
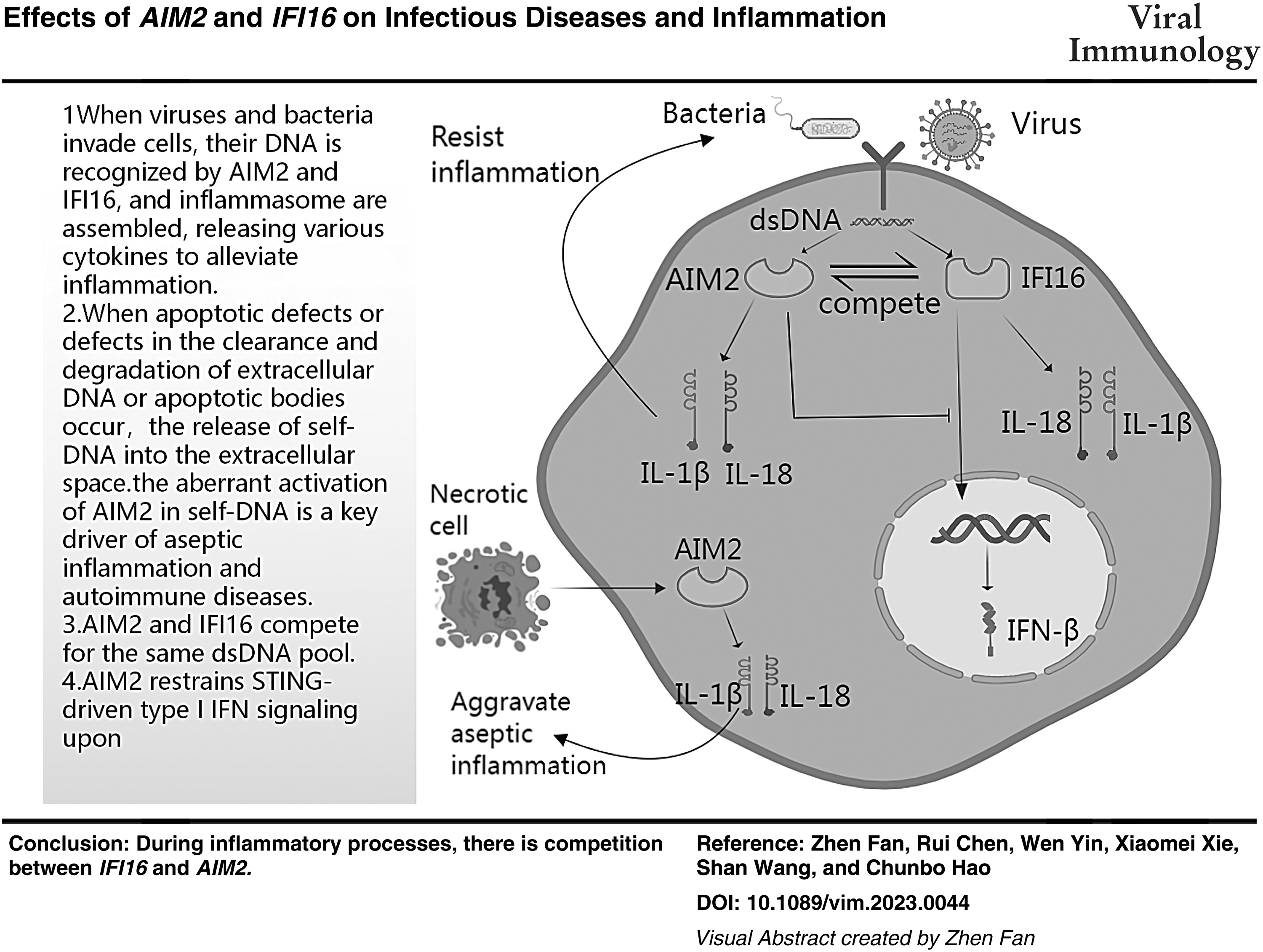
Abstract
Both absent in melanoma 2 (AIM2) and interferon-inducible protein 16 (IFI16) are intracellular innate immune receptors that recognize double-stranded DNA released during pathogenic infection, leading to the assembly of the inflammasome. The assembly of the inflammasome results in the secretion of bioactive interleukin (IL)-1β and IL-18 and induces cell death through an inflammatory process called pyroptosis. Although the AIM2 inflammasome is generally harmful in the context of some aseptic inflammatory illnesses, it plays a protective role in infectious diseases. During inflammatory processes, there is competition between IFI16 and AIM2. In this review, we explore the impacts of IFI16 and AIM2 in infectious disease and aseptic inflammation, respectively, and how they compete.
