
Abstract
Tumor necrosis factor superfamily 14 (TNFSF14) (LIGHT) is an interesting costimulatory molecule associated with T lymphocyte activation, and it mainly exerts its biological effects by binding to its receptors herpesvirus invasion mediator (HVEM) and lymphotoxin-β receptor. Research shows that TNFSF14 plays a critical regulatory role in immune responses to viral infection, but its role is different in different diseases. TNFSF14 can be a cytokine neutralization target during novel coronavirus infection, and anti-TNFSF14 monoclonal antibody treatment can reduce the risk of respiratory failure and mortality. When the host is infected with adenovirus, TNFSF14 can be used as an inflammatory biomarker to indicate whether there was an adenovirus infection in the host and the degree of disease caused by viral infection. When hosts suffer influenza virus infection, the TNFSF14-HVEM signaling pathway can stimulate the maturation and proliferation of memory CD8+ T cells, which helps the host immune system stimulate a second immune response against respiratory virus infection. TNFSF14 can act as an immune adjuvant and enhance the immunogenicity of the human papillomavirus (HPV) DNA vaccine when the host is infected with HPV. During hepatitis virus infection, TNFSF14 acts as a proinflammatory factor, participates in inflammation and causes tissue damage. In conclusion, TNFSF14 plays different and significant roles in diverse viral infections. This article reviews the current research on TNFSF14 in antiviral immunity.
Introduction
Since the outbreak of coronavirus disease 2019 (COVID-19), the immune response induced by viral infection has increasingly attracted the attention of scholars. Viral infection refers to the process in which a virus invades the host immune system through many pathways, proliferates inside a susceptible host cell and interacts with the organism. When the host is infected with different viruses, virions can be recognized by receptors on the host cell surface and then trigger the host immune response, which is essential for the host immune system to clear the virus (Getts et al, 2013).
However, in some circumstances, immunoregulatory mechanisms may fluctuate, leading to the breakdown of self-tolerance and the development of immune-mediated attacks against both viral and self-antigens (Getts et al, 2013), ultimately resulting in pathological lesions and dysfunctional cells, tissues, or organs. Viral infection is also one of the primary factors leading to autoimmune diseases (Getts et al, 2013). In addition to causing autoimmune diseases, viruses evade the host immune response and bind to corresponding antibodies, forming antigen-antibody complexes that deposit in local or systemic tissues to cause tissue damage.
Tumor necrosis factor (TNF) is a cytokine that was named after its initial discovery as an initiator of hemorrhagic tumor necrosis. The TNF superfamily (TNFSF), which includes ∼30 family members, are type II transmembrane proteins, the main members of which are TNF-α and TNF-β, and the latter is also known as lymphotoxin (LT). TNF superfamily 14 (TNFSF14), also known as LIGHT (homologous to LT), exhibits inducible expression and competes with HSV glycoprotein D to bind to herpesvirus invasion mediator (HVEM), a receptor expressed on T lymphocytes and is a member of the lymphotoxin-β (LTβ)-related TNFSF (Ware, 2008).
TNFSF14, which is a cell-surface molecule, is ubiquitously expressed on various cells at specific stages of differentiation, such as T cells, B cells, immature dendritic cells (DCs), natural killer (NK) cells, platelets, granulocytes, and monocytes (del Rio et al, 2010). As a TNF ligand, TNFSF14 binds to three tumor necrosis factor superfamily receptor (TNFSFR) members: HVEM (TNFSFR14), which is mainly expressed by T cells; LTβ receptor (LTβR), which is mainly expressed by stromal and nonlymphoid hematopoietic cells (Table 1), and soluble decoy receptor 3 (DcR3), which is predominantly expressed in lung tissue and the colon cancer cell line SW480 and regulates the biological function of TNFSF14 in vivo (Castellano et al, 2002).
Expression Pattern of Ligands and Receptors of the Tumor Necrosis Factor Superfamily 14-Related Pathways
Adapted from human lung fibroblasts (5) activated T lymphocytes (8) nonhematopoietic: most lineages (9) some nonhematopoietic cells (10) (31) and DX5+ NK cells (27).
