
Abstract
Epstein–Barr virus (EBV) is the first human oncogenic virus to be identified, which evades the body's immune surveillance through multiple mechanisms that allow long-term latent infection. Under certain pathological conditions, EBVs undergo a transition from the latent phase to the lytic phase and cause targeted dysregulation of the host immune system, leading to the development of EBV-related diseases. Therefore, an in-depth understanding of the mechanism of developing an immune response to EBV and the evasion of immune recognition by EBV is important for the understanding of the pathogenesis of EBV, which is of great significance for finding strategies to prevent EBV infection, and developing a therapy to treat EBV-associated diseases. In this review, we will discuss the molecular mechanisms of host immunological responses to EBV infection and the mechanisms of EBV-mediated immune evasion during chronic active infection.
Introduction
Epstein–Barr virus (EBV), a double-stranded DNA (dsDNA) virus belonging to the gamma herpesvirus family, was discovered by Epstein and Barr in 1964 while studying malignant lymphoma among children in Africa. EBV was the first human oncogenic virus to be discovered. EBV infection is linked to a variety of malignant tumors and malignant tissue proliferative diseases in humans, including malignant lymphoma, nasopharyngeal carcinoma, gastric cancer, and hemophagocytic syndrome (Morales-Sánchez and Fuentes-Pananá, 2014). Half of Hodgkin's lymphoma and Burkitt's lymphoma cases are caused by EBV infection (Saha and Robertson, 2019). Humans are generally susceptible to EBV infection, with ∼80–90% of the global population infected with EBV (Luzuriaga and Sullivan, 2010). Most EBV-positive adults are infected during their childhood, and some are latent carriers for life.
Infection with EBV is associated with lymphocytosis. EBV is commonly transmitted by infected saliva and infects the resting B lymphocytes and the epithelial cells (Nowalk and Green, 2016). During its life cycle, EBV undergoes two different types of infection modes, namely latent infection and lytic infection. During latent infection, EBV remains in the nucleus as episomal (free viruses) or integrated DNA without showing any clinical symptoms. The viral protein the immediate early gene (IEG) BamHI Z fragment leftward open reading frame-1 (BZLF1) initiates the EBV lytic cycle when EBV-infected memory B cells differentiate into plasma cells or when the virus enters differentiated epithelial cells. BZLF1, which is a member of the activator protein (AP-1) family of transcription factors, induces the high synthesis of lytic viral proteins required for viral particle assembly and release (Buschle and Hammerschmidt, 2020; Laichalk and Thorley-Lawson, 2005).
During the lytic phase, EBV produces infectious viral particles, allowing the virus to spread from cell to cell and host to host (Kieff and Rickinson, 2006; Rickinson and Kieff, 2006). During latent infection, EBV expresses six nuclear antigens (EBNAs), three latent membrane proteins (LMPs), two EBV-encoded RNAs (EBERs), and BamHI-A rightward transcripts (BARTs) (Tsao et al., 2015). EBV lytic activation is characterized by the expression of a large number of viral proteins, which can express more than 80 lytic genes. The current research found that some of the EBV lytic genes also play a key role in the occurrence of EBV-related tumors (Yap et al., 2022).
During the latent phase and lytic phase of viral infection, EBV evades immune recognition and clearance by the host immune system through multiple mechanisms. Following immune evasion, EBV can establish a latent infection that persists for life in reservoir cells or cause clinical symptoms, leading to the occurrence of malignant tumors. So, understanding the host immune response to EBV infection and the immune evasion mechanism of EBV is important for the treatment of EBV infection and EBV-related diseases.
Mechanism of EBV Infection and Host Immune Responses
Mechanism of EBV infection
EBV can infect B lymphocytes and epithelial cells, but it prefers to infect B lymphocytes, which can transform into malignant B cell lymphoma under certain circumstances (Bajaj et al., 2007). Specific complement receptors CR2 and CR21, which are present on the surface of B lymphocytes, can bind to EBV membrane glycoproteins complex gp350/220 with high affinity, facilitating various cellular processes, such as activating the nuclear factor kappa-B (NF-κB) signaling pathway, upregulating interleukin 6 (IL-6) levels, inducing receptor capping, and viral entry into B lymphocytes (Busse et al., 2010; Sugano et al., 1997). In addition, the fusion of the EBV envelope with host cells is critical for the entry of viral particles into B cells, which requires the interaction of both gp42 and gH/gL complex on EBV with the major histocompatibility complex II (MHC-II)-like molecules on the B cell surface.
Several envelope glycoproteins gH, gL, and gB are thought to be critical for EBV infection of epithelial cells. The current mechanism of EBV infection of epithelial cells involves the interaction of ephrin receptor A2 on the surface of epithelial cells with glycoproteins gH, gL, and gB of EBV, promoting the fusion of EBV with epithelial cells (Chen et al., 2018; Heldwein and Krummenacher, 2008). In addition to infecting epithelial cells and B lymphocytes, EBV has been shown to infect T lymphocytes and natural killer (NK) cells (Ayee et al., 2020), causing T cell lymphoma, NK cell lymphoma, NK cell chronic lymphoid hyperplasia, and hemophagocytic syndrome.
Inside infected cells, the life cycle of EBV consists of two phases: the latent phase and the lytic phase. Latent EBV infection is often associated with tumorigenesis. During the latent phase, EBV is present in the host cell nucleus as a ring-like appendage. It uses the viral protein EBNA1 to attach to the host genome chromosomes and proliferates as the host cell divides and proliferates (Damania et al., 2022). Only a limited number of viral genes, which are necessary to maintain their genome, are expressed in the latent phase. When native B cells are infected with EBV, repressive histone modifications restrict EBV gene expression. Following histone modification, CpG methylation of viral DNA occurs, and cellular gene expression of the virus is strongly inhibited (Murata et al., 2021).
The latent infection consists of four distinct phases (0, I, II, and III), which express latent genes, including six EBV nuclear antigens genes (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP), LMPs (LMP1, LMP2A, and LMP2B), two EBV-encoded RNAs (EBER1 and EBER2), and BARTs. EBNA1 and LMP-1 are also expressed during the lytic phase. Because only a small amount of viral protein is produced during the latent phase, it can be maintained without being recognized by the immune surveillance system. During the lytic phase, all viral genes are expressed, and viral particles are propagated in the host cells (Wang et al., 2019). Thus, the lytic phase can be divided into several stages: immediate early gene (IE) expression, early gene expression, late gene expression, viral assembly, and release of viral particles (Kenney and Mertz, 2014).
