
Abstract
COVID-19 is a globally infectious viral epidemic of great public health concern caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Angiotensin-converting enzyme 2 (ACE2) plays its role as the receptor for SARS-CoV-2 through binding with S protein and the binding results in ACE2 expression decrease. The change of ACE2 is supposed to elicit a series of cellular and molecular events. Other than as the receptor, ACE2's roles on infection by regulating other molecules need to be further studied during SARS-CoV-2 infection. In the present study, we established the ACE2 knockdown model using Vero E6 cells to study how ACE2 influenced the downstream signaling molecules. Analysis of transcriptome sequencing discovered that ACE2 alteration per se caused the alteration of immune factors, including some related to the viral infection-related signaling pathways. We found that ACE2 silencing induced the reduced interferon-induced transmembrane protein 3 (IFITM3) expression. Overexpression of IFITM3 promoted the SARS-CoV-2 pseudovirus infection of Vero E6 cells lacking the ACE2. It indicates that ACE2 can affect IFITM3 expression and function to affect the SARS-CoV-2 infection. Our results reveal possible mechanisms influencing SARS-CoV-2 infectivity and contribute to explaining the rapid spread and pathogenesis especially in the case of ACE2 low expression.
Introduction
By the end of February 2022, over 400 million confirmed cases of COVID-19 have been reported by the World Health Organization. Like SARS, COVID-19 is caused by a coronavirus, termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The majority of infected people suffer from mild or moderate respiratory symptoms. Some of them ultimately exhibit severe symptoms, indicative of acute respiratory distress syndrome associated with cytokine storm syndrome and immune dysfunction (Hirano and Murakami, 2020).
Angiotensin converting enzyme 2 (ACE2), a type I transmembrane protein on the surface of many cell types as the receptor for SARS-CoV, is used as the entry receptor by SARS-CoV-2 as well (Hoffmann et al, 2020; Wrapp et al, 2020). The ACE2 expression level is critically important to determine the entry and pathogenesis of SARS-CoV-2 (Hoffmann et al, 2020; Ou et al, 2020). Conversely, coronavirus infection can in turn affect the ACE2 expression. For instance, SARS-CoV infection causes ACE2 protein decrease in Vero E6 cells (Glowacka et al, 2010). Aligned with the results of SARS-CoV, SARS-CoV-2 infection of A549 cells also resulted in a decline of ACE2 expression (Blanco-Melo et al, 2020). Sui et al have verified that the S protein of SARS-CoV-2 directly inhibited ACE2 expression in most of the lung bronchoalveolar lavage specimens from naive rhesus macaques (Sui et al, 2021).
A decline of ACE2 expression during infection suggests a transcriptional or posttranscriptional regulation such as degradation or shedding (Saponaro et al, 2020). By now, the downstream factors sequentially affected by ACE2 alteration are rarely investigated. Therefore, it is necessary and urgent to elucidate the potential role of ACE2 other than as the entry receptor during the SARS-CoV-2 infection.
In the present study, we explored ACE2 influence on downstream signaling molecules utilizing ACE2 silencing system. Herein, interferon (IFN)-induced transmembrane protein 3 (IFITM3) was found to be significantly decreased by ACE2 knockdown. IFITM3, a 15 kDa protein belonging to the group of IFN-stimulated genes (ISGs), is an innate immune responder to viral infections by altering membrane rigidity and curvature to inhibit the fusion of virus and cell membranes (Li et al, 2013). It inhibits human coronaviruses, including SARS, MERS-CoV. as well as SARS-CoV-2 (Shi et al, 2021). Our study found that ACE2 and IFITM3 acted together to inhibit SARS-CoV-2 pseudovirus infection and ACE2 silencing converted the inhibitory effect of IFITM3 to the promotive effect. ACE2 affected both IFITM3 expression and function. The multifaceted roles of ACE2 raise implications for explaining the reasons for the rapid spread of this pandemic viral pathogen.
Materials and Methods
Small interfering RNA (siRNA)-mediated silencing of ACE2
Twenty-four hours after seeding, Vero E6 cells were transfected by 25 nM ACE2 specific siRNApool (siACE2) or negative control siRNA (siNC) with Lipofectamine RNAiMAX reagent (13778500; Invitrogen™) according to the manufacturer's instructions. All siRNAs were designed and synthesized by Tsingke Biotechnology (Nanjing, China) as shown in Table 1.
siACE2 Sequences Used in the Study
ACE2, angiotensin-converting enzyme 2.