BTLA, B and T lymphocyte attenuator; DC, dendritic cell; HVEM, herpesvirus invasion mediator; LTβR, lymphotoxin-β receptor; NK, natural killer; TNFSF14, tumor necrosis factor superfamily 14.
In general, the TNFSF14-HVEM interaction transmits costimulatory signals, mediates T cell activation and proliferation, and enhances the production of proinflammatory factors. LTβR acts as an indirect stimulatory receptor and activates T lymphocytes by interacting with TNFSF14. TNFSF14 binds to the receptor DcR3 and acts as an inhibitor. As an important immunoregulatory factor, TNFSF14 has multiple identities and plays different immunomodulatory roles by binding to different receptors in different diseases. This article reviews advances in the dual roles of TNFSF14 in antiviral immunity to mediate the full use of immune molecules to eliminate viruses.
TNFSF14/LIGHT-related pathways
LIGHT has three known mechanisms of action: sensitization of LTβR, activation of HVEM, and disruption of the HVEM-B and T lymphocyte attenuator (BTLA) inhibitory pathway (HVEN-BTLA inhibitory signaling is blocked when T cells transiently express TNFSF14) (Ware, 2008). TNFSF14 plays different roles through different pathways, thereby exerting distinct immunoregulatory effects on T cells and the immune response.
TNFSF14-HVEM signaling pathway
One critical function of TNFSF14 is to provide a costimulatory signal for complete activation of naive T cells. TNFSF14, which is a member of the LTβ-related TNF family, modulates T cell activation through HVEM and indirectly through LT-β (Ware, 2008). HVEM is widely expressed in hematopoietic cells such as T lymphocytes, monocytes, NK cells, DCs, and B lymphocytes (Duhen et al, 2004). Studies have shown that HVEM has three ligands: herpes simplex virus glycoprotein D and the TNF family members LTα and LIGHT. The extracellular domain of HVEM contains three cysteine-rich regions, and the fourth C-terminal CRD forms two of the characteristic three disulfide bonds of the molecule (Ware, 2008).
The CRD2 and CRD3 domains interact with TNFSF14 to produce a signal independent of the CD28 costimulatory pathway, thereby recruiting intracellular tumor necrosis factor receptor-associated factors (TRAF) (TNFR-associated factors) and activating NF-κB pathway-related serine kinases, which mainly acts on T cell activation and clonal expansion stage (del Rio et al, 2010). HVEM-TNFSF14 co-stimulation induces a Th1-type immune response that is dominated by the secretion of interferon-γ, granulocyte macrophage colony stimulating factor, interleukin-12 and stimulates specific CD8+ CTL reactions (Tamada et al, 2000).
TNFSF14-HVEM-BTLA signaling pathway
HVEM can also act as a ligand that binds to BTLAs to generate co-inhibitory signals through bidirectional signaling to inhibit T lymphocyte activation. BTLA, whose binding site is located in the first CRD domain of the N-terminal of HVEM, is an inhibitory receptor with a type I transmembrane glycoprotein structure (Ware and Sedy, 2011). After binding to HVEM, BTLA acts as a neutralizing kinase by recruiting tyrosine phosphatase SHP-1/2 (Src homology 2-containing tyrosine phosphatase 1/2), thereby transmitting inhibitory signals to T cells (Shui et al, 2011). Thus, HVEM can act as a molecular switch between proinflammatory and inhibitory signaling (Ware, 2008). BTLA is mainly expressed in lymphocytes and myeloid cells, with the most abundant expression in peripheral B cells. In addition, BTLA expression was also detected in CD11b+ macrophages and DX5+ NK cells (Steinberg et al, 2009).
TNFSF14, which generates a soluble expression form with biological activity by proteolytic cleavage, is a type II transmembrane protein (Sedgmen et al, 2006). Soluble TNFSF14 binds to HVEM with high affinity but does not compete with BTLA, allowing simultaneous binding of both ligands to HVEM. Studies have shown that soluble TNFSF14 and LTα can enhance the binding of BTLA to HVEM, which infers that these three molecules can form a trimolecular complex (Ware and Sedy, 2011). In contrast, the membrane-expressed form of TNFSF14 interferes with HVEM-BTLA binding through a noncompetitive mechanism, presumably due to steric hindrance, suggesting that TNFSF14 may be a regulator of HVEM-BTLA axis inhibitory signaling.
