proteinase K was added to lyse cells overnight at 55°C. DNA was extracted with phenol-chloroform. Supernatants were collected after centrifugation at 4°C, 400 rpm for 10 min, and treated with 100 μg/mL RNase A for an hour at 37°C. Supernatants were extracted again and precipitated with a half volume of ammonium acetate (7.5 M), 1 μL glycogen, and twice the volume of ethanol. DNA was resuspended in nuclease-free water. Viral or cellular DNA was quantified by qPCR with specific primers as shown in Table 4.
Primers for Quantitative Polymerase Chain Reaction
qF, qPCR forward; qR, qPCR reverse.
Statistical analysis in this study was performed using Student's t-test and ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001, and ns, nonsignificant).
Data analysis
GraphPad Prism v7.02 software and t-test were used to analyze data. P-value of < 0.05 = *, P-value of < 0.01 = **, P-value of < 0.001 = ***, were considered statistically significant, and ns = no-significant. All experiments were performed at least two or three times on separate days in two biological and two technical replicates, but only the representative data from a single experiment are shown.
Experimental Results and Analysis
MS analysis and immunoprecipitation reveal cellular proteins that interact with pUL43 or pUL24
UL24 and UL43 are both members of the US22 family of HCMV, and a study has reported that the viral proteins they encode have interactions (47). Therefore, we hypothesize whether UL24 and UL43 can cooperate and play some critical roles in the process of viral-cell infection similar to other members of the US22 family such as IRS1 and TRS1. We constructed Flag-tagged GFP, UL24, and UL43 plasmids and transfected them into HEK293T cells to explore this idea. After 48 h of transfection, the cells were collected and lysed with cell lysate, and then added to the magnetic beads with Flag antibody.
The products were finally eluted and separated by SDS-PAGE protein gel, and silver-stained. Figure 1A and B show that the UL43 and UL24 have precipitated many specific bands. After that, we expanded the number of cells for immunoprecipitation and Coomassie brilliant blue staining, and specific bands were collected for MS. Based on MS results, the particular protein that interacts with pUL43 was TRBP, and the particular proteins that interact with pUL24 were Dicer, SAMHD1, and USP24 (Supplementary Table S1 of MS data) but in this study, we mainly concentrated on viral gene interaction with cellular Dicer-TRBP pathway.
For further confirmation, we performed immunoprecipitation experiments to detect whether they can interact with cellular TRBP or Dicer by using specific antibodies. The HCMV virus gene US31 was used as a negative control in the experiment and a Flag-tagged US31 plasmid was constructed. We also built an HA-labeled UL43 plasmid to verify the interaction between pUL24 and pUL43. Because Dicer and TRBP are both essential members of the RISC, and the complex contains small nucleic acid molecules, to rule out that the interaction between proteins is mediated by nucleic acid rather than the direct action of protein, we treated the sample with RNase A enzyme. After 48 h of transfection, the cells were collected and lysed with cell lysate, and then added to magnetic beads labeled with Flag antibody for immunoprecipitation. As shown in Figure 1C, the results of eluted products processed by western blotting have shown that pUL24 and pUL43 can interact with each other, and interact with Dicer and TRBP.
Furthermore, to prove that these two cellular proteins can interact specifically with viral pUL24 or pUL43 proteins, we also used TRBP to test whether pUL24 and pUL43 can be co-precipitated. At first, we constructed a TRBP plasmid with HA-tag and then it transfected with US31, UL24, or UL43 into HEK293T cells. Cells were lysed 48 h after transfection and treated with the RNase A enzyme. Then incubate the protein A magnetic beads with HA antibody to form a HA antibody-labeled magnetic bead, add the processed cell supernatant to the magnetic beads, undergo immunoprecipitation, and finally separate the protein on the magnetic beads with the lysis solution, and the product is eluted. As shown in Figure 1D, western blotting results showed that TRBP could co-precipitate pUL24 and pUL43. Altogether, pUL24 or pUL43 can interact with Dicer and TRBP through specific interactions between proteins and are not mediated by nucleic acids.
To further confirm that in the case of HCMV infecting cells, whether these two viral proteins can also interact with Dicer and TRBP. We first carried out viral modification in the BAC containing the entire genome of HCMV, respectively, adding the HA-tag to the C-terminal of the UL24 virus gene, and adding the Flag-tag to the C-terminal of UL43 to construct two new BACs, namely pBAC-AD-UL24-HA and pBAC-AD-UL43-Flag. BAC was electroporated to MRC5 cells to infect the cells with these tagged viruses. After 48 h of infection, the cells are collected and tested in related experiments. As shown in Figure 1E, we infected MRC5 cells with the HCMV AD-GFP, and AD-UL24-HA, collected cell samples 48 h after infection, and then used HA antibody-coated magnetic beads for immunoprecipitation.