Introduction
The innate immune system is the body's initial line of defense against the presence of intruders; this system initiates processes to eliminate the risk of infection. Pattern recognition receptors (PRRs) (Table 1), a class of receptors that are widely present on the surface of innate immune cells, the cytoplasm, and the nucleus that can directly recognize foreign pathogens and their products or certain molecular patterns commonly expressed in aberrant and senescent cells in the host, are responsible for microbial sensing.
List of Abbreviations
AIM2, absent in melanoma 2.
Based on protein domain homology, most PRRs may be categorized into five families: Toll-like receptors (TLRs), C-type lectin receptors (CLRs), nucleotide binding domains, nucleotide binding leucine-rich repeat (LRR) receptors (NLRs), RIGI-like receptors (RLRs), and absent in melanoma 2 (AIM2)-like receptors (ALRs). These families are split into two major categories: cell membrane receptors and intracellular receptors that are not linked to the cell membrane. TLRs and CLRs, which are found on the cell surface or in endosomes, are members of the first class. These receptors bind to microbial ligands in the extracellular space and endosomes. The latter category includes NLRs, RLRs, and ALRs, which are found intracellularly and probe the existence of intracellular pathogens (Brubaker et al., 2015).
In humans and mice, 4 and 13 AIM2-like receptors (ALRs) are expressed, respectively; only interferon-inducible protein 16 (IFI16) and AIM2 act as inflammasomes, recognizing cytoplasmic and nuclear DNA from pathogens and injured cells (Li et al., 2021).
ALR Family
Absent in melanoma 2
AIM2 was found in a melanoma tumorigenicity suppressor screen after chromosome 6 was introduced into the UACC903 human melanoma cell line (Gariglio et al., 2011). The AIM2 molecule contains 337 amino acid residues and has the typical structural characteristics of PYHIN family proteins. The N-terminus contains the pyrin domain (PYD), and the C-terminus contains the HIN-200 domain (Cresswell et al., 2005).
The PYD domain is a death fold structure comprising six independent spiral bundles. This shape is widespread in proteins involved in apoptosis or inflammation, and it promotes protein-protein interactions that result in the creation of massive signaling complexes. AIM2's PYD domain connects directly to ASC's PYD domain. The interaction of PYD domains of these two proteins results in the formation of helical filaments, allowing the AIM2 inflammasome to oligomerize. Two successive oligosaccharide/oligonucleotide binding folds (OB folds) in the HIN-200 domain bind double-stranded DNA (dsDNA). The OB fold includes 70–80 amino acid residues that form a five- or six-stranded β-barrel. The strands are linked by loops of varied lengths that create the protein's ligand-binding domain. The OB fold has the ability to bind oligosaccharides, oligonucleotides, proteins, and metal ions (Kumari et al., 2020).
The AIM2 inflammasome senses DNA in the cytoplasm and plays an important role in host defense against bacterial and viral diseases. AIM2 is expressed in the cytoplasm, where it interacts with microbes and its dsDNA in a sequence-independent manner. The PYD domain of AIM2 can induce activation through self-aggregation. Following pathogenic stimulation, host DNA is released into the cytoplasm, resulting in decreased membrane integrity, which activates AIM2-dependent innate immunity. Microbial DNA and pathogen-associated molecular patterns (PAMPs) are released into the cytoplasm of host cells during microbial infection and recognized by particular PRRs.
AIM2 identifies DNA from intracellular pathogens, such as viruses and bacteria, in addition to host DNA. AIM2-dependent effector cytokines and pyroptosis help protect the host against microbial invasion (Sharma et al., 2019). There are now two known activation routes for the AIM2 inflammasome: “canonical” and “noncanonical.” The binding of DNA to AIM2 to induce inflammasome assembly is referred to as “canonical” activation since it is quick and does not need type I interferon (IFN) activity. During most bacterial infections, “noncanonical” activation occurs when intracellular bacteria escape vacuoles and expose small amounts of DNA, resulting in the activation of cyclic GMP-AMP synthase and IFI204, which together drive the activation of type I IFNs.