Establishment of stable cell lines
Lentiviral particles were produced by cotransfection of HEK293T cells with the lentiviral vector and the packaging plasmids psPAX2 and pMD2.G at a ratio of 4:2:1, using the Xfect Transfection Reagent (631318; Takara). After 48 h, the lentiviral supernatants were harvested, centrifugated at 300 g for 10 min, and filtered by 0.45 μm filter to collect the supernatant and then aliquoted and storied at −80°C. Vero E6 cells were transduced with pLKO.1-shACE2-puro and pLKO.1-shNC-puro lentivirus and maintained in culture medium supplemented with 5 μg/mL puromycin for about 1 week.
Production and luciferase assay of pseudovirus
SARS-CoV-2 pseudovirus was generated as previously described (Ou et al, 2020). The VSVG encoding plasmid (pVSVG) was obtained from Addgene (Cambridge, MA). pVSVG expressing SARS-CoV-2 S protein was constructed using the packaging plasmid (pVSV-ΔG-Luciferase), in which the glycoprotein (G) was deleted. Lenti-X™ 293T cells were grown to 70% confluency before transfection with pVSV-ΔG-Luciferase (or pVSVG), pWPXL, and pSPAX2 using Xfect Transfection Reagent (631318; TaKaRa) according to the manufacturer's instructions. The supernatant was harvested 48 h later, centrifugated at 300 g for 10 min and filtered by 0.45 μm filter to collect the pseudoviruses, and then aliquoted and storied at −80°C.
Cells were infected with pseudoviruses at 37°C for 24 h and replaced with the fresh culture medium. After 24 h, the luciferase activity of infected cells was measured using the Bright-Luciferase Reporter Assay System (E2650; Promega). Relative luminescence unit of luciferase activity was detected on a Varioskan™ LUX multimode microplate reader (Thermo Fisher Scientific, Vantaa, Finland).
Transient transfection
Vero cells were seeded onto the 12-well plate before transfection, and vector or pcDNA3.4-IFITM3 was transiently transfected using Xfect Transfection Reagent (631318; Takara) for 48 h to overexpress IFITM3 proteins.
RNA extraction and quantitative polymerase chain reaction
Total RNA was extracted using the TaKaRa MiniBEST Universal RNA Extraction Kit (9767; TaKaRa) and reverse-transcribed using PrimeScript RT Master Mix (RR047A; TaKaRa) for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) according to the manufacturer's instruction. Quantitative real-time PCR (qPCR) was performed on CFX Manager (Bio-Rad, Hercules, CA) using SYBR Green PCR Master Mix (RR820A; TaKaRa), with the use of β-actin as an internal control. The sequences of real-time PCR primer pairs were shown in Table 2.
Oligonucleotides Used in the Study
qPCR, quantitative polymerase chain reaction.
Flow cytometry analysis
5 × 105 cells were suspended in FACS buffer (PBS supplemented with 2% FBS), centrifuged at 300 g for 5 min, and resuspended in FACS buffer. Cells were treated with Fc blocking on ice for 10 min, and then incubated with primary antibody rabbit-anti-ACE2 (21115-1-A; ProteinTech) at the indicated dilution and cells on ice for 30 min with intermittent gentle agitation. Cells were washed twice with FACS buffer and incubated with the secondary antibody AlexaFluor647 goat anti-rabbit IgG (No. A32733; Fisher) for 30 min on ice. Cells were washed twice with FACS buffer, centrifuged, and resuspended in FACS buffer. After live/dead cell staining containing SytoxBlue (No. S34857; Thermo Fisher) at 1:1,000 dilution for 5 min, cells were washed and filtered into new FACS tubes. Flow cytometry was performed on a BD FACSAria III instrument, and all the data were analyzed by FlowJo v10 software.
Western blot
Western blot assays were performed as described previously (Ren et al, 2016). Primary antibodies used in this study were as followed: rabbit anti-ACE2 (ab15348; Abcam), rabbit anti-IFITM3 (11714-1-AP; Proteintech), and mouse anti-β-Tubulin (T0023; Affinity). The secondary antibodies were as follows: Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (No. 115-035-003; Immuno Research) and Goat anti-Rabbit IgG (H+L) Secondary Antibody (No. 31210; Invitrogen).
Immunofluorescence analysis
Immunofluorescence was performed based on our published method (Wang et al, 2019). After blocking with 3% BSA, samples was stained overnight with primary antibodies (rabbit anti-IFITM3 Proteintech, 11714-1-AP or mouse anti-ACE2 Proteintech, 666991-Ig) diluted in PBS in a wet chamber at 4°C. After washing, slides were incubated with secondary antibodies, Alexa Fluor 488-labeled Goat Anti-Mouse IgG (H+L) (A0428; Beyotime) and Cy3-labeled Goat Anti-Rabbit IgG (H+L) (A0516; Beyotime) and 500 ng/mL DAPI (KGA215; KeyGEN BioTECH). Images were acquired by confocal microscope (Leica DMi8).