The western blotting results showed that in AD-UL24-HA-infected cells, UL24-HA could co-precipitate TRBP, and Dicer compared with AD-GFP-infected cells (control). Also, in Figure 1F, we used the AD-UL24-HA virus to infect cells, and Protein A magnetic beads were used and incubated with IgG (control) and TRBP antibodies. The supernatant was added to the processed magnetic beads. The results of co-immunoprecipitation and immunoblotting showed that the antibody incubated with IgG could not co-precipitate pUL24, but the antibody incubated with TRBP antibody could co-precipitate pUL24 and Dicer. These two experimental results show that pUL24 expressed by the virus can interact with the endogenous TRBP and Dicer of the cell.
Similarly, to identify whether pUL43 can interact with endogenous TRBP and Dicer, such as pUL24 in the infected cells. As shown in Figure 1G, MRC5 cells were infected with the viruses AD-GFP and AD-UL43-Flag. Cell samples were collected 48 h postinfection (hpi), and then used Flag antibody-coated magnetic beads for immunoprecipitation. The results of western blotting showed that, in the cells infected with AD-UL43-Flag, UL43-Flag was able to co-precipitate TRBP and Dicer compared with the cells infected with AD-GFP.
Furthermore, in Figure 1H, we infected cells with the AD-UL43-Flag virus and the Protein A magnetic beads were incubated with IgG or TRBP antibodies, and the supernatant of the infected cell lysate was added to the processed magnetic beads. The blotting results showed that the antibody incubated with IgG could not co-precipitate pUL43, but the antibody incubated with TRBP could co-precipitate pUL43 and Dicer. These two experimental results show that pUL43 expressed by the virus can interact with the endogenous TRBP and Dicer in the cell.
In summary, the experimental data indicate that in HCMV infection, the viral UL24 or UL43 protein can interact with the endogenous TRBP and Dicer.
Double knockout of UL24 and UL43 does not affect the virus growth, entry, and infectivity
According to the aforementioned experimental results, we found that pUL24 or pUL43 can interact with TRBP and Dicer, because TRBP and Dicer are two essential components of the RISC, which can regulate the production of miRNAs and thus regulate the expression of some genes; therefore, we speculate that whether these two viral proteins can work together to participate in the regulation of the production of miRNAs through the interaction with TRBP and Dicer that may ultimately play some essential biological functions. Previous studies have shown that UL24 and UL43 are nonessential factors for the growth of the HCMV and any deletion of them will not affect virus replication (32,35). However, there is no related report of double deletion; we knocked out UL24 and UL43 in the wild-type virus genome at the same time to detect whether the replication of the virus will be affected. The red recombination system was applied to perform viral gene knockout, as reported in the previous study (46).
We first electrotransformed the IsceI-KanS containing the UL24 C-terminal homologous fragment into E. coli GS1783 containing wild-type pBAC-AD/Cre-GFP for homologous recombination, then induced the enzyme to excise the IsceI-KanS, and the BAC deleted UL24 was generated, named pBAC-AD-d24. Besides, using the same method as UL24 knockout, the IsceI-KanS containing the C-terminal homologous fragment of UL43 was electroporated into E. coli GS1783 having pBAC-AD-dUL24 for homologous recombination, and finally BAC double deleted UL24 and UL43 were got and named pBAC-AD-dd24-dd43.
We finally got the double deletion virus AD-GFP-dd24-dd43 by electroporation of BAC into MRC5 cells. We use the wild-type virus AD-GFP (or AD-WT) and the double deletion virus AD-GFP-dd24-dd43 (Or AD-Mut) with a low MOI of 0.1 and a high MOI of 1. The MRC5 cells were incubated with the virus for 2 h, and then removed from the supernatant and replaced with a fresh medium. Finally, the cell supernatant was collected at a specific time postinfection and used to test the virus titer. As shown in Figure 2A, B, we found that the replication ability of the UL24/UL43 double-deleted virus was close to that of the wild-type virus.