Following activation, type I IFNs are secreted extracellularly and bind to the type I IFN receptor in an autocrine manner, leading to the expression of GBPs and IRGB10 downstream through the type I IFN/IRF1 axis. Bacterial/tonoplast rupture caused by GBP2-, GBP5-, and IRGB10-induced bacteriolysis leads to the release of vast quantities of bacterial DNA, which AIM2 eventually recognizes. In conclusion, AIM2 inflammasome assembly results in proteolysis and the activation of interleukin (IL)-1β, IL-18, and GSDMD. GSDMD forms pores on the cell membrane to secrete IL-1β/IL-18 (Fig. 1).
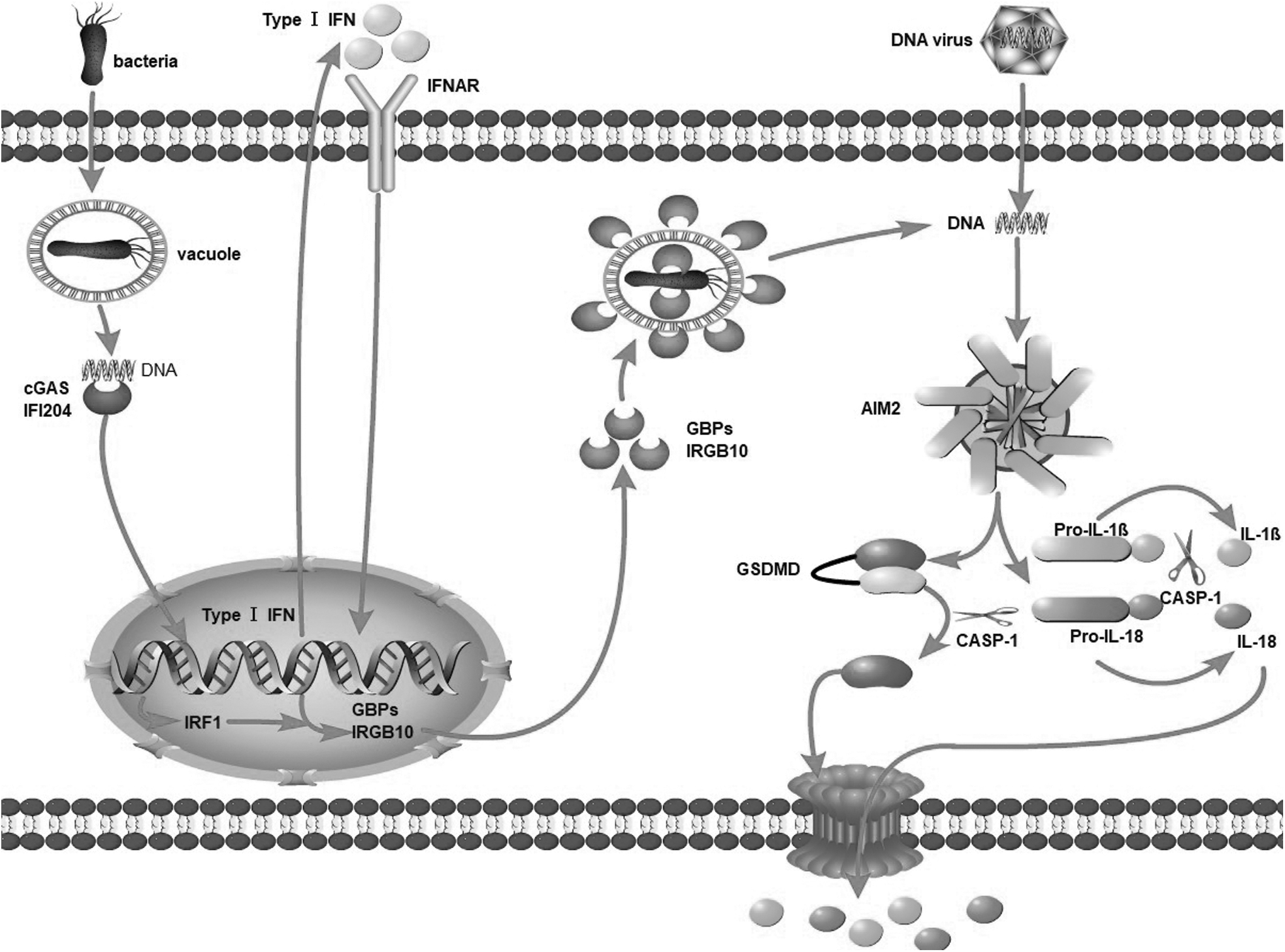
Inflammasome activation through the AIM2 pathway: (1) The canonical activation pathway: Viral DNA enters cells, is immediately identified by AIM2, and causes the AIM2 inflammasome to become active. (2) Noncanonical activation pathway: Intracellular bacteria escape vacuoles and expose a small amount of DNA, resulting in the activation of cGAS, which drives the activation of type I IFNs. This causes the production of GBP2/GBP5 and IRGB10 and ruptures the bacterial/tonoplast, exposing large amounts of DNA to AIM2. GSDMD and IL-1β/IL-18 are activated proteolytically as a result of the AIM2 inflammasome being assembled. GSDMD creates breaches in the cell membrane that promote IL-1β and IL-18 secretion. AIM2, absent in melanoma 2; cGAS, cyclic-GMP-AMP synthase; GBP, guanylate-binding protein; GSDMD, gasdermin D; IFN, interferon; IFNAR, IFN α and β receptor; IFI204, IFN activated gene 204; IL, interleukin; IRF1, IFN regulatory factor 1; IRGB10, immunity-related GTPase family member b10.
The pathway that is activated downstream of AIM2 DNA induction, whether by “normative” or “non-normative” activation, includes the creation of an inflammatory complex and caspase-1 activation, and the maturation of IL-1β/IL-18 and GSDMD.