Exogenous expression of either the IEG BZLF1 or BamHI R fragment leftward open reading frame-1 (BRLF1) can also induce viral reactivation. BZLF1 and BRLF1 encode the transcription factor Z (also known as Z, ZTA, and ZEBRA) and R protein (also known as R and RTA), respectively. The transcription factors Z and R proteins can cooperatively activate the promoters of early (E) lytic genes that encode the viral replication proteins. Early lytic genes encode proteins for viral replication, and as viral genes replicate, late viral genes are expressed to encode proteins required for viral DNA replication (Kenney and Mertz, 2014).
Host immune mechanism against EBV
The antiviral immune mechanism includes both innate immune response and adaptive immune response. The innate and adaptive immune mechanisms of host immunity against EBV are described in the following sections.
Innate immune mechanisms of host immunity to EBV infection
The innate immune system recognizes pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) through pattern-recognition receptors (PRRs), which are the first line of defense against pathogen invasion. Following recognition of PAMP and DAMP, PRRs induce the production of interferons (IFNs), cytokines, and chemokines, all of which generate nonspecific immune responses, and respond rapidly to foreign pathogens (Takeuchi and Akira, 2010). Innate immune activation is mainly mediated via the following PRR pathways: Toll-like receptor (TLR) pathways, retinoic acid–inducible gene-I–like receptors (RLRs) pathways, NOD-like receptor (NLR) pathways, and absent in melanoma (AIM) 2-like receptor (ALR) pathways.
Through these PRR pathways, the immune cells trigger the activation of the NF-κB pathway, type I interferon (I-IFN) pathways, and other inflammatory signaling pathways to induce the production of large amounts of proinflammatory factors, antiviral factors, and chemokines, which cooperatively induce adaptive immune responses and exert their antiviral effects (Cui et al., 2014). I-IFN response is one of the most important antiviral defense mechanisms of host cells. Among these receptor pathways, TLRs and RLRs are particularly important in the production of I-IFNs and different types of cytokines, whereas NLRs are important for the maturation of IL-1β through the activation of caspase-1 (Babamale and Chen, 2021; Fekete et al., 2018) (Fig. 1). The role of each receptor pathway in the development of innate immune responses after EBV infection is described in the following sections.

Host immunity to EBV via the PRR pathways. TLR2 and TLR4 are located on the cell surface, whereas TLR3, TLR7, and TLR9 are located in intracellular vesicles. Recognition of EBV by TLR2 stimulates the release of MCP-1 and IL-8 release from mononuclear macrophages in an innate immune response to EBV. IFN-α, produced by TLR4 upon recognition of EBV, stimulates macrophages to express IL-27, which controls the adaptive immune response. EBER can induce the release of IFNs and inflammatory factors via TLR3. TLR7 senses ssRNA and then produces IRF-5, a key mediator of cytokine induction after TLR stimulation, which can coactivate cytokine promoters with NF-κB. TLR9 can recognize and bind to CpG DNA of EBV, producing IL-8, IL-6, and IFN-α. Host cell RNA polymerase III transcribes EBV DNA into RNA ligands that can be recognized by RIG-I, whose C-terminal end is responsible for dsRNA recognition and whose N-terminal end activates downstream signals via the mitochondrial adapter IPS-1, thereby activating the transcription factor NF-κB and encoding IRF3 to induce an innate immune response; NLRP3 inflammasome is a polyprotein complex formed by NLRP3, ASC, and caspase-1. NLRP3 inflammasome activation promotes IL-1β and IL-18 secretion via pro-IL-1β. IFI16 is a sequence -independent natural sensor ALR that recognizes dsDNA of EBV in the nucleus and produces Caspase-1, which induces the release of IL-1β and IL-18 and the maturation of IL-33. ALR, AIM (absent in melanoma) 2-like receptor; ASC, apoptosis-associated speck-like protein containing a CARD; CpG DNA, the cytosine-phosphate-guanine containing DNA; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; EBER, EBV-encoded RNAs; EBV, Epstein–Barr virus; IFI16, interferon-γ–inducible protein 16; IFN, interferon; IL, interleukin; IPS-1, interferon β promoter stimulator 1 polymorphisms; IRF, interferon regulatory factor; MCP-1, monocyte chemotactic protein-1; NF-κB, nuclear factor kappa-B; NLRP3, NOD-like receptor family pyrin domain containing 3; pro-IL-1β, pro-interleukin-1β; PRRs, pattern-recognition receptors; RIG-I, retinoic acid–induced gene-I; ssRNA, single-stranded RNA; TLR, Toll-like receptor.
Toll-like receptors
There are many different TLRs, and in humans, 10 different TLRs have been found (named TLR1 to TLR10), of which TLR1, TLR2, TLR4, TLR5, and TLR6 are located on the cell surface, whereas TLR3, TLR7, TLR8, and TLR9 are located in intracellular vesicles. TLR10, a pseudogene, does not have any functions in recognition of pathogens and subsequent signal transduction (Thompson and Locarnini, 2007). TLRs are type-I transmembrane glycoproteins composed of extracellular, transmembrane, and intracellular signal transduction regions. TLRs are expressed in a variety of immune cells, such as macrophages, NK cells, and dendritic cells (DCs). TLRs activate the NF-κB pathway and induce the secretion of inflammatory factors, chemokines, and I-IFNs through both medullary differentiation factor 88 (MyD88)–dependent and MyD88-independent signal transduction pathways (Wang et al., 2015).
During the replication of EBV in vivo, the viral proteins and nucleic acids can activate monocytes/macrophages (MC), DCs, and B lymphocytes to express different TLRs. TLR2, TLR3, TLR4, TLR7, TLR8, and TLR9 are known to recognize EBV antigens (Lünemann et al., 2015; Martin et al., 2007). TLR2 recognizes the major envelope glycoprotein complex (gp350/220) of EBV and activates the complement receptors CR2 (CD21) and CR1 (CD35) on B cells, thereby stimulating the release of monocyte chemotactic protein-1 (MCP-1) from mononuclear macrophages. Cytokines, such as IL-8, are also released by innate immune cells in response to EBV (Fingeroth et al., 1984; Gaudreault et al., 2007; Ogembo et al., 2013; Tanner et al., 1987). EBER is abundantly expressed in EBV-infected cells; therefore, EBER is often used as a marker for EBV infection. EBER can induce the release of IFNs and inflammatory factors through the TLR3 signaling pathway (Iwakiri et al., 2009).
Recognizing pathogens, TLR4 stimulates the production of IFN-α, which promotes the expression of IL-27 by macrophages, which is involved in the initiation of T helper (Th)1 response and controls the magnitude and duration of the adaptive immune responses (Pirhonen et al., 2007). Plasmacytoid dendritic cells (pDCs) predominantly express TLR7 and TLR9. EBV can promote cytokine release from pDCs following interaction with TLR9. TLR9 plays an important role in recognition of EBV by pDCs, inducing the release of IFN-α, IL-6, and IL-8 (Fiola et al., 2010). Interferon regulatory factor 5 (IRF-5) is a downstream mediator of the TLR7 signaling pathway. EBV infection causes the induction of IRF-5 in type III latent B cell lines, and IRF-5 shows tumor-suppressive and antiviral properties (Martin et al., 2007). Therefore, following infection with EBV, the cells can release a variety of inflammatory factors through a number of TLR signaling pathways, which generate antiviral immune responses.