LTβR-TNFSF14 signaling pathway
LTβR-TNFSF14 is another costimulatory signaling pathway. LTβR is part of a complex communication system connecting lymphocytes with surrounding parenchymal and stromal cells (Steinberg et al, 2009). Ware (2008) noted that T lymphocytes in a naive, activated, or memory state can express most TNF receptors but not LTβR. Thus, LTβR is not considered to be a direct costimulatory molecule but indirectly affects T cell activation by modulating the differentiation of antigen presenting DCs, mast cells, tissue macrophages, and stromal cells (Ware, 2008). For most TNFR members, both the LTβR and HVEM signaling pathways involve in the recruitment of TNFR-related factors and the activation of downstream transcription factors.
Owing to the differences in the expression patterns of LTβR and HVEM and their ability to bind to different signaling molecules, although these two TNFSF14 receptors have similarities, the downstream signaling pathways they trigger are different (Steinberg et al, 2009). After TNFSF14 binds to LTβR, LTβR triggers downstream signaling pathways by recruiting TRAF, thereby inducing the activation of genes involved in proinflammatory responses, including macrophage inflammatory protein-1/2 (MIP-1/2), adhesion molecules, and chemokines (Steinberg et al, 2009) (Fig. 1).
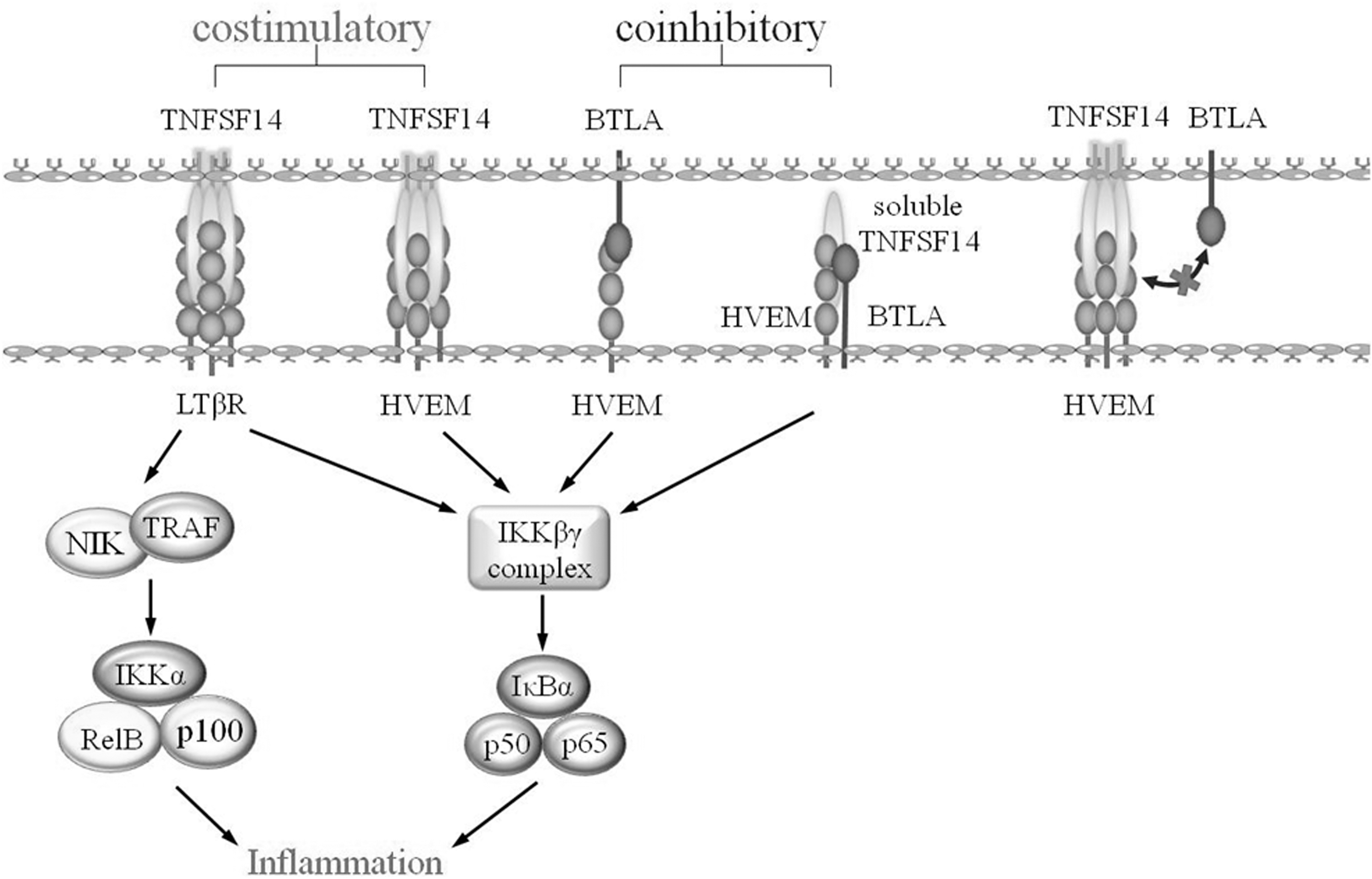
TNFSF14-related pathways. BTLA, B and T lymphocyte attenuator; HVEM, herpesvirus invasion mediator; LTbR, lymphotoxin-β receptor
The role of TNFSF14 in the immunity of virus infection
Respiratory virus infection
According to previous research, in autoimmune diseases, intestinal inflammation, or infectious diseases, TNFSF14 generally acts as a proinflammatory factor. For example, COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since March 2020, COVID-19 has become a global pandemic (Hui et al, 2020), and more than 600 million people have been infected with the SARS-CoV-2 as of September 1, 2022 [WHO Coronavirus (COVID-19) Dashboard]. Although vaccine development and many new treatments continue to improve, as the emergence of mutant strains and cytokine release syndrome are considered to be the major causes of morbidity and mortality in COVID-19 (Huang et al, 2020), a growing number of research teams have begun to focus on the immune response to SARS-CoV-2 to find effective therapeutic targets for neutralizing the cytokines involved in the immune response.