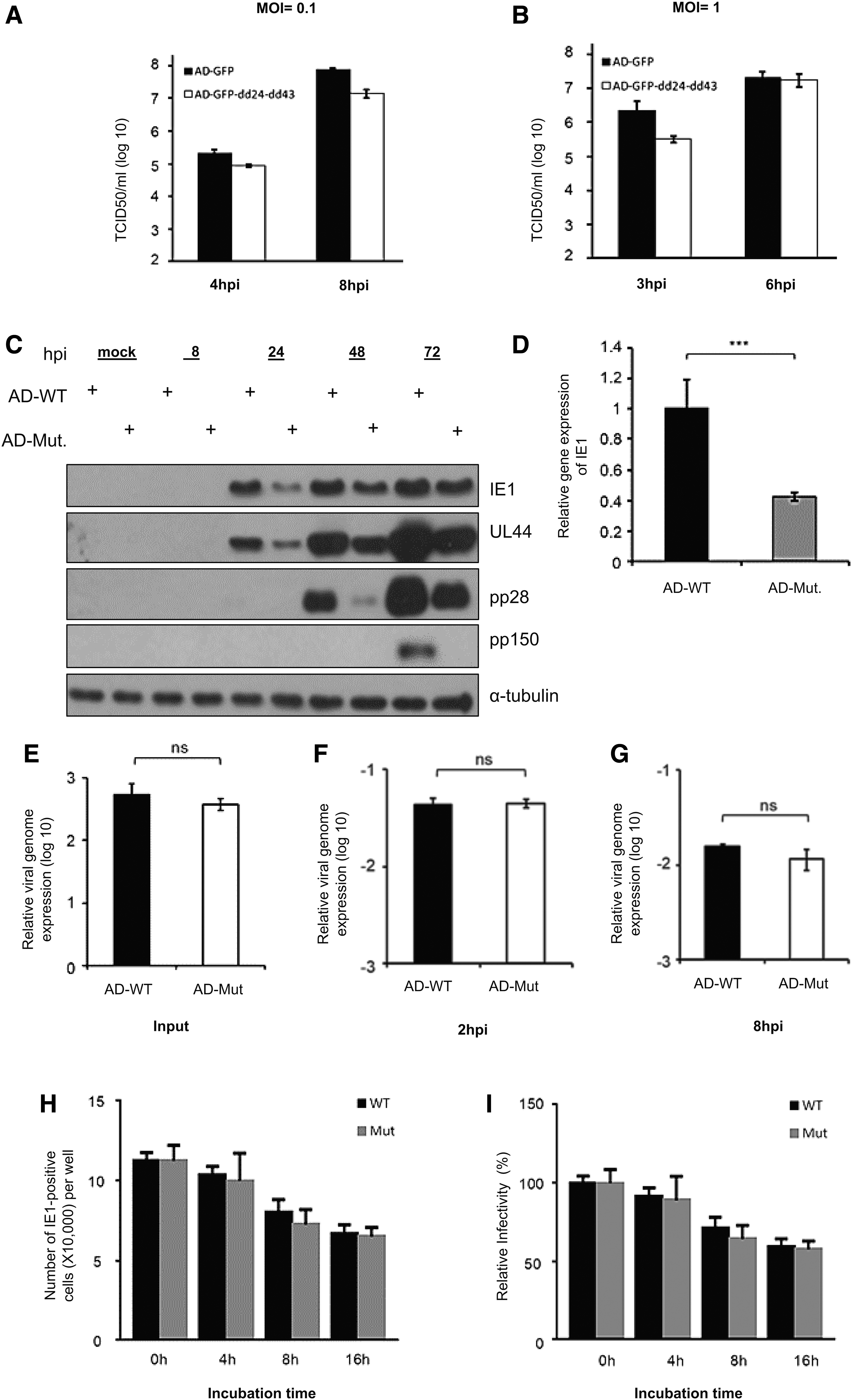
Double knockout of CMV UL24 and UL43 genes does not affect the HCMV growth, entry, and infectivity in experimental strain (AD169).
Altogether, these results indicate that the double-knockout and single-knockout phenotypes of these two genes are the same and do not affect the virus growth and show that these two viral genes are indeed nonessential genes for the development of the HCMV virus.
We also tested the HCMV early gene IE1, the early gene UL44, and the late gene UL99 (pp28) and UL32 (pp150) expression. We infected MRC5 cells with MOI of 1 and cells collected for western blotting at a specific time point. In Figure 2C, we found that the expression of viral protein IE1 and even other tested genes were downregulated in infected cells with a double-deletion virus. We further tried the transcription of early viral genes, infected MRC5 cells with an MOI of 1, collected RNA from the cells 8 h after infection, and performed RT-qPCR. As shown in Figure 2D, the mRNA level of IE1 was also downregulated in the early stage of the disease. These data suggest that UL24 and UL43 may affect the early stage of virus replication.
Next, we had to explore whether the double knockout of these two viral genes affects virus entry into cells or virus viability. To test that, MRC5 cells were infected with the wild-type or deleted viruses with an MOI of 1 for 2 h. The fresh medium was replaced and the cells were collected at the indicated time points and data analyzed by quantitative PCR. The input was the virus stock solution. In Figure 2E–G, we find almost no difference between the wild-type and the deleted virus genome. This shows that the double knockout of UL24 and UL43 (AD-Mut) does not affect the ability of the virus to enter the cell.