The AIM2-ASC complex can activate caspase-8, resulting in apoptosis. Although AIM2 can detect the DNA associated with many diseases, it cannot differentiate microbial DNA from host DNA due to the lack of sequence specificity, which determines whether it plays a beneficial or detrimental function in the host ( Wang et al., 2020; Wang et al., 2019; Zhu and Lu, 2016).
Interferon-inducible protein 16
IFI16 belongs to the interferon-inducible protein family. The N-terminus of the IFI16 molecule contains a PYD domain, and the C-terminal HIN200 domain has 2 OB fold regions, which mediate the recognition and binding of DNA. The IFI16 protein interacts with the ASC protein through the PYD to generate a functioning IFI16-ASC inflammasome. The presence of a PYD domain within the protein's N-terminus implies that it participates in the apoptotic pathway by regulating the activity of particular transcription factors in the nucleus, which are implicated in cell death. The HIN-200 domain of the IFI16 molecule can recognize both dsDNA and single-stranded DNA (ssDNA).
IFI16 is widely distributed in the nucleus and cytoplasm and not only acts as a nuclear DNA sensor but also serves as a cytoplasmic DNA sensor (Johnstone et al., 1998; Kumari et al., 2020;). IFI16 was previously shown to induce inflammasome activation in human cells in response to Kaposi's sarcoma-associated herpesvirus infection. Mechanistically, IFI16 appears to directly recognize dsDNA in the nucleus to form inflammasome complexes, which then translocate into the cytoplasm, where IFI16 mediates pyroptosis through the activation of caspase-1 (Mondini et al., 2010) (Fig. 2). In addition, IFI16 can also mediate the expression of IFN through the STING pathway.