Perlin et al (2020) measured serum levels of free TNFSF14 in 47 COVID-19 patients by a validated immunoassay and found that the concentration of free TNFSF14 in COVID-19 patients was significantly higher than that in age- and sex-matched healthy controls. Arunachalam et al (2020) measured plasma cytokine concentrations in COVID-19 patients and a healthy control group with the Olink multiplex inflammation panel and an enzyme-linked immunosorbent assay system. The researchers found that the results of the two methods were consistent with the increased level of TNFSF14 in the plasma of COVID-19 patients and was closely related to disease severity.
Haljasmägi et al (2020) also found that plasma TNFSF14 levels in hospitalized patients with COVID-19 continued to increase in a longitudinal analysis of plasma proteins in 40 hospitalized patients with COVID-19. Excessive levels of TNFSF14 may lead to the accumulation of neutrophils, macrophages, and T cells that promote tissue destruction, resulting in pathological injury to body tissues (Perlin et al, 2021). Therefore, free TNFSF14 may be considered a cytokine that initiates pathological changes during airway viral infections (Perlin et al, 2020).
Human adenovirus is a double-stranded DNA virus that mainly causes respiratory infections, and a common disease is adenovirus pneumonia. In recent years, multiple teams have conducted extensive research on the molecular mechanism of TNFSF14 in human adenovirus-induced pneumonia. TNFSF14, a noncanonical stimulator of the NF-κB pathway, can activate the NF-κB pathway and promote the expression of downstream inflammatory factors after binding to its receptors HVEM and LTβR, thereby triggering an inflammatory response (D'Ignazio et al, 2018). Therefore, TNFSF14 is a proinflammatory factor that can stimulate T lymphocyte activation and trigger innate immune responses.