Furthermore, we performed virus viability tests on wild-type (AD-WT) and mutant viruses (AD-Mut). We treated the wild-type and deleted viruses with the 37°C for 4, 8, and 16 h. Then, the untreated and processed viruses were used to infect MRC5 cells at an MOI of 1. After 24 h of infection, we collected and incubated the cells with the virus IE1 antibody and the corresponding secondary antibody, and the IE1 positive cells were counted under the fluorescent microscope. As shown in Figure 2H, the number of IE1 positive cells gradually decreases with 37°C treatment time extension.
Still, interestingly, we found that the number of IE1 positive cells in the mutated virus was almost similar to that of the wild-type virus regardless of whether it was processed with 37°C. We also checked the relative infectivity of the virion, as shown in Figure 2I. The results were consistent with Figure 2H. It was clear that treatment with 37°C has reduced the virus infectivity percentage, but there was no difference between the wild type and the deletion type in the reduction levels. In summary, these results indicate that the double knockout of UL24 and UL43 genes did not affect the ability of the virus entry and the virus infectivity.
Double knockout of UL24 and UL43 in clinical virus strains also does not affect virus replication, entry, and virus infectivity
In addition to the HCMV experimental virus strain (HCMV AD-GFP), we also want to know whether the double knockout of UL24 and UL43 genes in the HCMV clinical virus strain (TB40E-Mcherry = TB-WT) can similarly affect the virus replication and its cellular entry. At first, we generated UL24/UL43 double deletion virus named TB40E-Mcherry-dd24-dd43 (TB-Mut). Then to analyze the growth ability of this clinical mutant virus (TB-Mut), we infected the MRC5 cells with the wild type and the deleted virus for 2 h at an MOI of 0.1 or 1.
The medium was replaced with a fresh one. Finally, the cell supernatant was collected at a specific time point postinfection. It was used to infect MRC5 cells and the virus titer was measured by tissue culture infectious dose 50% (TCID50) assay. As shown in Figure 3A, B, the growth ability of the Mut virus was almost identical to that of the wild-type virus. These results indicate that the double knockout of UL24 and UL43 in clinical virus strain, similar to the experimental virus strain, does not affect the replication of the virus, thus confirming that the UL24 and UL43 are nonessential genes for clinical virus strains too.
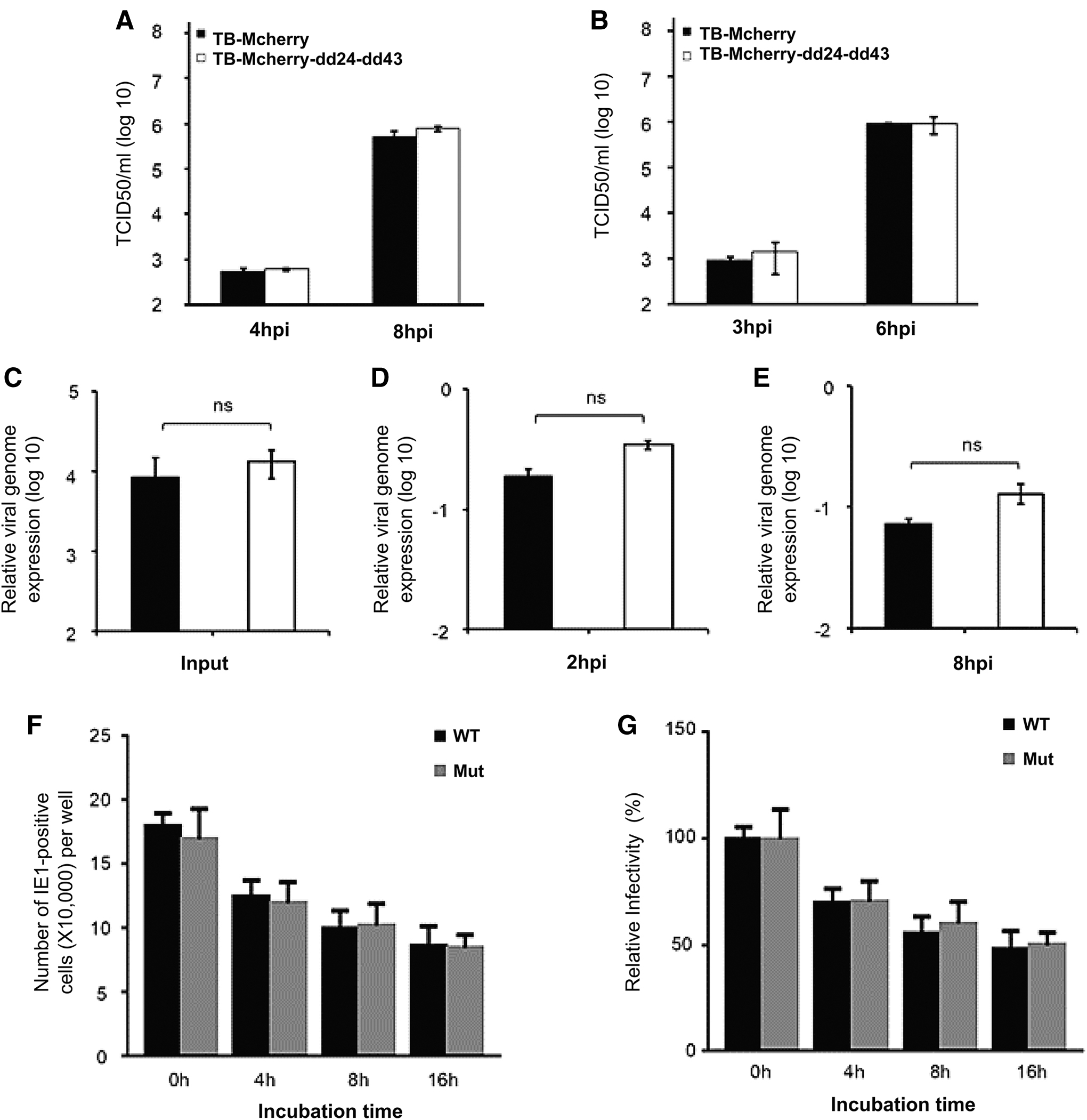
Double knockout of CMV UL24 and UL43 genes does not affect the HCMV growth, entry, and infectivity in the clinical strain (TB40E).
Next, we checked whether the knockout of these two viral genes affects virus entry into cells. The MRC5 cells were infected with wild-type and deleted clinical viruses at an MOI of 1 for 2 h, then the supernatant was removed and replaced with fresh ones. The cells were collected at the specified time point postinfection, and the viral genome (viral DNA) was extracted and analyzed by qPCR. The virus stock solution was used as input. As shown in Figure 3C–E, there was almost no difference in the amount of wild-type virus genome and deleted virus genome, which shows that the double knockout of UL24 and UL43 does not affect the ability of clinical strains to enter cells as well.