Retinoic acid–inducible gene-I-like receptors
RLRs belong to a family of RNA decapping enzymes containing the DExD/H-box helicase domain. RLRs are intracellular PRRs that can detect RNAs in the cytoplasm. RLRs play an important role in developing intrinsic antiviral immunity by recognizing viral RNAs in the cytoplasm and stimulating a downstream signaling cascade that induces the production of IFN and inflammatory factors. The currently identified members of the RLRs family are retinoic acid–induced gene-I (RIG-I), melanoma differentiation–associated gene-5 (MDA-5), and laboratory of genetics and physiology-2 (LGP-2) (Loo and Gale, 2011). Among them, RIG-1 plays a role in the process of EBV infection (Samanta et al., 2006). Host cell RNA polymerase III transcribes EBV DNA into RIG-I, activating RNA, thereby activating the RLR signaling pathways, transcribing I-IFN, and eliciting innate immune response (Chiu et al., 2009). RIG-I recognizes 5′-triphosphate RNAs, so EBERs as 5′-triphosphate RNAs can interact with RIG-I (Samanta et al., 2006). In addition to antiviral immune effects, there is more evidence that activating the RIG-I pathway can be used in the treatment of a wide range of tumors (Ranoa et al., 2016; Wu et al., 2017).
NLRs and ALRs
Nucleotide-binding oligomerization domain NLRs and ALRs are cytoplasmic inflammatory vesicle sensors that recognize exogenous molecules, including DNA. NLRs recognize pathogenic and risk-associated molecular patterns, such as lipopolysaccharides, peptidoglycans, viral DNAs, and RNAs. NOD-like receptor family pyrin domain containing 3 (NLRP3) is an important member of the NLRs family. NLRP3 inflammasome is a polyprotein complex formed by NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1. It can sense the PAMPs from pathogenic microorganisms and its own DAMPs (Das et al., 2021). NLRP3 inflammasome activation promotes IL-1β and IL-18 secretion via pro-interleukin-1β (pro-IL-1β) (Kelley et al., 2019). LMP1 can modulate IL-1β production through NLRP3 and exert antiviral effects on EBV (Cai et al., 2017).
The ALR family member interferon-inducible protein-16 (IFI-16) is a sequence-independent natural sensor that recognizes dsDNA of EBV in the nucleus and produces Caspase-1 and IL-1β (Chen et al., 2012). IFI-16 can persist throughout the latent phase of EBV infection, inducing the release of IL-1β and IL-18 and the maturation of IL-33 (Ansari et al., 2013).
Innate immunity plays an important antiviral role during EBV infection. However, innate immunity also can act as a double-edged sword, as the release of inflammatory factors and the induction of programmed cell death may lead to the release of large amounts of virulence factors, thus promoting the spread of infection and the development of EBV-associated autoimmune diseases (Pontejo et al., 2018).
Adaptive immune mechanisms of host resistance to EBV infection
Adaptive immunity is involved in the production of immunoglobulins and the activation of lymphocytes following stimulation by antigens, such as pathogenic microorganisms. Adaptive immunity is responsible for developing antigen-specific immune responses, including cellular immune responses and humoral immune responses, both of which play an important role in the defense against viral infections. The main members of the adaptive immune response are T cells and antigen-presenting cells (APCs).
T cells are lymphocytes that participate in cellular immunity. T cells recognize viral antigens presented by APCs and generate an immune response to them. The mechanism by which CD4+ T cells control EBV infection is not fully understood. On the one hand, the cytotoxic activity of CD4+ cells can eradicate EBV-transformed B cells expressing EBNA-1. On the other hand, CD4+ T cells can also limit viral replication by recognizing viral antigens presented by major histocompatibility complex II (MHC-II) on APCs and releasing cytokines, such as IFN-γ, tumor necrosis factor (TNF)-α, and IL-2, all of which are responsible for the eradication of EVB-infected cells in the lytic phase via cellular immune surveillance and targeted killing (Lawler and Stevenson, 2020).
Following interaction with MHC-II molecules presenting EVB epitopes from APCs, naive CD4+ T cells differentiate into different types of T helper cells (Th1, Th2, and Th17) under different cytokine environments (Takeuchi and Saito, 2017). T helper cells activate NK cells and CD8+ T cells by secreting large amounts of cytokines. CD8+ T cells are also known as cytotoxic T lymphocytes (CTLs). Following activation, CD8+ T cells bind directly and specifically to EBV-infected target cells and eradicate viral reservoir cells (Del Vecchio et al., 2021). CTL plays an important role in the control of viral infections by recognizing viral peptides presented by MHC class I molecules. Current studies suggest that CTLs recognize abnormal B lymphocytes infected by EBV through cell surface T cell receptors (TCRs) and kill EVB-infected B cells by releasing several cytotoxic factors, including TNF, TNF-related apoptosis-inducing ligand (TRAIL), granzyme, and perforin (Freeman et al., 2021).
In EBV infection, APCs are also known as EBV-infected B cells. B cells are mainly involved in humoral immunity. T helper cells can promote the differentiation of B cells into plasma cells, which can produce distinct types of antibodies, such as immunoglobulin G (IgG), IgA, and IgM, and can neutralize viruses. EBV is able to stimulate the humoral immune responses of the body, especially to some lytic cycle proteins (Taylor et al., 2015). However, the neutralizing antibodies produced by humoral immunity appear 2 weeks after primary infection and are therefore less useful to protect from early infection of viruses.
In addition to T cells and B cells, NK cells are one of the important effectors of the innate immune system of the body, enabling the killing of tumor cells and virus-infected cells, regulating the activity of other immune cells, and promoting tissue growth (Freud et al., 2017). NK cells, which can recognize and kill virus-infected cells via cell-surface receptor–ligand interaction, release cytokines, mediate antibody-dependent cellular cytotoxicity (ADCC), and exert antiviral effects, are regulated by an intricate balance of inhibitory and activating receptors (Valipour et al., 2019). NK cells eliminate their target cells mainly through four pathways (Cichicki et al., 2016; Guillerey et al., 2016; Voskoboinik et al., 2006): (1) release of perforin and granzyme, which mediate cytotoxic effects by inducing apoptosis; (2) initiation of caspase cascade through Fas/FasL interaction, mediating apoptosis of target cells; (3) release of cytokines (such as TNF-α), which exert their cytotoxic effects by binding to the corresponding receptors on the surface of target cells; (4) expression of surface IgG-Fc receptors, which can eliminate target cells through ADCC.