The experimental results obtained from genetically engineered mice showed that the expression of TNFSF14 on human T cells leads to abnormal lymphocyte activation, multiorgan inflammation, and tissue destruction (Shaikh et al, 2001). Fan et al used airway foreign body blockage as the control, collected bronchoalveolar lavage and plasma samples from patients with human adenovirus-induced pneumonia and the control group, used the Luminex method to measure 37 common inflammatory proteins, and found that the concentration of TNFSF14 in patients with adenovirus-induced pneumonia was significantly higher than that in the control group (Fan et al, 2020).
Xu et al (2019) sequenced the transcriptomes of peripheral blood mononuclear cells from human adenovirus type 55 (HAdV55)-infected patients and healthy volunteers. In HAdV55-infected patients, the expression levels of TNFSF14 in neutrophils and macrophages were closely related to the severity of infection and the degree of lung tissue damage (Xu et al, 2019). TNFSF14 can be considered an inflammatory marker of adenoviral pneumonia, but the molecular mechanism of its involvement in adenoviral pneumonia is unclear, and further experiments are required to verify how it exerts its immunoregulatory effects.
More interestingly, Desai et al (2018) prepared recombinant influenza virus by gene recombination technology and infected HVEM−/− mice, wild-type (WT) mice, and TNFSF14−/− OT-I transgenic mice and found that the development of lung memory CD8+ T cells was an important component of the immune response to respiratory viruses (Abboud et al, 2016). The researchers found that TNFSF14 was expressed on the membrane of activated CD8+ T cells, and TNFSF14 played a crucial role in regulating memory CD8+ T cells in the lung after respiratory virus infection. TNFSF14−/− CD8+ T cells exhibited no gross defects in their activation, proliferation, functionality, or trafficking to the lungs in response to respiratory virus infection.
However, fewer TNFSF14−/− CD8+ T cells survived after the peak effector response, resulting in severe memory CD8+ T cell damage (Desai et al, 2018). TNFSF14 might play a core regulatory role in the formation of memory CD8+ T cells and could also play an essential role in the host secondary immune response to respiratory virus infection. Furthermore, the researchers demonstrated that the TNFSF14-HVEM interaction has an essential survival or antiapoptotic effect when differentiated effector CD8+ T cells mature and transition into long-lived memory cells (Desai et al, 2018). These findings can deepen our understanding of TNFSF14, which not only appears to be an inflammatory factor during viral infection but also stimulates memory cell production and proliferation. However, the molecular mechanism by which TNFSF14 converts effector cells into memory cells is still unclear, and whether it has similar effects on other types of viral infections needs to be explored further.
Human papillomavirus infection
Human papillomavirus (HPV) infection is one of the key factors that causes cervical cancer. HPV vaccination can effectively prevent HPV infection and block the development of cervical cancer (Satyaprakash et al, 2009). To expand on the original intention of vaccines to prevent HPV infection, different research institutions have proposed the concept of therapeutic HPV vaccines, using therapeutic vaccines to eliminate HPV infections that have already occurred and related malignant tumors caused by precancerous lesions (Lin et al, 2010). DNA vaccines have attracted much more attention because they are safe and effective and can induce antigen-specific cellular immunity and humoral immunity at the same time (Hung et al, 2007).
However, since DNA does not have the inherent ability to amplify or spread in transfected cells such as viral vectors, one of the most contentious issues limiting the development of DNA vaccines is their weak immunogenicity (Monie et al, 2009). Li (2011) divided cervical cancer model mice into six groups, and three experimental groups were treated with TNFSF14 alone, HPV-E7 DNA vaccine alone, and TNFSF14/E7 DNA vaccine combined treatment, whereas the three control groups were treated at the same time. The researchers found that the tumor microenvironment of mice in the combined treatment group was changed, the expression of immunosuppressive cytokines was reduced, and the expression of proinflammatory cytokines was increased, which damaged tumor immune tolerance in the tumor microenvironment and was conducive to activating an effective immune response against tumor cells, resulting in tumor cell killing (Li, 2011).
Therefore, we have reason to believe that TNFSF14 will be a robust HPV DNA vaccine adjuvant to help induce a powerful antitumor immune response and achieve a more efficacious tumor-killing effect (Li, 2011). Thomas et al concluded that TNFSF14 stimulation promoted CD40L-induced B cell proliferation and Immunoglobulin G and Immunoglobulin M (but not IgA) secretion by simulating B cell contact with activated TNFSF14-expressing T lymphocytes and costimulation with TNFSF14 (Duhen et al, 2004). CD40L is a potent B cell stimulator, but stimulation alone cannot induce TNFSF14 expression (Duhen et al, 2004).