Furthermore, we tested the virion stability of wild-type and deleted viruses in the clinical strain. The wild-type virus and the deleted virus were treated at 37°C for 4, 8, and 16 h, then the MRC5 cells were infected with the untreated and the treated virus separately with an MOI of 1. At 24 hpi, the virus IE1gene was incubated with primary and the corresponding fluorescent secondary antibody. Finally, the IE1 positive cells were calculated under the fluorescence microscope.
As shown in Figure 3F, the number of IE1 positive cells gradually decreases with the 37°C treatment time extension. Still, there was no significant difference between the number of IE1 positive cells with both the wild type and mutant virus. Also, to check the relative virus infectivity. As shown in Figure 3G–F, the treatment with 37°C has reduced the infectivity of the virus without a significant difference between the wild type and the deletion type. In summary, these results indicate that HCMV clinical viruses were consistent with experimental strain in that the double knockout of UL24 and UL43 does not affect the viral entry and infectivity.
Double knockout of UL24 and UL43 does not affect the expression of some common HCMV miRNAs related to immune escape
miRNAs are noncoding RNAs with a size of about 22 nucleotides and can participate in the regulation of many signaling pathways in cells. Initially, the Pfeffer study group identified nine HCMV-encoded miRNAs (10). Later on, 26 miRNAs were discovered through a series of studies, and they are all produced by RISC processing (43). These miRNAs perform various functions after the virus infects cells, such as regulating the cell cycle, the expression of a specific host or viral genes, viral DNA synthesis, the generation of virus assembly centers, and the production of immune regulation-related inflammatory factors (7,19,23 –25,28,29).
According to our results, the UL24 and UL43 can interact with Dicer and TRBP, and their knockout (AD-Mut) does not affect the virus growth and the virus entry. Also, based on previous studies that many of the miRNAs encoded by HCMV are involved in regulating the immune response and helping the virus escape (5). And most cells make many different miRNAs at different times during the cell cycle, and viruses also encode miRNAs, especially appreciated in the case of HCMV. Therefore, we speculate that pUL24 and pUL43 may cooperate to regulate some viral miRNAs.
They may regulate some signaling pathways to escape the immune system's recognition of the virus. There are currently four known HCMV miRNAs that are mainly involved in immune regulation, which are miR-UL112-3p, miR-US5-1, miR-UL148D, and miR-US25-1-5p; therefore, we speculated whether the knockout of UL43 and UL24 genes affects the expression of these miRNAs. To perform that, MRC5 cells were infected with wild-type viruses at an MOI of 1, and the cells were collected to detect the expression of these miRNAs.