Immune Evasion Mechanism of EBV
Although the immune system eradicates viruses, EBVs have unique ways to evade recognition by the immune system and remain in the body. Immune evasion by EVB is achieved primarily through different ways, which are discussed in the following sections. The roles of lytic and latent genes on immune evasion are given in Tables 1 and 2.
Lytic EBV Gene Products Interfering with Host Immunity
BRLF1, BamHI R fragment leftward open reading frame-1; BZLF1, BamHI Z fragment leftward open reading frame-1; CSF-1, colony-stimulating factor 1; EBV, Epstein–Barr virus; gp, glycoprotein; IFN, interferon; IL, interleukin; IRF, interferon regulatory factor; MHC, major histocompatibility complex; mRNA, messenger RNA; NF-κB, nuclear factor kappa-B; NK, natural killer; RIG-I, retinoic acid–induced gene-I; TLR, Toll-like receptor; TNF, tumor necrosis factor; TRAF, TNF receptor–associated factor; TRIM25, tripartite motif–containing protein 25; vIL-10, viral homolog of IL-10.
Latent EBV Gene Products Interfering with Host Immunity
3′UTR, 3′ untranslated region; BARTs, BamHI-A rightward transcript; CREB-BP, CREB-binding protein; CXCL11, C-X-C motif chemokine ligand 11; EBNA, Epstein–Barr virus nuclear antigen; IL-1R, IL-1 receptor; ISG15, IFN-β stimulate gene 15; LMP, latent membrane protein; MICA, MHC class I chain-related chain A; MICB, MHC class I chain-related chain B; NLRP3, NOD-like receptor family pyrin domain containing 3; PD-L1, programmed death receptor ligand 1; Th, T helper; ULBP, UL16 binding protein.
Inhibition of the PRR pathways, blocking the release of associated inflammatory factors
The TLRs pathway is the predominant immune pathway in the PRR pathways. The TLR signaling pathways induce the production of I-IFNs and proinflammatory factors that are involved in initiating antiviral responses and subsequently instigate adaptive immune responses. According to the different life cycle of EBV, EBV has different intervention measures for TLRs pathway. During the latent phase, EBV can establish and maintain latent infection in vivo by reducing the expression of TLRs and evading host innate immunity through different mechanisms. Human B cells express high levels of TLR1, TLR6, TLR7, TLR9, and TLR10. EBV-LMP1 is a strong inhibitor of TLR9 transcription. LMP1, overexpressed in B cells, reduces the activity of TLR9 promoter and decreases the expression of TLR9 messenger RNA (mRNA) and protein, thereby impeding the host innate immune response (Bussey et al., 2019; Fathallah et al., 2010; van Gent et al., 2011).
During the lytic phase, EBV produces large amounts of lytic proteins. These lytic proteins can antagonize the TLR pathway. EBV lytic protein BGLF5 can reduce TLR2 and TLR9 expression (van Gent et al., 2015; van Gent et al., 2011). EBV lytic phase protein BGLF5 is a viral nucleic acid exonuclease that acts not only as a DNase in vitro, but also as an Mn2+-dependent RNase to reduce TLR9 mRNA, thereby downregulating TLR9 levels (Bussey et al., 2019; van Gent et al., 2011). EBV can also evade host immune system by suppressing the expression and function of antiviral factors that are downstream of TLR signaling pathways (Younesi et al., 2010). In addition, ubiquitination plays an important role in regulating TLR signaling. The EBV-encoded protein BPLF1 is a deubiquitinase and late lytic protein.
BPLF1 interacts with TNF receptor–associated factor (TRAF) 6 to deubiquitinate TRAF6 and inhibit TLR-mediated activation of NF-κB signaling or TRAF6-mediated activation of downstream products, thereby promoting viral lytic DNA replication (Saito et al., 2013; van Gent et al., 2014).
Like other PRRs, RIG-I can recognize viral RNA as well as replication and transcription products from viruses. Therefore, evading recognition by RIG-I is one of the strategies used by viruses for their survival and immune evasion. I-IFN induced by RIG-I is a key component in the host innate immune response and plays an antiviral and immunomodulatory role. During latency, LMP1 inhibits RIG-I–mediated transactivation of the IFN-β promoter, phosphorylation of interferon regulatory factor 3 (IRF3), and production of IFN-β stimulate gene 15 (ISG15). In addition, LMP1 promotes the degradation of RIG-I via the protease pathway (Xu et al., 2018). EBV-miR-BART6–3p, a microRNA (miRNA), inhibits the RIG-I-like receptor signaling pathway and I-IFN response. In addition, miR-BART6–3p, miR-BART3, and miR-BART19 promote EBV infection by targeting the 3′UTR (3′ untranslated region) of RIG-I mRNA and inhibiting EBV-triggered IFN-β response (Bouvet et al., 2021; Lu et al., 2017).
The RIG-I pathway can promote apoptosis of EBV-infected cells by producing inflammatory factors or activating inflammatory vesicles. During the lytic phase, EBV BCL-2 homolog BHRF1 can prevent the activation of IFN-β by inducing mitochondrial fission, inhibiting the activation of the IFN-β promoter, and blocking the nuclear translocation of IRF3 (Vilmen et al., 2021). The EBV IEG BRLF1 interacts with an RNA polymerase III subunit and inhibits the function of inflammatory vesicles involved in the RIG-I pathway, alleviating the antiviral effects of the RIG-I pathway (Long et al., 2021). In addition, the function of RIG-I also depends on the ubiquitination of the tripartite motif-containing protein 25 (TRIM25). On the one hand, BPLF1 can inhibit the ubiquitination of TRIM25, thus affecting the downstream signal transduction of RIG-1 and can also interact with 14-3-3 protein and E3 ligase TRIM25, sealing TRIM25 in a trimeric complex, reducing TRIM25-mediated I-IFN responses (Gupta et al., 2019).
NLRP3 inflammasomes play an important role in the NLRs pathway. At present, EBV-encoded miRNA is considered to be the key regulator of NLRP3 inflammasomes. EBV can upregulate Has-miR-372-5p, resulting in reduced expression of NIMA-related kinase 7 (NEK7), a human homolog of Aspergillus NIMA. This ultimately allows NEK7 to regulate NLRP3 inflammasome activation and reduce IL-1β production, realizing immune evasion (Chen et al., 2021). In addition, EBV miR-223 and miR-BART15 can target the same site in the NLRP3 3′UTR to block the accumulation of NLRP3 and inhibit IL-1β production by inflammatory vesicles (Haneklaus et al., 2012).
Therefore, during the latent phase of EBV, EBV can reduce the immune damage of EBV by reducing the expression of TLRs, promoting the degradation of RIG-1, and affecting the production of I-IFN and the activation of NLRP3 inflammasomes. In the lytic phase, EBV can not only inhibit the expression of PRRs, but also affect the downstream signal transduction of PRRs through de-ubiquitination, thus reducing the release of inflammatory mediators and promoting the replication of EBV (Fig. 2).