In sharp contrast, B cell costimulation by CD40L and TNFSF14 can induce TNFSF14 expression (Duhen et al, 2004). TNFSF14 positively stimulates its own expression; TNFSF14 expressed on T lymphocytes and CD40L stimulate B lymphocytes to express TNFSF14, and then TNFSF14 expressed by B cells in turn stimulates T lymphocyte responses. These experimental results suggest that TNFSF14 can induce and enhance the humoral immune response, but whether it plays a positive role in other viral infections is unclear and needs to be further discussed.
Hepatitis virus
Viral hepatitis is inflammation of the liver caused by hepatitis viral infection. This disease is mainly divided into five types: intestinal transmission by hepatitis A and hepatitis E and blood transmission by hepatitis B, hepatitis C, and hepatitis D. The T cell-mediated immune response plays a decisive role in hepatocyte injury induced by autoimmune hepatitis, viral infection, and hepatotoxin (Shui et al, 2011). The binding of TNFSF14 and HVEM can generate costimulatory signals and lead to the activation of T lymphocytes, thereby enhancing the T cell-mediated immune response, promoting the secretion of inflammatory factors, and causing inflammatory damage. Li et al infected TNFSF14-KO mice and WT mice with murine hepatitis virus strain 3 for 72 h and performed hematoxylin–eosin staining on liver sections in these two strains of mice.
TNFSF14-KO mice had a lower degree of liver damage than WT mice (Li et al, 2015). Alanine aminotransferase, a significant indicator of impaired liver function, was significantly downregulated in the plasma of TNFSF14-KO mice compared with WT mice (Li et al, 2015). TNFSF14 may play a key role as a proinflammatory factor in viral hepatitis. By measuring the mRNA expression levels of HVEN and LTβR in the spleen and liver in infected mice, it was shown that the TNFSF14-HVEM and TNFSF14-LTβR pathways promote hepatitis virus infection of hepatocytes and participate in liver function damage in viral hepatitis. Li et al (2020) established a mouse renal fibrosis model with unilateral urethral obstruction (UUO) and showed that the expression of TNFSF14 and its receptors HVEM and LTβR was significantly increased in the UUO-induced mouse fibrosis model and patients with fibrotic nephropathy.
Sphingosine kinase 1 (Sphk1), an enzyme that produces sphingosine-1-phosphate (S1P), is highly expressed in inflammation-related renal disease and could be involved in renal inflammation and fibrosis. A study showed that TNFSF14 could upregulate the expression of Sphk1, hence TNFSF14 may be a profibrotic factor (Li et al, 2020). Anand et al proposed a new view that free TNFSF14 plays a crucial role in the pathogenesis of hepatic inflammation, which is consistent with the increase in plasma TNFSF14 levels in renal fibrosis (Anand et al, 2006). Viral hepatitis may induce liver cirrhosis in the later stage. Based on the involvement of TNFSF14 in the pathogenesis of renal fibrosis (Li et al, 2020), we suggest that TNFSF14 can act as a profibrotic factor, but whether TNFSF14 is also involved in liver cirrhosis needs to be further studied to reveal its underlying molecular mechanism.
Conclusion
Because of the continuous challenge of viral infection, it is important to study the immune mechanism of viral infection and host antiviral immunity, and find effective and safe intervention targets to inhibit the damage caused by viral infection. Many host molecules play different roles in immune responses to viral infection, among which TNFSF14 has become a key factor in immune responses to viral infection in recent years (Fig. 2). TNFSF14 plays different roles in different kinds of viral infections. TNFSF14 not only acts as a proinflammatory factor but can also play an anti-inflammatory role. We need to conduct in-depth research on the molecular mechanism of TNFSF14 in viral infections and focus on different disease types to clarify the detailed molecular mechanism of TNFSF14 to identify symptomatic treatment strategies to achieve the better therapeutic effect.