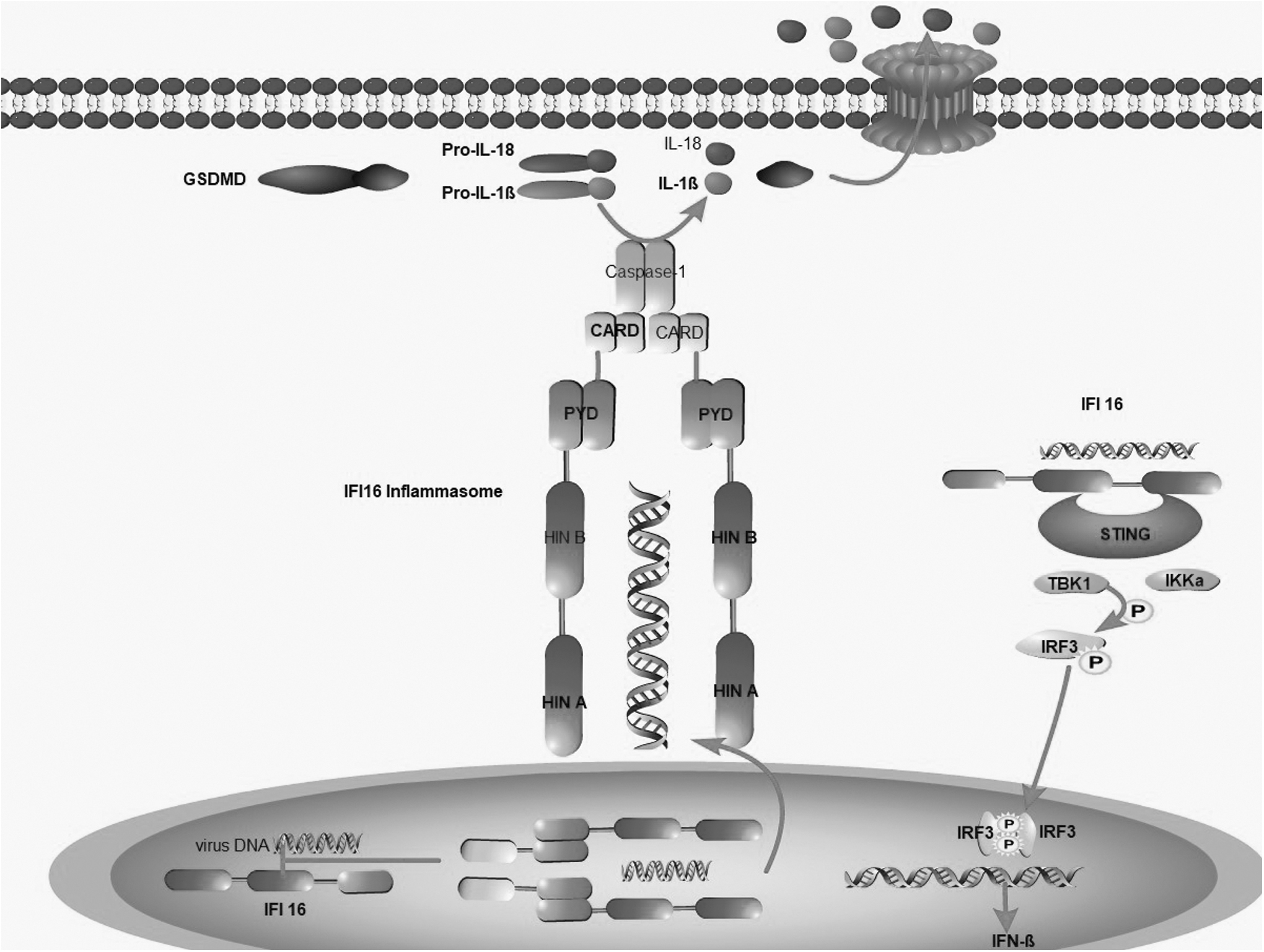
IFI16 inflammasome activation pathway: (1) Viral DNA is recognized by IFI16 in the nucleus, allowing the assembly of the IFI16 inflammasome, which is then translocated to the cytoplasm, causing the proteolytic activation of GSDMD and IL-1β/IL-18. GSDMD creates breaches in the cell membrane that promote IL-1 and IL-18 secretion. (2) After IFI16 recognizes exogenous dsDNA, it binds to STING, recruits TBK1, and activates NF-κB and IRF3, which in turn go into the nucleus to trigger IFN-β production. dsDNA, double-stranded DNA; GSDMD, gasdermin D; IFI16, interferon-inducible protein 16; IFN, interferon β; IRF3,interferon regulatory factor 3; IKKα, inhibitor of kappa B kinase α; STING, stimulator of interferon genes; TBK1, recombinant tank binding kinase 1; NF-κB, nuclear factor kappa-B.