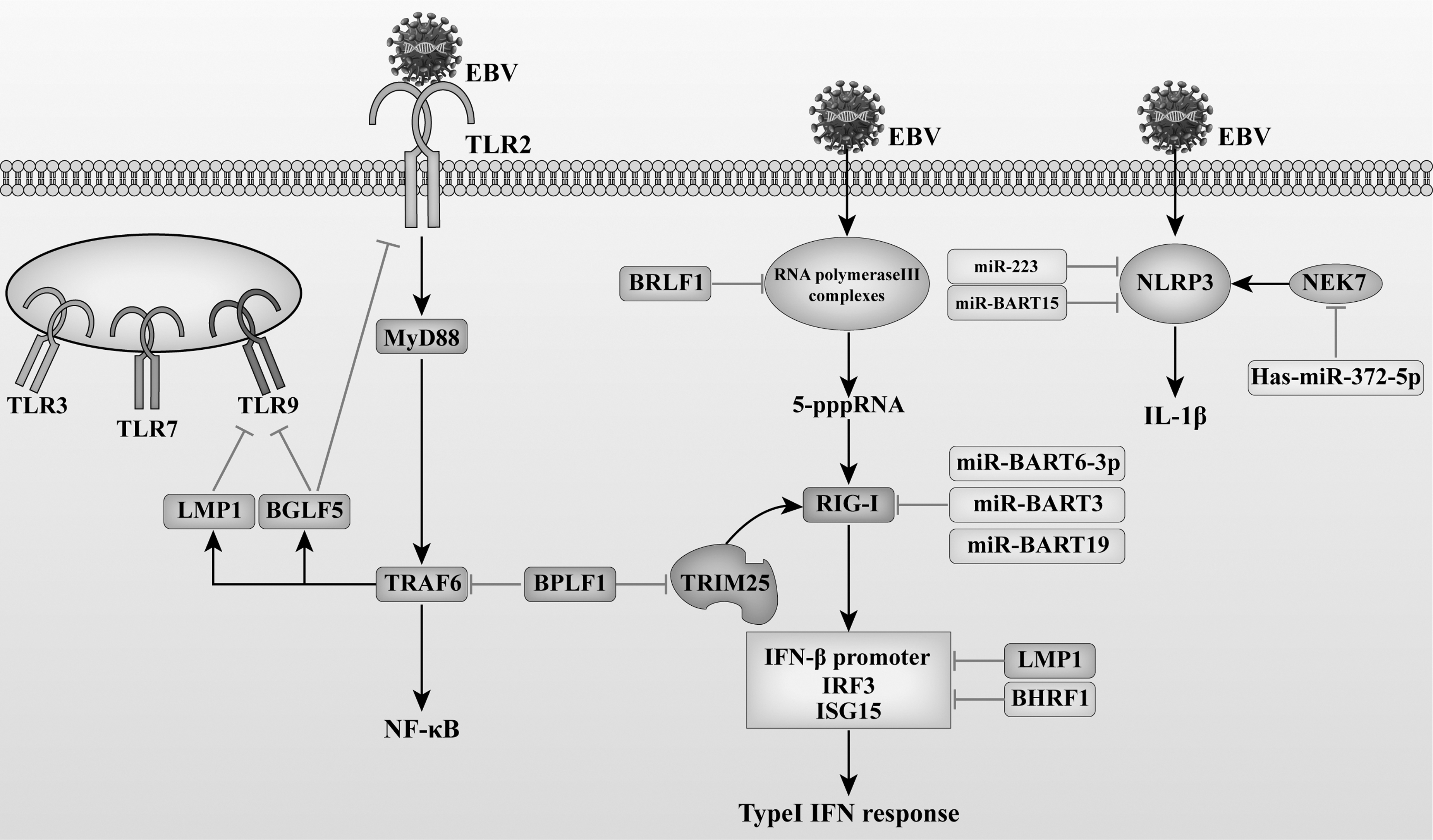
EBV interferes with PRR pathways. LMP1 reduces the activity of TLR9 promoter, and decreases the expression of TLR9 mRNA and protein. LMP1 also inhibits RIG-I–mediated transactivation of the IFN-β promoter, phosphorylation of IRF3, and production of ISG15. EBV lytic phase protein BGLF5 is a viral nucleic acid exonuclease. BGLF5 can reduce TLR2 and TLR9 expression. The EBV-encoded protein BPLF1 is a deubiquitinase and late lytic protein. BPLF1 interacts with TRAF6 to deubiquitinate TRAF6 and inhibits TLR-mediated activation of NF-κB signaling or TRAF6-mediated activation of downstream products; EBV-miR-BART6–3p inhibits the RIG-I-like receptor signaling pathway and type I interferon response. miR-BART6–3p, miR-BART3, and miR-BART19 promote EBV infection by targeting the 3′UTR of RIG-I mRNA and inhibiting EBV-triggered IFN-β response. EBV BCL-2 homolog BHRF1 can prevent the activation of IFN-β by inducing mitochondrial fission, inhibiting the activation of the IFN-β promoter, and blocking the nuclear translocation of IRF3. The EBV immediate early gene BRLF1 interacts with an RNA polymerase III subunit and inhibits the function of inflammatory vesicles involved in the RIG-I pathway, alleviating the antiviral effects of the RIG-I pathway. BPLF1 can inhibit the ubiquitination of TRIM25, thus affecting the downstream signal transduction of RIG-1, and can also interact with 14-3-3 protein and E3 ligase TRIM25, sealing TRIM25 in a trimeric complex, reducing TRIM25-mediated type I IFN responses. NEK7 can promote the activation of NLRP3 inflammasomes and release IL-1β. EBV can upregulate Has-miR-372-5p, resulting in reduced NEK7 expression and thus immune evasion. 3′UTR, 3′ untranslated region; BART, BamHI-A rightward transcript; BRLF1, BamHI R fragment leftward open reading frame-1; ISG15, IFN-β stimulate gene 15; LMP1, latent membrane protein 1; mRNA, messenger RNA; NEK7, NIMA-related kinase 7; TRAF, tumor necrosis factor receptor–associated factor; TRIM, tripartite motif–containing protein.