We extract RNA from cells and use the specific reverse transcription primers of these miRNAs, as shown in Table 3, to obtain their cDNA by the Stem-loop RT PCR method, and finally qPCR was used for quantitative detection. As shown in Figure 4A–D, the expression of these miRNAs was very high at 48 hpi. Therefore, we have used the 48 hpi as the time point to compare the differences between these miRNAs in wild-type and mutated virus-infected cells. As shown in Figure 4E–H, the expression levels of these four miRNAs under different virus infections have no significant difference. This result indicates that the double knockout of UL24 and UL43 does not affect the production of these miRNAs.
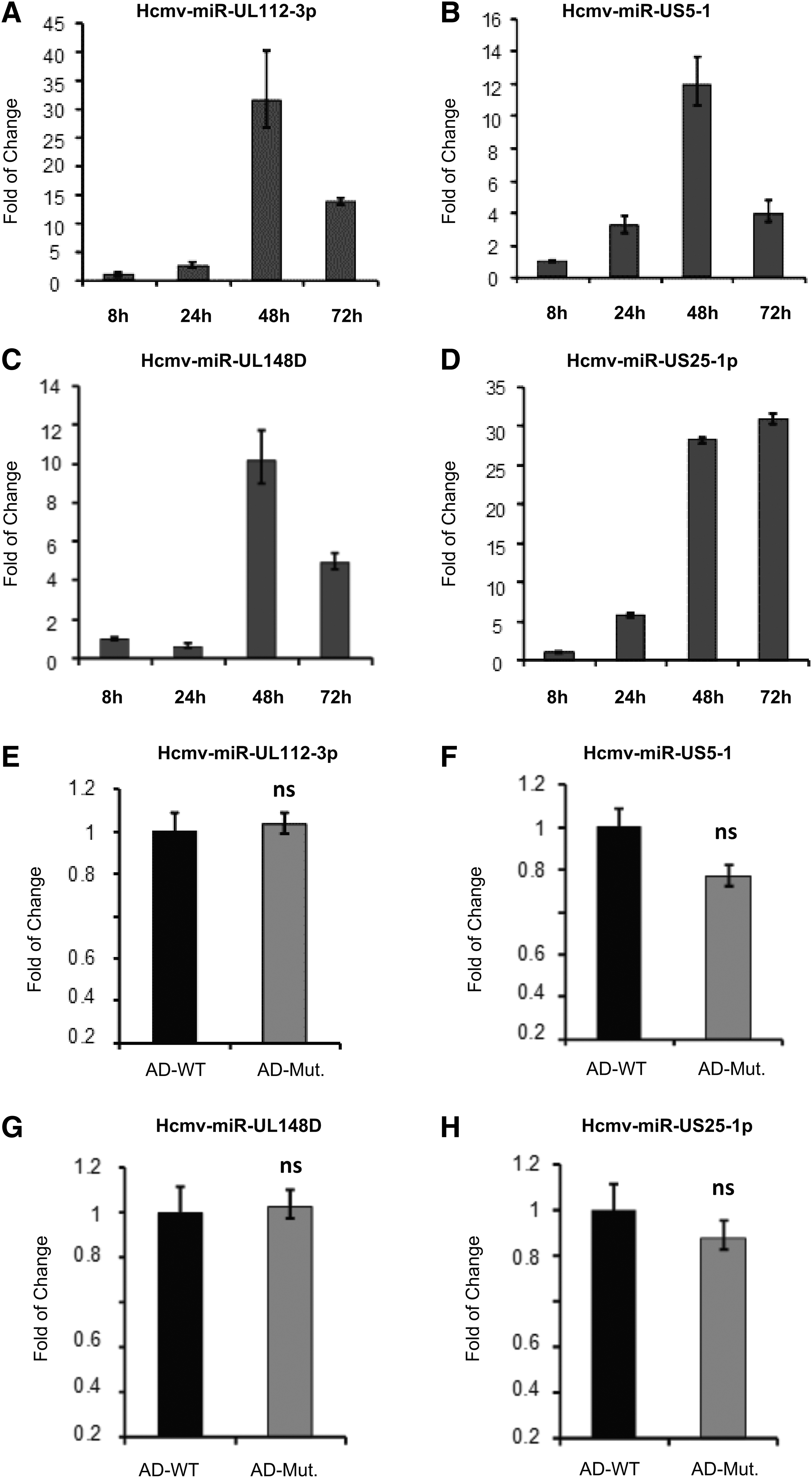
Double knockout of CMV UL24 and UL43 genes does not affect the expression of HCMV miRNAs.