Type I IFN treatment
Vero-shNC or Vero-shACE2 cells were seeded in 12-well plates, respectively. The next day, cells were treated with IFNβ (100 U/mL, 300-02BC; PeproTech). At 24 h poststimulation, cells were harvested for RT-qPCR and western blot to analyze ACE2 and IFITM3 expression, respectively.
Alignment of RNA-seq data and differential expression analysis
Differential expression genes were identified by DESeq2 package. Those genes with |log2 (Fold change) |>2 and significant p-value <0.05 were considered as differentially expressed genes (DEGs) between the siNC group and siACE2 group. Volcano plots were generated with the R ggplot2 (v.3.1.1) package. The R package Pheatmap (1.0.8) software was used for bidirectional cluster analysis of DEGs. Pathway analysis was performed to examine the significant pathways of the DEGs according to KEGG. Gene set enrichment analysis (GSEA) was performed to analyze the inflammation-related gene sets Broad Institute Box plots were generated using the ggplot2 (v.3.1.1) package in R software.
Statistical analysis
All the statistical analyses were performed using GraphPad Software8.0.1. The comparisons between two groups were performed using the Student's t-test. Multiple comparisons were evaluated by either one-way or two-way analysis of variance among the treatment groups. Levels of significance were determined as follows: ***p < 0.001, **p < 0.01, and *p < 0.05.
Results
ACE2 knockdown model was established by siRNA silencing in Vero E6 cells
To screen out the appropriate cell line to establish the ACE2 knockdown cell model, we examined the endogenous ACE2 expression levels among A549, HuH-7, Caco-2, Calu-3, and Vero E6. High expression was detected in Vero E6 by RT-qPCR (Fig. 1A) and western blot (Fig. 1B), while lowly expressed ACE2 in other cell lines were clearly observed. Nearly 80% of Vero E6 were positive for ACE2 by flow cytometry (Fig. 1C), in comparison with the isotype group. Using the same gating strategy, only A549 cells showed partially positive (37.4%). Positive populations in HuH-7, Caco-2, and Calu-3 cells were rarely detected. To determine whether ACE2 expression was affected by SARS-CoV-2 pseudovirus infection bearing S protein, lysates from SARS-CoV-2 pseudovirus infected Vero E6 cells were analyzed by both qPCR and western blot.
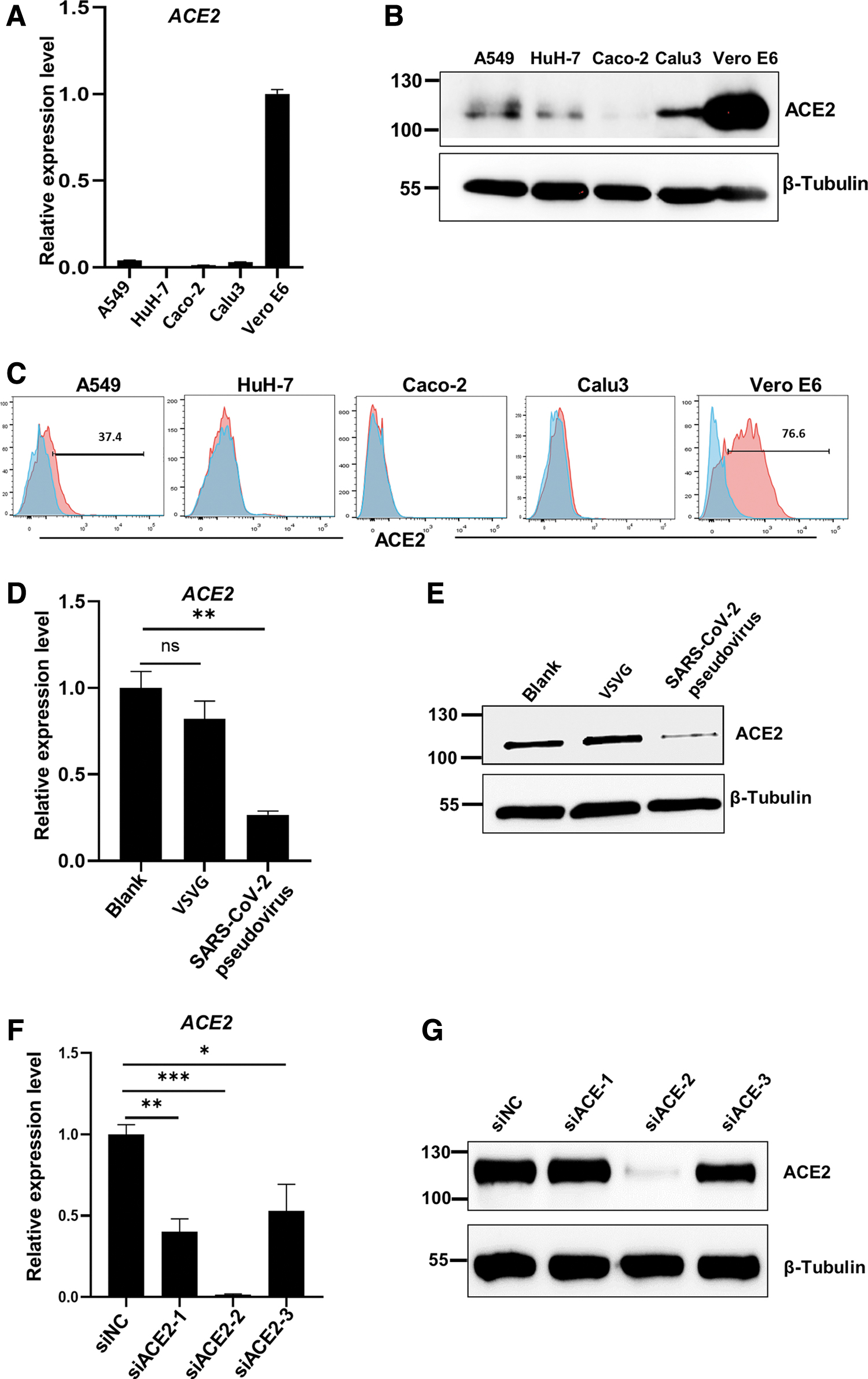
ACE2 knockdown model was established by siRNA silencing in Vero E6 cells.