Regulation of the production of IRFs and I-IFNs and their signaling pathways
Type I interferons (I-IFNs) are expressed in humans, including IFN-α (including all 13 isoforms), IFN-β, IFN-ɛ, IFN-κ, IFN-ω, IFN-δ, and IFN-ζ (Pestka et al., 2004). I-IFN plays an important role in developing antiviral immune responses by demonstrating a direct antiviral activity. However, viruses have evolved with many strategies to interfere specifically with the production of hormones and their downstream mediators. The study found that EBV-miRNAs can target and regulate the genes involved in the IFN signal pathway, including T-bet, C-X-C motif chemokine ligand 11 (CXCL11), CLEC2D, RIG-I, and CREBBP, to inhibit the expression and release of IFNs and the activation and immunotoxicity of immune response. EBV miR-BART20-5p directly targets and inhibits T-bet expression in aggressive EBV+ nasal NK/T cell lymphoma, whereas EBV miR-BART8 can indirectly block the IFN-γ/STAT1 pathway and can suppress the occurrence of IFN-γ–activated immune responses (Lin et al., 2013).
CXCL11 is a chemokine induced by IFN. EBV miR-BHRF1-3 directly or indirectly regulates the expression of CXCL11 and inhibits the recruitment and activation of NK cells and Th1 cells (Xia et al., 2008). EBV-encoded miR-BART16 is a novel viral immune evasion factor that interferes with the I-IFN signaling pathways. The miR-BART16 directly targets CREB-binding protein (CREB-BP), a key transcriptional coactivator involved in I-IFN signaling pathways, thereby downregulating the expression of CREB-BP in EBV-transformed B cells and gastric cancer cells. EBV-miR-BART16 also inhibits the production of IFN-stimulating factors and blocks the antiproliferative effect of IFN-α on latently infected Burkitt lymphoma/leukemia (BL) cells (Hooykaas et al., 2017). During the lytic phase, the EBV IEG BZLF1 can interfere with the secretion of IFN-α mediated by the JAK/STAT signaling pathway by inducing the expression of suppressor of cytokine signaling factor 3 (SOCS3) and SOCS1 (Michaud et al., 2010).
The activation of IRFs IRF3 and IRF7 is essential for inducing type I IFN and innate antiviral responses. BZLF1 can also inhibit the activation of IRF7, thus blocking the activation of IFN-α4, IFN-β, and Tap-2 by IRF7 (Hahn et al., 2005). Another early immediate protein, BRLF1, has been shown to downregulate the transcription of IRF3 and IRF7, leading to reduced protein expression and thus achieving regulation of IFN. BRLF1 may affect the expression of IRF by binding to the GC-rich regions of IRF3 and IRF7 or by affecting the extension of RNA (Bentz et al., 2010). The EBV early protein BFRF1, which is associated with viral maturation, blocks the kinase activity of IKKi and inhibits phosphorylation, dimerization, and nuclear translocation of IRF3, thereby inhibiting IFN-β activity (Wang et al., 2020).
EBV tegument protein BGLF2 blocks type I IFN-induced phosphorylation of TLK-2, STAT-1, and STAT-3, inhibits the expression of IFN-stimulated genes (ISGs) IRF1, IRF7, and MxA, inhibits I-IFN-mediated signaling, suppresses host antiviral immune responses, promotes the reactivation of EBV in latently infected cells, and protects virus-infected cells from I-IFN–mediated immune responses (Liu et al., 2020). The EBV-encoded BCL2 homolog BHRF1 inhibits the activation of the IFN-β promoter and blocks the nuclear translocation of IRF3 (Vilmen et al., 2021). The host cytokine colony-stimulating factor 1 (CSF-1) stimulates macrophage differentiation and IFN-α secretion (Chitu and Stanley, 2006). BRAF1 is a 29 kDa monomer, which can be used as the receptor of CSF-1 and form a hexamer ring to bind CSF-1. By blocking cytokines, interfering with the receptor binding surface, and inducing the conformational change of CSF-1, BRAF1 interferes with the signal transduction mediated by CSF-1, thus reducing IFN-α secretion (Shim et al., 2012).
Thus, we can find that during the latent phase, EBV mainly uses miRNAs to block the signaling pathway of IFNs to suppress the expression of IFNs. During the lytic phase, EBV lytic proteins mainly regulate the production of IFNs by affecting the activation of IRFs.
Interference with NF-κB signaling pathway
The NF-κB family of transcription factors is a major regulator of innate immunity. In the early stages of viral infection, activation of NF-κB can induce downstream inflammatory signaling pathways, which in turn release numerous inflammatory factors and protect the host from viral infection. Virus-encoded factors can facilitate viral replication but prevent virus-induced apoptosis by manipulating the NF-κB signaling pathway (Khatiwada et al., 2017; Lu et al., 2021). BZLF1 and NF-κB mutually inhibit each other. During the lytic phase, BZLF1 inhibits the release of inflammatory factors TNF-α and IFN-γ by binding to the promoter of TNF-α and preventing NF-κB activation (Li et al., 2015). Phosphorylation of NF-κB is thought to be a key mechanism driving NF-κB activity. The p65 subunit of NF-κB is phosphorylated at multiple sites, including Ser536 (Oeckinghaus and Ghosh, 2009).
EBV tegument protein BGLF2 can interact with NF-κB subunits p65 and p50 and inhibits NF-κB activation by inhibiting the phosphorylation of p65 at Ser536 and blocking the nuclear translocation of NF-κB (Chen et al., 2019). Ubiquitously expressed transcript (UXT) is a coactivator of the NF-κB enhancer and is important for the nuclear function of NF-κB. BGLF4 is a member of herpesvirus kinase, which can attenuate the transcriptional activity of NF-κB by interacting with and phosphorylating UXT and reducing the interaction of UXT with NF-κB (Chang et al., 2012). During latency, EBNA1 was likewise found to inhibit the typical NF-κB pathway by inhibiting IKKalpha/beta phosphorylation (Valentine et al., 2010).
The IL-1 signaling pathway is a key initiator of inflammation and host innate immune responses after viral infection (Mills and Dunne, 2009; Sims and Smith, 2010). It has been shown that miR-BHRF1–2–5p binds directly to the m-RNA of IL-1 receptor (IL-1R) and blocks the expression of IL-1R, thereby inhibiting IL-1β-triggered activation of NF-κB (Skinner et al., 2017).
From this, we can find that influencing NF-κB activation by affecting NF-κB phosphorylation is the main way in which EBV interferes with the NF-κB signaling pathway.
Downregulation of host cell gene expression
During the latent phase, EBV expresses ∼10 gene products, whereas during the lytic replication phase, >90 gene products are expressed. Silencing the lytic phase genes is a critical part in establishing a lifelong latent infection. EBV evades immune recognition by silencing the expression of many genes during the latent phase. Upon reactivation, EBV expresses IEGs, which trigger a cleavage cascade that leads to the shedding of progeny viruses. LMPs family includes LMP1, LMP2A, and LMP2B. LMP1 catalyzes the sulfonylation of cellular proteins through its C-terminal activation region 3 (CTAR3), which can lead to the transcriptional repression of EBV lytic genes and thus maintain the latent state (Bentz et al., 2015). LMP2 is a nononcogenic gene that exists in two alternative forms, LMP2A and LMP2B.