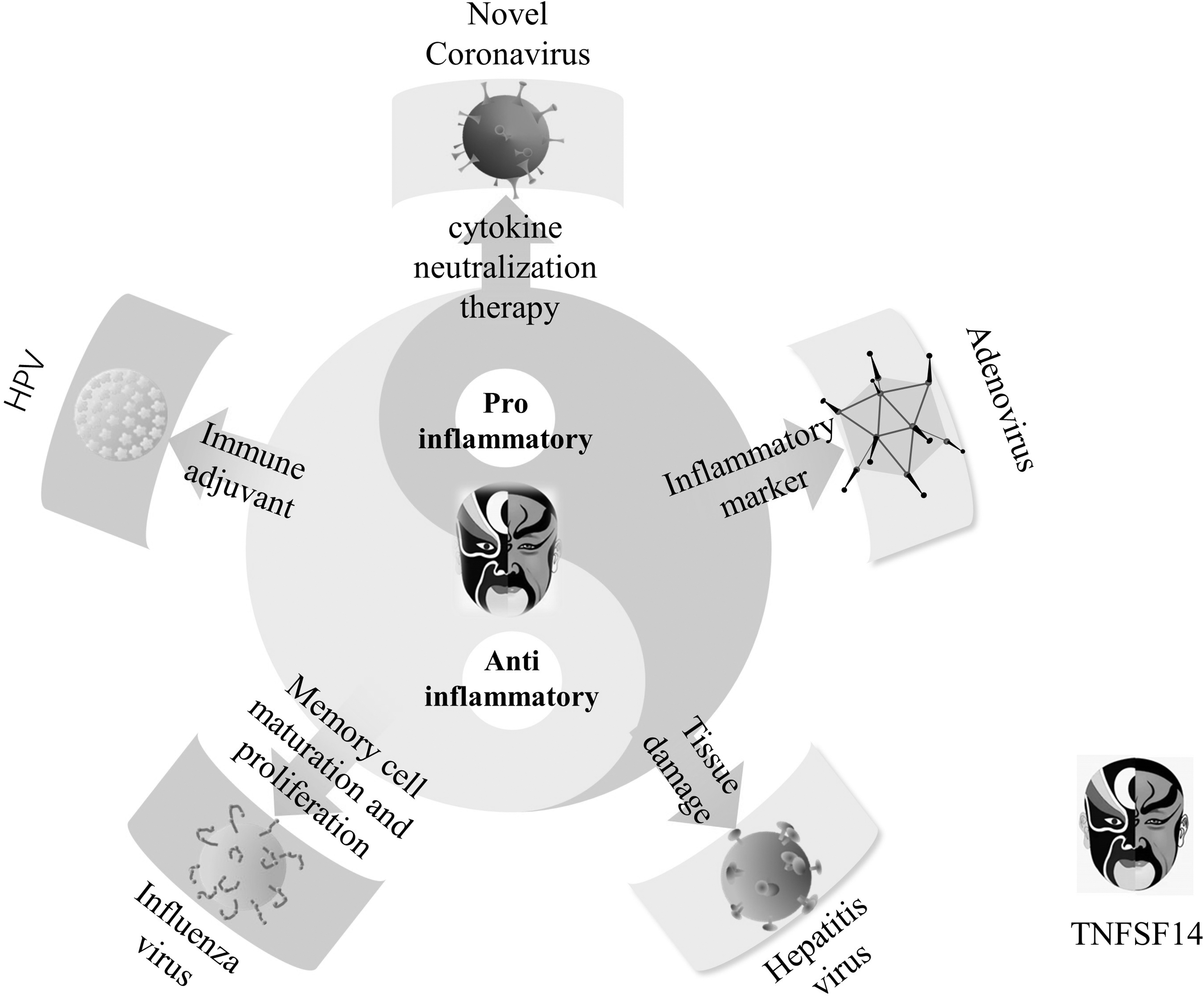
TNFSF14 plays dual roles in regulating the immune system during different types of virus infection. TNFSF14, tumor necrosis factor superfamily 14.
Footnotes
Authors' Contributions
Conception, modification, and editing was performed by J.C and C.-C. An earlier draft of the review was written by Y-H. Figures were prepared by Y.-W.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation (No. 81401311).