The results showed that ACE2 was obviously decreased by SARS-CoV-2 pseudovirus infection, compared to the blank group, while ACE2 expression in the VSVG-treated group was not significantly changed, compared to the blank group (Fig. 1D, E). However, it is still not clear which downstream molecules associated with regulation on the SARS-CoV-2 infection are affected by the decrease of ACE2 expression. To address this issue, we generated ACE2 knockdown model utilizing siRNA to silence ACE2 expression in Vero E6, which simulated the downregulation of ACE2 expression induced by SARS-CoV-2 pseudovirus infection and simultaneously excluded interference from the changes of other molecules caused by infection. Vero E6 cells were transfected with ACE2-specific siRNApool (siACE2, siRNA targeting ACE2) or negative control siRNA (siNC, a scramble siRNA without any targeting gene).
The yields of ACE2 mRNA by qPCR exhibited that the ACE2 mRNA levels were significantly reduced in all three siRNA-treated groups, especially in the siACE2-2 group, in comparison to the siNC group (Fig. 1F). Western blot results also confirmed that the ACE2 protein was significantly decreased by siACE2-2 (Fig. 1G). Herein, the siRNA-induced ACE2 knockdown system was successfully established.
IFITM3 was decreased in ACE2-silenced Vero E6 cells, and downregulated by SARS-CoV-2 pseudovirus infection in normal Vero E6 cells as well
To investigate the changes of molecules related to silenced ACE2, we executed transcriptome sequencing of Vero-siACE2 and Vero-siNC groups by next-generation sequencing. In total, after ACE2 knockdown, we identified 167 and 299 significantly (log|FC| > 2 and false discovery rate (FDR) <0.05) up- and downregulated transcripts, respectively (Fig. 2A). Heatmap, KEGG enrichment analysis and GSEA were performed to determine the biological processes and signaling pathways associated with the DEGs. These analytical results demonstrated that the genes involved in immune signaling pathways, including both innate and adaptive immunity, were enriched (Fig. 2B–D). Impressively, the antivirus-related pathways, including viral protein interaction with cytokines and IFN-related processes, could be induced in the absence of infection, implying that the ACE2 alteration per se is sufficient to induce the immune responses.

IFITM3 was downregulated in ACE2-silenced Vero E6 cells, and decreased in the cells infected by SARS-CoV-2 pseudovirus as well.
Accordingly, the expression of a number of immune-related cytokines and inflammatory molecules was found to be altered by ACE2 knockdown. The decrease of IFITM3, CXCL17, CXCL12, IL33, OX40, and CD52 in the siACE2 group was confirmed by qPCR (Fig. 2E). It has been reported that both ACE2 and IFITM3 belong to ISGs. Therefore, we speculated that there were interactions between ACE2 and IFITM3 when Vero E6 cells were infected by SARS-CoV-2. Subsequently, the decrease of IFITM3 induced by ACE2 knockdown was further focused upon and verified by western blot (Fig. 2F).