Studies have shown that most STING-dependent DNA sensors rely on the STING-TBK1-IRF3 signaling pathway to transmit immune signals and induce the expression and secretion of IFN. After IFI16 recognizes exogenous dsDNA, IFI16 binds to the endoplasmic reticulum protein STING, thereby recruiting TBK1 molecules and activating transcription factors such as nuclear factor kappa-B (NF-κB) and IRF3. The entry of NF-κB and IRF3 into the nucleus can induce the expression of IFN-β (Cresswell et al., 2005) (Fig. 2).
AIM2 and IFI16 compete with each other
AIM2 was the first cytoplasmic member of the ALR family to be recognized as an innate immune sensor, recognizing dsDNA in a sequence-independent manner. IFI16 can bind to both dsDNA and ssDNA (Briard et al., 2020). Wang et al. described IFI16-β, a new transcript isoform of human IFI16 with a domain architecture similar to mouse p202. IFI16-β, like p202, has two HIN domains, but no PYD. IFI16-β is located mostly in the cytoplasm. In HeLa cells, IFI16-β basically colocalizes with ectopically produced AIM2 or with endogenous AIM2 in differentiated THP-1 cells. IFI16-β interacts with and activates STING, resulting in type I IFN production.
Importantly, they all compete for the same dsDNA pool. Therefore, the signaling strength of a single pathway depends on the expression level of the sensor and its binding affinity for cytoplasmic dsDNA. Wang et al. discovered that biotinylated dsDNA90mer pulls down both AIM2 and IFI16-β, with IFI16-β having a higher affinity for dsDNA than AIM2. IFI16-β may compete with AIM2 for dsDNA binding in cells. Furthermore, IFI16-β chelates cytoplasmic dsDNA, rendering it unreadable by AIM2 (Wang et al., 2018).
Veeranki et al. found that the expression of AIM2 and IFI16 was induced by treating cultured human monocytes with type I or type II IFN. AIM2 and IFI16 were mainly detected in the cytoplasm. IFI16 protein binds to part of the AIM2 protein in the cytoplasm and prevents the formation of the AIM2-ASC complex. In addition, increased IFI16 protein levels inhibited AIM2-ASC inflammasome-mediated activation of caspase-1 (Veeranki et al., 2011). Similarly, Wang et al. corroborated the previous results and discovered that forced expression of either IFI16-β resulted in a decrease in ASC being identified in the AIM2 immunoprecipitates.
Furthermore, IFI16-β inhibits AIM2 inflammasome activation in a dose-dependent manner, and knocking down IFI16-β increases IL-1β secretion induced by dsDNA, but not dsRNA (Wang et al., 2018). The defective expression of IFI16 or defective protein function may suppress anti-inflammatory responses, thereby increasing proinflammatory responses (Marchesan et al., 2017). Yan et al. found that AIM2 deficiency increases STING-driven type I IFN signaling and promotes increases in the level of phosphorylated TBK1 and IRF3. The enhancement effect depends on the concentration of IFN-β mRNA and DNA.
It is worth mentioning that STING-TBK1-IRF3 pathway is the downstream signal pathway of IFI16 inflammatory body activation (Yan et al., 2018) (Fig. 3). vAIM2 and IFI16 are negative regulators of each other. However, AIM2 and IFI16 play different roles in different diseases. These regulators may cooperate with each other to control the activation of AIM2 or IFI16 to prevent the exacerbation of severe pathogenic inflammation. Understanding this process will help us better understand how the negative regulation of the activation of inflammasomes by AIM2 and IFI16 may reveal new strategies for anti-inflammatory treatment.