LMP2 acts as a B cell receptor (BCR) mimic that prevents the reactivation of latent EBV by blocking tyrosine phosphorylation, allowing the continued survival of virally infected B cells (Fukuda and Longnecker, 2005). EBNAs family includes EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP, of which EBNA1 is the only protein that is expressed during both latent and lytic phases. EBNA1 can maintain EBV latent infection by regulating the expression of host and viral genes (Frappier, 2015). EBNA2 can inhibit the transcription of microRNA-34a (miR-34), indirectly increasing the expression of miR-34a target gene programmed death receptor ligand 1 (PD-L1). High expression of PD-L1 on EBV-infected cells facilitates immune evasion from T cells expressing PD-1 (Anastasiadou et al., 2019). EBV-miRNA also plays an important role in controlling viral lytic replication. BZLF1 encodes IE virus transcription factor Zta.
It has been confirmed that there is an miRNA interaction site in 3′UTR of BZLF1. miR-BHRF1-3 can reduce the fragmentation and replication of EBV by targeting 3′UTR of BZLF1 (Fachko et al., 2022). BALF5 is an EBV DNA polymerase encoded by virus mRNA. EBVmiR-BART2 can target BALF5 to inhibit viral DNA replication, thus maintaining the latency period of the virus (Barth et al., 2008). Current studies have shown that, during the latent phase, the EBV expresses miRNA20–5p to downregulate the expression of two early genes BZLF1 and BRLF1, thereby repressing the activation of the lytic cycle and the production of progeny viruses, which are essential for immune evasion and the establishment of latent infection (Jung et al., 2014). Finally, EBV can also use interferon-γ–inducible protein 16 (IFI16) to maintain the latent infection of EBV and prevent the reactivation of EBV (Pisano et al., 2017).
In recent studies, IFI16 was found to cooperate with the constitutive heterochromatin machinery (HCM) to silence the key EBV lytic switch proteins, thereby allowing for the persistent latent presence of EBV in B cells (Xu et al., 2022).
Inhibition of host immune cells (NK cells)
NK cells, a part of the innate immune system, play a key role in eradicating invading pathogens, and NK cells are known to inhibit EBV replication by releasing IFN-γ. BART-encoded microRNAs (miR-BARTs) are expressed in all phases of the viral life cycle in EBV-infected cells, and miR-BARTs are responsible for altering and maintaining viral latency. Among miR-BARTs, miR-BART2–5p can downregulate the expression of stress-induced immune ligand major histocompatibility complex class I chain-related chain (MIC) B (MICB) to evade recognition by NK cells (Wang et al., 2018). The receptors on the surface of NK cells can recognize a variety of ligands on the surface of target cells, including atypical MHC class I, MHC class I chain-related chain A (MICA), MICB, UL16 binding proteins 1–6 (ULBP1–ULBP6) and some viral proteins.
It has been demonstrated that LMP2A can reduce the expression of MICA and ULBP4, thus, affecting the recognition of EBV-infected cells by NK cells (Ma et al., 2016). It is demonstrated that the LALLFWL and LLLAL sequences of the first transmembrane structural domain of LMP1 display strong homology to the immunosuppressive structural domain (LDLLFL) of the retrovirus-encoded transmembrane protein p15E; therefore, the LMP1-derived LALLFWL peptide potentially has a strong inhibitory effect on T cell proliferation and NK cytotoxicity (Dukers et al., 2000). EBNA1 is required to maintain the EBV genome in latently infected cells, and it also plays an important role in suppressing NK cell–mediated cell death of infected neoplastic cells. EBNA1 downregulates the expression of NKG2D ligands ULBP1 and ULBP5 and regulates the expression of c-Myc, resulting in reduced NK cell–mediated killing of EBV-infected cells.
EBNA1 reduces apoptosis of newly infected B cells allowing more B cells to survive (Westhoff Smith et al., 2021). In late EBV lytic infection, the virus can express BHRF1 (vBcl-2 protein). BHRF1 protects EBV-infected B cells from BZLF1-mediated killing by NK cells (Williams et al., 2016). Receptors on NK cells recognize a variety of ligands on the surface of target cells, including atypical MHC class I, MICA, MICB, ULBP1–ULBP6, and other viral proteins. It has been demonstrated that LMP2A can reduce the expression of MICA and ULBP4, thus affecting the recognition of EBV-infected cells by NK cells (Ma et al., 2016). EBV expresses BCRF1, which is a viral homolog of IL-10 (vIL-10). vIL-10 inhibits NK cell–mediated killing of EBV-infected B cells (Jochum et al., 2012).
Through the above research, we found that EBV can affect the immune recognition of NK cells by reducing the ligand expression on the surface of target cells during the latency. In the lysis, NK immune damage is reduced by encoding viral homologues.
Regulation of host cell apoptosis
Host cell apoptosis affects viral replication and viral protein expression. To enable better intracellular proliferation, the EBV uses a number of measures to limit the apoptosis of the host cell. LMP1, the major oncoprotein of EBV, mimics a constitutively active CD40 receptor that can interact with TRAF proteins to induce multiple downstream signaling pathways (including NF-κB, PI3K, and STAT3), promote B cell proliferation, and develop resistance to apoptosis (Ma et al., 2015). EBNA1 not only reduces the activity of NK cells, but its glycine–alanine repeat region also inhibits the proteasomal degradation of EBNA1, facilitating immune evasion (Yin et al., 2003). EBV regulates B cell apoptosis by various means, such as miRNA-mediated apoptosis. It has been demonstrated that miR-BHRF1 can block the apoptosis of human B cells infected with EBV, improve the survival of target cells, and enable the better proliferation of EBV in the host (Kalla and Hammerschmidt, 2012).
BART miRNAs regulate at least a few hundred apoptosis-related target genes. The miR-BART5–3p directly targets the 3′UTR of the tumor suppressor gene tp53 and inhibits tp53 expression, thereby maintaining the latent phase and promoting tumorigenesis (Zheng et al., 2018). EBV miR-BART miRNAs can also maintain Burkitt lymphoma by inhibiting Caspase 3 (Vereide et al., 2014).
Interference with antigen processing and presentation
Cell-mediated immunity plays a key role in developing host immunity to viral infections, and antigen processing and presentation are important parts of T cell–mediated immunity. In cellular immunity, MHC class I and class II molecules present antigens and activate CD8+ and CD4+ T cell–mediated responses. EBV can impede T cell–mediated immune responses in several ways, including inefficient cross-presentation of viral antigens by MHC molecules. The late protein EBV gp150 forms an immune-evasive glycan shield on the surface of infected cells that prevents antigenic expression of HLA class I, class II, and CD1d molecules, thus enabling immune evasion and persistent infection (Gram et al., 2016).