To determine whether IFITM3 expression was affected by SARS-CoV-2 pseudovirus infection, cell lysates from SARS-CoV-2 pseudovirus-infected cells were analyzed by qPCR and western blot. Both ACE2 and IFITM3 were significantly reduced in the SARS-CoV-2 pseudovirus-infected group in contrast to the nontreated cells, while VSVG infection did not induce the ACE2 and IFITM3 reduction (Fig. 2G, H). Altogether, these results indicate that both ACE2 silencing and SARS-CoV-2 pseudovirus infection reduced IFITM3 expression in Vero E6 cells.
IFITM3 overexpression inhibited the SARS-CoV-2 pseudovirus infection of Vero-shNC cells, while promoted the infection of Vero-shACE2 cells
We applied a lentiviral transfection system to generate the stably silenced cell line (Vero-shACE2) using shRNA as schemed in the Figure 3A. IFITM3 expression in the shACE2 cells decreased as ACE2 was silenced, which was consistent with the results in siRNA silencing model (Fig. 3B–D). The SARS-CoV-2 pseudovirus infection was predominantly impaired in shACE2 cells (Fig. 3E), which is in accord with the concept that ACE2 is the critical receptor for SARS-CoV-2 infection.
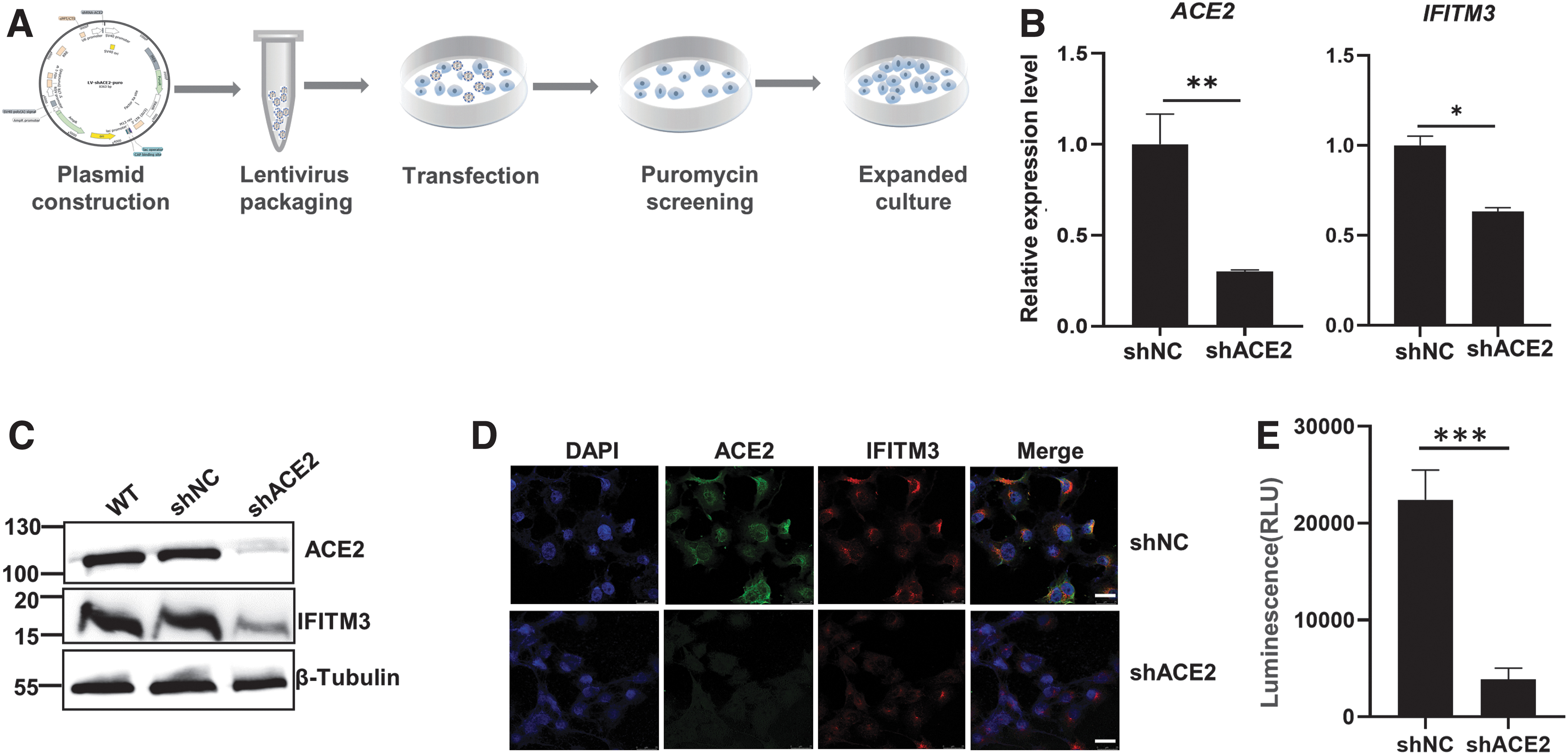
ACE2 knockdown inhibited SARS-CoV-2 pseudovirus infection in Vero E6 cells.