RNA-seq demonstrates that the double knockout of UL24 and UL43 regulates the expression of miR-UL59 and its downstream target gene ULBP1
To further analyze the potential role of UL24 and UL43 in HCMV virus infection of host cell miRNAs biogenesis, and to find the target miRNAs regulated by these viral genes, the extracted total RNA was performed for RNA-seq. The MRC5 cells were infected with AD-WT and AD-Mut, respectively, with an MOI of 1. After 48 hpi, cell collection and RNA extraction with 1 mL Trizol were performed and sent for RNA-seq. Based on the results of RNA-seq, we found that the expression of miR-UL59 was different. For further verification, we infected the MRC5 cells with AD-WT and AD-Mut again at an MOI of 1. Each virus was treated with two identical treatments. After 48 hpi, we used Trizol to collect cells and extract RNA and the Stem-loop method to design miR-UL59 reverse transcription primers and quantitative qPCR primers (Table 5) (6,41). Experimental results showed that the double knockout of UL24 and UL43 resulted in the downregulation of miR-UL59 (Fig. 5A).

Double knockout of UL24 and UL43 genes regulates the transcript level of miR-UL59 and ULBP1.
Primers for Quantitative Polymerase Chain Reaction
There are limited research reports on miR-UL59, and it is described that the downstream target gene of miR-UL59 is ULBP1 (UL16-binding protein 1) (11). ULBP1 is one of the ligands of the natural killer group 2, member D (NKG2D), which is an activating receptor expressed on immune effector cells to recognize different MHC-I-related ligands, including MIC and ULBP proteins. Infection or stress response can induce the expression of the NKG2D ligand, leading to the activation of effector cells and ultimately killing the ligand-related target cells (13,37,40,49). Besides, it has previously been reported that the membrane glycoprotein UL16 of HCMV can bind to three NKG2D ligands, namely MICB, ULBP1, and ULBP2. And UL16 is also very important for the immune escape of HCMV (13).
To check the impact of these two genes on the ULBP1 expression level, we similarly infected the MRC5 cells with AD-WT and AD-Mut at an MOI of 1. At 48 hpi, we applied Trizol to cells, and cells were ultimately collected and the ULBP1 mRNA levels were measured by quantitative qPCR using specific primers as shown in Table 5 (6,41). Experimental results showed that the double knockout of UL24 and UL43 has resulted in the upregulation of ULBP1 mRNA levels (Fig. 5B).
As in the experimental virus strain, the double knockout of UL24 and UL43 would inhibit the expression of miR-UL59, and upregulate the expression of its target gene ULBP1, we checked whether this phenotype is in clinical virus strains unanimous. Therefore, we spread MRC5 cells in a 12-well plate and then infected the cells with wild-type and mutant deletion viruses with an MOI of 1. After 48 h of infection, the cells were harvested with Trizol, and RNA was extracted. Next, quantitative PCR primers were used to detect the expression of miR-UL59. As shown in Figure 5C, we identified that the expression of miR-UL59 was downregulated after the virus-infected cells.
As shown in Figure 5D, we also found that the target gene of miR-UL59, ULBP1, was downregulated. The mRNA level was higher in cells infected with the deletion virus than in cells infected with the wild-type virus, and the results were consistent with the results of the experimental virus strains, indicating that knocking out UL24 and UL43 in clinical strains can also inhibit the expression of miR-UL59 and upregulate the mRNA of ULBP1 Level. As one of the ligands of NKG2D, ULBP1 is essential for cellular immunity. Therefore, we speculate that although UL24 and UL43 of the HCMV virus do not affect virus replication, they may regulate the expression of some miRNAs in the cell and thus participate in immune regulation to promote the immune escape of the virus.
To confirm these results, we further infected the MRC5 cells with AD-WT and AD-Mut at an MOI of 1. After 48 hpi, cells were collected and the ULBP1 protein levels were normalized against that of α-tubulin and the protein levels were measured by immunoblotting (Fig. 5E).
Altogether, our data suggest that UL24 and UL43 may modulate the expression of ULBP1 through the regulation of the miR-UL59 expression level.
Discussion
UL24 and UL43 are the late viral proteins that make up HCMV virus particles. Previous studies have shown that the viral genes that encode these two proteins are unnecessary for virus replication, and their deletion does not affect virus growth (35). A recent study proved that the pUL24 and pUL43 proteins encoded by these two viral genes could interact (47). It is reported in the literature that some genes in the US22 family can play a synergistic effect during viral infection. For example, UL28 and UL29 can activate the expression of early genes, and TRS1 and IRS1 can regulate gene transcription to promote viral replication.
Moreover, even if some genes expressing interlayer protein are not necessary for virus growth, they can inhibit the expression of antiviral genes, thereby inhibiting the immune response and helping the virus escape, such as UL82, and UL83. Therefore, we speculate whether, in HCMV infection, the UL24 and UL43 can play a synergistic role in regulating specific intracellular reactions. First, we constructed the plasmids of these two viral genes and transfected them into cells for expression. Through co-MS and immunoprecipitation analysis, we found that the Dicer and TRBP proteins can interact with the viral UL24 and UL43 proteins in HEK293T cells.