Transporter associated with antigen processing (TAP) plays a key role in the antigen presentation of MHC class I molecules by transporting cytoplasmic antigenic peptides to the endoplasmic reticulum lumen, where it provides the processed peptides to MHC class I molecules to form peptide–MHC complexes that can be transported to the cell surface. The peptide–MHC I complex can be recognized by CTL, inducing protective CTL-dependent cellular immunity (Abele and Tampé, 2004). During the lytic phase, EBV lytic proteins also inhibit viral antigen presentation and reduce the body's immune recognition. It is shown that the EBV-encoded protein BNLF2a can bind to TAP to inhibit the antigen transport function of TAP, reduce the large ER-resistant pool of MHC class I molecules to present viral antigen, and reduce the large ER-resistant pool of MHC class I molecules to present viral antigen, rendering CD8+ T lymphocytes unable to recognize virally infected B cells (Jochum et al., 2012).
The immune evasion protein BDLF3, which is expressed late in the lytic cycle, can affect CD8+ T cell–mediated antigen recognition by targeting the ubiquitination and subsequent degradation of MHC class I molecules. Of interest, BDLF3 can also act on MHC class II molecules, thus affecting CD4+ T cell–mediated cellular immunity (Quinn et al., 2016). EBNA1 is expressed during EBV latency and is a target of EBV-specific CD8+ T cells. On the one hand, the amino terminus of EBNA1 contains a glycine–alanine repeat sequence structure that allows EBV to evade recognition by CD8+ cytotoxic T cells by blocking antigen presentation by MHC class I molecules (Tellam et al., 2004). On the other hand, EBV miRNAs can directly target the peptide transporter subunit of TAP2 and reduce the expression levels of TAP1 subunits, MHC I molecules, and EBNA1.
In addition, miRNAs can reduce IL-12 secretion and decrease the recognition of infected cells by EBV-specific CD8+ T cells (Albanese et al., 2016). EBV-encoded LMP1 and LMP2, which are important LMPs, are anchored in the lipid raft region of the cell membrane and participate in multiple intracellular signaling pathways by interacting with various signaling molecules, such as TRAF family proteins, caspase family proteins, and STAT family proteins in the host cell, which in turn affects cell migration and apoptosis. A current study confirms that LMP1 can inhibit the function of immune cells, allowing EBV to evade immune surveillance. T cell expresses costimulatory molecule CD137, whose ligand CD137L is expressed on APCs. LMP1 can induce CD137 production that is internalized by CD137L-expressing APCs, and the CD137-CD137L complex is subsequently degraded, thereby attenuating the CD137-mediated T cell immune response (Yoshimori et al., 2014).
EBV LMP2A can alter the gene expression of B cells and reduce the expression of MHC class II molecules by regulating the B cell–specific transcription factors E47 and PU.1, thus enabling immune evasion (Rancan et al., 2015; Westhoff Smith et al., 2021). The pDC can produce large amounts of type I IFN, which are the key immune cells for establishing immunity to most viral infections. EBV can infect pDC through viral binding to the MHC class II molecule HLA-DR. Although EBV can induce pDC activation, it cannot induce maturation, thus preventing pDC from producing a complete cellular response (Severa et al., 2013).
Interference with the release and activity of cytokines
EBV infection stimulates immune cells to produce a number of cytokines that act as messengers between cells and coordinate signal-dependent immune responses. EBV can stimulate immune escape by up- or downregulating the expression of relevant cytokines from the host cells. EBV infects immature pDCs and is able to induce activation but not the maturation of pDCs, leading to the impaired TNF-α secretion, thus preventing the host immune system from initiating a potent T cell immunity and enabling EBV to evade immune attack by T cells (Severa et al., 2013). EBV evades the host immune system by suppressing NK cell activity and reducing host cell apoptosis. EBV-infected cells express mi-RNAs, which can also interfere with cytokine release, promoting viral immune escape. NLRP3 inflammasome plays an important role in inflammatory responses, controlling the activation of cystatinase 1 in macrophages and promoting the release of IL-1β and IL-18.
miR-223 suppresses NLRP3 expression through a conserved binding site within the 3′UTR of NLRP3. EBV miR-BART15 targets the miR-223 binding sites in the 3′UTR of NLRP3 mRNA and thereby inhibits NLRP3 expression, which consequently inhibits IL-1β production (Haneklaus et al., 2012; Zamani et al., 2020). CXCL11, also known as “interferon-induced T-cell alpha chemokine,” is a cytokine belonging to the CXC chemokine family. It can selectively interact with CXCR3 receptor (C-X-C Motif chemokine receptor 3) expressed on T cells promoting the clearance of viruses by immune cells. miR-BHRF1–3 downregulates CXCL11 expression, thereby inhibiting the release of CXCL11 (Xia et al., 2008). Treg is a subtype of CD4+ T lymphocytes that inhibits the activity of cytotoxic CD8+ T cells.
Chemokine (C-C motif) ligand (CCL) 17 and CCL22 recruit Treg into the tumor microenvironment (TME) by activating the CCR4 chemokine receptor. In this process, LMP1 plays a central role in the direct upregulation of chemokines, which ultimately leads to immune evasion (Jorapur et al., 2022). The vIL-10 encoded by the BCRF1 gene of EBV, a viral homolog of human IL-10, can affect the antiviral immune response of the body (Sin and Dittmer, 2012). vIL-10 inhibits the secretion of pro-inflammatory cytokines such as IFN-γ and IL-2. vIL-10 impairs NK cell–mediated killing of infected B cells, interferes with CD4+ T cell activity, and modulates cytokine responses. At the beginning of EBV infection, it significantly reduces the immune activity of EBV-infected cells, which is conducive to the establishment of EBV latent infection (Jochum et al., 2012).
In summary, EBV has evolved with strategies to achieve long-term latency in their host or during lytic proliferation. EBV evades immune surveillance by inhibiting the expression of TLRs, regulating numerous signaling pathways, reducing host cell apoptosis, affecting antigen presentation, and inhibiting the cytotoxic activity of effector cells, all of which collectively contribute to the malignant transformation of EBV-infected host cells. An in-depth understanding of EBV-specific immune responses of the host and EBV-mediated host immune evasion is important in understanding the reactivation mechanism of EBV in the host cells and the induction of malignancy, as well as in providing a theoretical basis for the later development of vaccine and immunotherapeutic strategies.
Footnotes
Authors' Contributions
Y.Y. collected and analyzed related articles, then wrote the article. W.K. and L.Y. collected related articles. Y.D. supervised data and article. H.C. supervised analysis of related articles and revised writing of the article. All authors read and approved the final version of the article.
Author Disclosure Statement
The authors have no competing interests.
Funding Information
The authors did not receive support from any organization for the submitted work.