At 48 h posttransfection of IFITM3-expressing plasmid, IFITM3 expression was significantly increased, while ACE2 expression was unchanged by IFITM3 overexpression (Fig. 4A). Western blot and Immunocytochemistry results confirmed that IFITM3 was dramatically overexpressed at the protein level (Fig. 4B, C). In the shNC groups, IFITM3 overexpression inhibited the infection in accord with the previous studies. Unexpectedly, in the Vero-shACE2 group, the data showed that IFITM3 overexpression significantly promoted SARS-CoV-2 pseudovirus infection (Fig. 4D). Therefore, IFITM3 turns its inhibitory effect on SARS-CoV-2 infection to promotive effect in the condition of ACE2 defect. ACE2 and IFITM3 act together to regulate SARS-CoV-2 infection of the host cells.
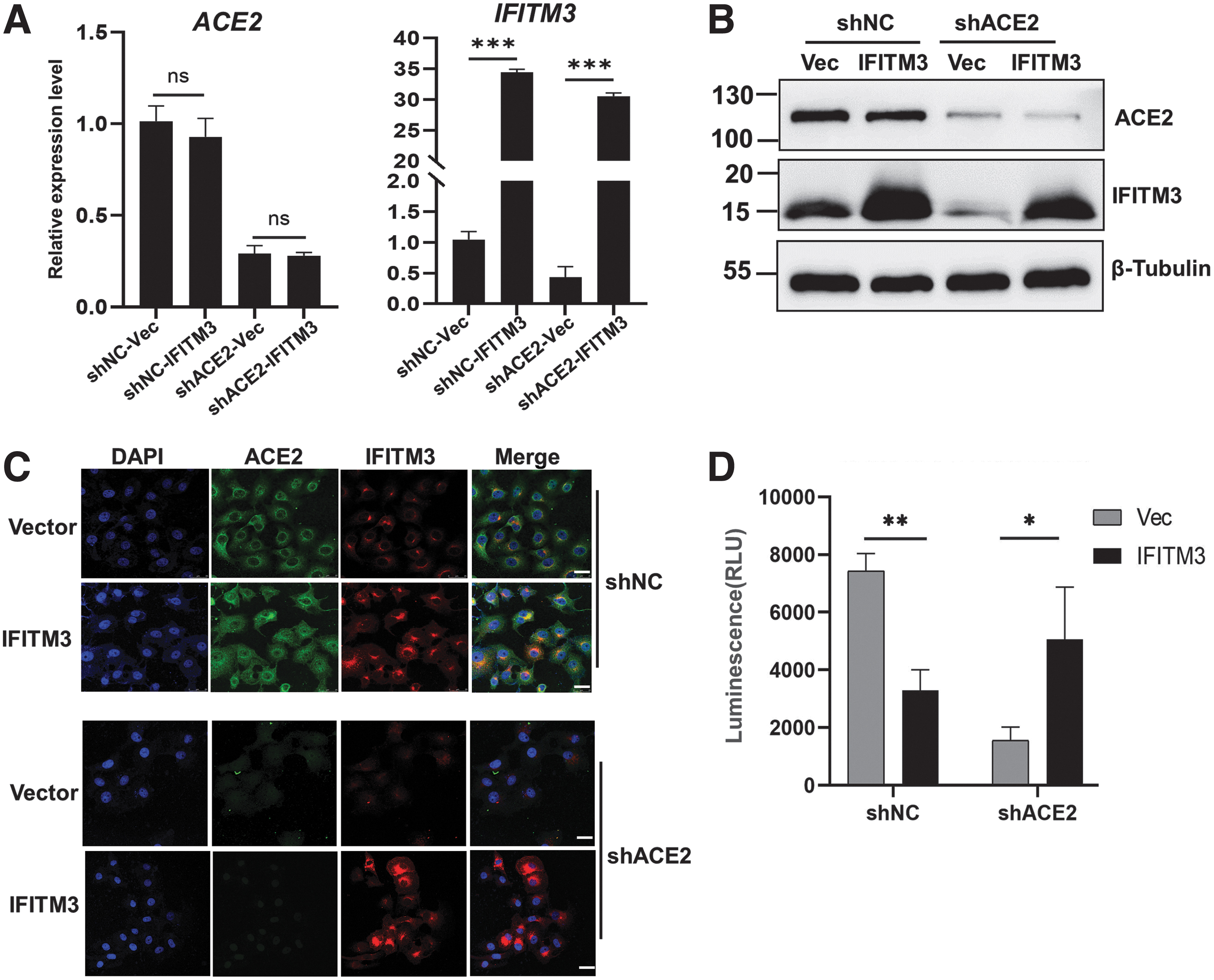
IFITM3 overexpression inhibited the SARS-CoV-2 pseudovirus infection in Vero-shNC cells, while promoted the infection in Vero-shACE2 cells.