To prove this interaction, we added the HA-tag to the C-terminus of UL24 or the Flag tag to the C-terminus of UL43 in the HCMV virus genome. Then these two viruses were used to infect MRC5 cells separately. Based on the immunoprecipitation assay, the data showed that pUL24 or pUL43 could also interact with Dicer and TRBP. These results indicate that endogenous interactions between these viral and cellular proteins also exist in the HCMV infected cells.
Furthermore, to check whether they have a synergistic effect, we knocked out UL24 and UL43 genes together (double deletion) to observe the phenotype. At first, through viral genome modification, these two viral genes were knocked out to construct a mutant virus. The results of virus growth analysis showed that the knockout of these late viral genes did not affect the virus growth, entry, and infectivity. In addition to the experimental HCMV strains, we also obtained the same results with the clinical HCMV strains.
Besides, studies have reported that Dicer and TRBP are cellular proteins associated with processing, microRNA biogenesis, and composing RISC (50). Therefore, we checked whether the encoded pUL24 and pUL43 could regulate the production of some miRNAs through the interaction with Dicer and TRBP, thereby regulating some cellular immune responses to facilitate the virus escape. Since studies have been reported that HCMV tegument proteins can inhibit the expression of some cellular proteins or surface antigens from escaping the immune system, for example, a study has been reported that UL82, the interlayer protein of HCMV, can inhibit STING-mediated signaling pathways to avoid antiviral immune responses (18).
Also, the protein pp65 encoded by the viral gene UL83 can inhibit the expression of antiviral genes (4). This raises the question of whether these two genes may involve in regulating the antiviral immune response. Based on the results of previous experiments, we expected that UL24 and UL43 might regulate the production of downstream miRNAs. About 26 miRNAs encoded by HCMV, of which the most studied antiviral related are miR-UL112-3p, miR-US5-1, miR-UL148D, and miR-US25-1-5p. We identified that the expression of these four miRNAs was not affected in cells infected with the mutant-type virus compared with wild-type one.
To find the target HCMV miRNAs of these two viral genes, we performed RNA-seq and identified that UL24 and UL43 regulate the expression of miR-UL59. Experimental data showed that compared with wild-type virus, the double deletion virus type has resulted in the downregulation of miR-UL59 expression, a newly discovered HCMV miRNA. A study has reported that the target gene of miR-UL59 is ULBP1. ULBP1 is one of the ligands of NKG2D, an activating receptor expressed on immune effector cells that can recognize different MHC I-related ligands, including MIC and ULBP proteins. Infection or stress response can induce the expression of the NKG2D ligand, leading to the activation of effector cells and ultimately killing the ligand-related target cells. Therefore, we checked the expression of ULBP1 in infected cells. As a result, in both experimental and clinical virus strains, ULBP1 mRNA and protein levels in the double-deleted HCMV infected cells were apparently higher than that of wild-type virus-infected cells.
In summary, our current research data show that the double knockout of UL24 and UL43 does not affect the growth, entry, and relative infectivity of the virus, but leads to the downregulation of HCMV miR-UL59, and upregulation of its corresponding target ULBP1 level. In another manner, the UL24 and UL43 may reduce the expression of ULBP1 ligands on the cell surface reducing the cytotoxicity mediated by infected cells to NK cells, thereby avoiding NK cell recognition and helping the immune escape of infected cells. However, further experiments will be necessary to explore whether UL24 and UL43 are related to NK cell-mediated cytotoxicity in the future.
Footnotes
Acknowledgments
We thank all the Herpesvirus and Molecular Virology Research Unit members for helpful discussion and Roger Everett (University of Glasgow) for the pLKO-based lentiviral expression system.
Authors' Contribution
Formal analysis (lead), investigation (supporting), conceptualization (supporting), writing—original draft (lead), project administration (lead), and software (lead) by S.S. Investigation (lead), conceptualization (lead), formal analysis (lead), software (supporting), and methodology (lead) by H.H. Visualization, review, and editing (equal) by H.N. Software (supporting), review, and editing (equal) by P.J. Visualization, review, and editing (equal) by N.S. Writing—review and editing (equal) by D.O. Conceptualization (lead), validation(lead), and supervision by Z.Q. The authors have read and agreed to the published version of the article. S.S. and H.H. contributed to this study equally, and are first co-authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Ministry of Science and Technology of China (2016YFA0502101), National Natural Science Foundation of China (Grants 81371826 and 81572002) to Z.Q., the Chinese Academy of Sciences “100 Talents” program to Z.Q., CAS-TWAS Fellow. The funders had no role in study design, data collection, analysis, decision to publish, or article submission.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.