ACE2 was not involved in IFNβ-induced IFITM3 production
As a member of the IFN-induced transmembrane protein (IFITM) family, IFITM3 can be induced by type I IFN with higher sensitivity (Gobillot et al, 2018). To examine whether type I IFN is involved in ACE2 regulation on IFITM3 expression, we used recombinant human IFNβ to induce IFITM3 production and detected the IFITM3 expression in the shACE2 group. At 48 h posttreatment with IFNβ (100 U/mL), the expression of ACE2 and IFITM3 of mRNA and protein was tested by RT-qPCR and western blot in Vero-shNC and Vero-shACE2 cells, respectively. qPCR results exhibited that IFITM3 could be significantly increased by IFNβ to the same extent in both Vero-shNC and Vero-shACE2 groups (Fig. 5A). The same results were observed by western blot analysis at the protein level (Fig. 5B).

ACE2 was not involved in IFNβ-induced IFITM3 production.
However, ACE2 expression was not changed by IFNβ treatment neither at the mRNA level nor at the protein level. The results demonstrate that IFNβ could induce IFITM3 production with or without ACE2, indicating that ACE2 and IFNβ regulations on IFITM3 expression are relatively independent processes.
Discussion
It is commonly recognized that ACE2 plays its role as the receptor for SARS-CoV-2 to enter the host cells during the COVID-19 pandemic. Besides that, we found that ACE2 affected IFITM3 expression in Vero E6 cells and IFITM3 promoted the SARS-CoV-2 pseudovirus infection in the absence of ACE2. To better reflect ACE2 function, Vero E6 was utilized, which has high endogenous ACE2 expression in comparison to other cell lines. This is in accord with the report that Vero E6 is the most susceptible to SARS-CoV-2 among 12 common cell lines without artificially exogenous ACE2 overexpression (Yeung et al, 2021). Our advantage is the establishment of a different cell model from others’, while most of current studies were conducted using cell lines artificially overexpressing the ACE2, which is actually not appropriate for study on endogenous ACE2 function.
Previous studies proved that the artificial overexpression of genes conferred the different expression pattern from their endogenous counterparts (Prelli Bozzo et al, 2021; Yao et al, 2013). Endogenous ACE2 better reflects the regulatory mechanism, which naturally happens without artificial interference.
As the receptor for SARS-CoV-2 infection, ACE2 binding with S protein is the initial step of the whole pathogenesis. The SARS-CoV-2 S protein binding causes the decrease of ACE2 (Kuba et al, 2005; Wan et al, 2020). Consistent with most of previous reports, our study showed that ACE2 was decreased at both mRNA and protein levels after the SARS-CoV-2 pseudovirus infection. To investigate the effect of ACE2 decrease on the downstream factors, we utilized the ACE2 knockdown system, which could better manifest the ACE2 function excluding the interference from the changes of other receptors (CD147, NRP1, DPP4, etc.) caused by SARS-CoV-2 infection (Guo et al, 2021; Masre et al, 2020).
Although Lu et al had reported the siRNA silencing of ACE2 resulted in the reduction of SARS infection of Vero E6 cells, there are no reports about silenced ACE2 influence on other factors during coronavirus infection using the ACE2 silencing system (Lu et al, 2008; Masre et al, 2021).
With the help of bioinformatic tools, the gene expression profiling displayed that in addition to the RAS-related genes, hundreds of other genes were changed by ACE2 knockdown. Many genes related to immune responses were identified in the ACE2-silenced group. The reported immune dysfunction during SARS-CoV-2 pathogenesis might be attributed to the change of immune status caused by ACE2 alteration. Although in our ACE2 knockdown model no virus infection was utilized, immune defense responses to virus, including NK-mediated cytotoxicity, IFN-related singling pathways, and viral protein with cytokines were clustered. It suggests that the ACE2 alteration per se is enough to initiate the viral infection-related signaling pathways. Among the DEGs, IFITM3 is one of the most affected genes by ACE2 silencing.
To our knowledge, it is the first report that ACE2 can affect IFITM3 expression. Both our study and Huang's work have demonstrated that the overexpression of IFITM3 cannot affect ACE2 expression, suggesting ACE2 possibly localizes in the upstream of IFITM3 (Huang et al, 2011), which is also in accord with the fact that ACE2 is responsible for SARS-CoV-2 early entry at the cell surface or early endosomes, while the endosomal IFITM3 mainly restricts the late entry of SARS-CoV-2 into host cells (Zhao et al, 2020).
IFITM3 is known to be a factor which inhibits the fusion between the viral and cellular membranes and inhibitory for a variety of virus entry (Gobillot et al, 2018; Ren et al, 2020). Bozzo's study reported that the overexpression of IFITMs, particularly IFITM3, reduced the entry of SARS-CoV-2 spike-mediated pseudoparticles into human lung epithelial cells by two orders of magnitude, which suggested that IFITM3 played important roles during SARS-CoV-2 infection. In contrast, IFITM3 did not affect VSVG-dependent entry, indicating that SARS-CoV-2 receptor-related singling pathways were involved in IFITM3 regulatory processes (Bozzo et al, 2020). In present study, we obtained consistent results as reported that overexpressed IFITM3 exerted an inhibitory effect on the SARS-CoV-2 pseudovirus infection of Vero E6 cells with wild-type ACE2.
Interestingly, we simultaneously found that IFITM3 overexpression significantly promoted SARS-CoV-2 pseudovirus infection of stable ACE2-silenced cells. It suggests that ACE2 is necessary for IFITM3 inhibitory effects on SARS-CoV-2 infection. ACE2 and IFITM3 could act together to influence the SARS-CoV-2 infection. During SARS-CoV-2 infection, S protein binding induces the ACE2 decrease in host cells, which might be a defense strategy for host cells to reduce the further infection. In ACE2 low or deficient condition, as a possible mechanism for enhancing infection, SARS-CoV-2 might counteract the defense by subverting IFITM3 to convert its antiviral to promotive effects on infection, which needs further experimental verification.
As an important factor induced by type I IFN, IFITM3 promotive effects on SARS-CoV-2 infection might be a strategy for SARS-CoV-2 to impair the type I IFN signaling pathway, which provides a probable explanation why type I IFN does not work well against SARS-CoV-2 infection. The same phenomenon was also found in the process of other coronavirus infections that HCoV-OC43 utilized IFN-induced IFITM3 to facilitate its entry into host cells (Zhao et al, 2014). To date, the study on the interactions among SARS-CoV-2, ACE2, and IFITM3 is still not enough and especially further confirmation with authentic SARS-CoV-2 viruses is required.
Conclusion
Collectively, our work demonstrates that ACE2 is not only a receptor for SARS-CoV-2 but also a regulator for IFITM3 during the SARS-CoV-2 pseudovirus infection. In the absence of ACE2, overexpression of IFITM3 could promote the infection. Hence, these two molecules act together to influence SARS-CoV-2 infection and pathogenesis. Clarifying the ACE2 function in the context of COVID-19 could help understand the physiopathology of the disease. These findings contribute to explaining the rapid spread especially in the cases of ACE2 low expression, provide a clearer picture of the pathogenic mechanism of COVID-19, and have significant implications in future therapeutic strategies to combat COVID-19.
Footnotes
Acknowledgments
We thank all the members in our laboratory for their helpful discussion. We thank Feiyang Luo and Kang Li for providing plasmids pCDNA3.4 and pLKO.1-shNC-puro, respectively.
Authors' Contributions
J.W.W. conceived and designed the study. S.H.L. performed the experiments. S.L.L. performed the bioinformatic analysis. S.H.L., X.Z.Z., and S.Z.C. generated the figures and graphs. J.W.W. wrote the article. Q.L.P. revised the article. All the authors read and approved the final version of the article.
Ethical Approval Statement
These studies did not directly involve human subjects or animals. Therefore, approval by an Institutional Review Board or Animal Care Committee was not required. These studies complied with all applicable regulations.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (Grant No. 81972023), the Natural Science Foundation of Chongqing City (Grant No. cstc2021jcyj-msxm0172).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.