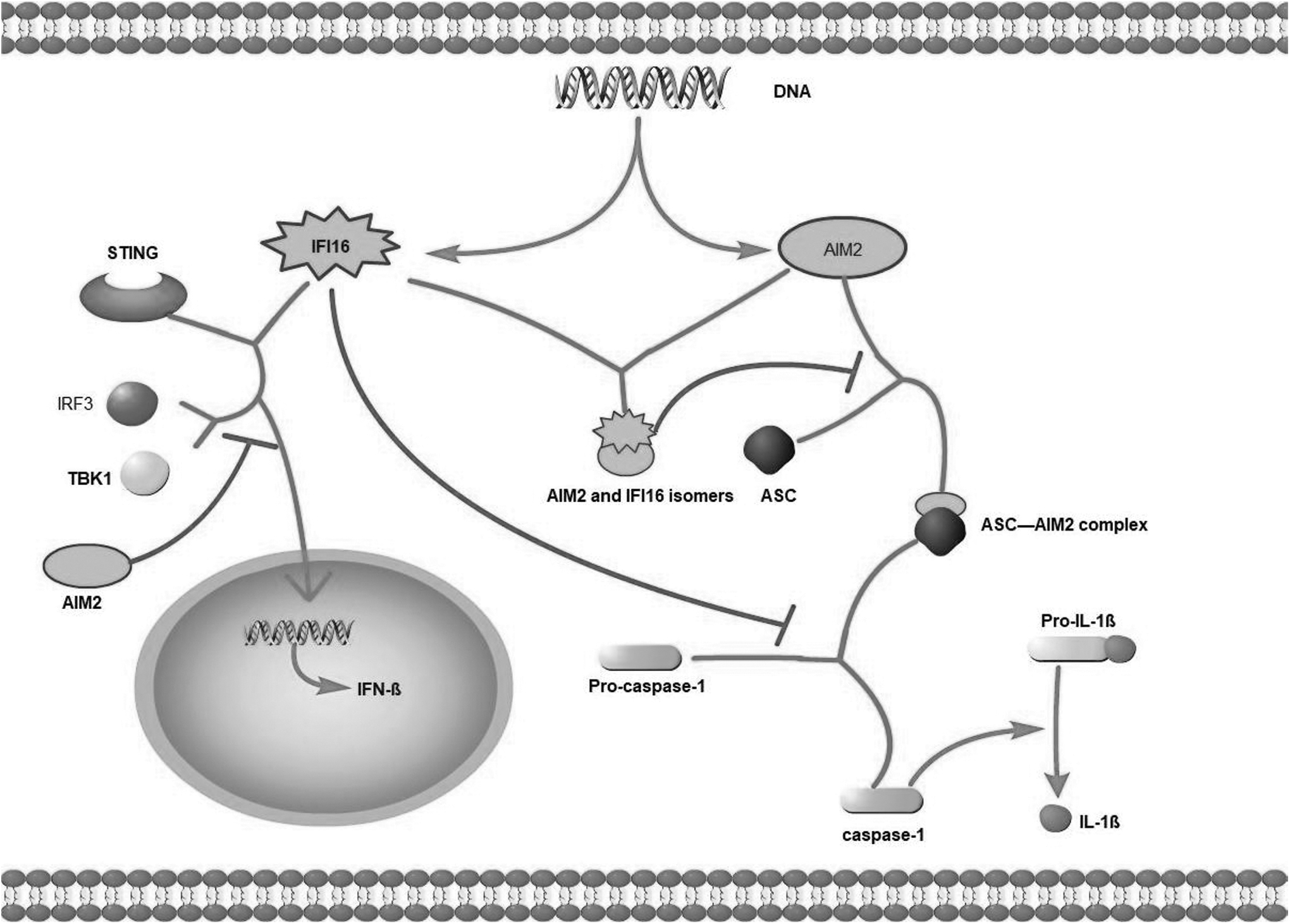
AIM2 and IFI16 compete. (1) AIM2 and IFI16 compete for the same dsDNA pool. (2) IFI16 protein binds to part of the AIM2 protein in the cytoplasm and prevents the formation of the AIM2-ASC complex. (3) IFI16 protein inhibited AIM2-ASC inflammasome-mediated activation of caspase-1. (4) AIM2 restrains STING-driven type I IFN signaling; moreover, phosphorylated TBK1 and IRF3, which are the activated forms of both molecules, are inhibited by AIM2.
The Role of AIM2 and IFI16 in Infectious Diseases
Herpes simplex virus infection
The herpes virus family includes many DNA viruses that may cause infection when the immune system is compromised. This family is further classified into three subfamilies: α-, β-, and γ-herpesviruses. Herpes simplex virus types 1 (HSV-1) and 2 (HSV-2) are members of the α herpes virus subfamily in the genus HSV. They are neurotropic viruses that infect the trigeminal and dorsal root ganglions. HSV seropositivity is quite high. HSV-1 infection can cause a variety of skin and mucosal illnesses, including cold sores, keratitis, and herpetic vitiligo. HSV-2 infection mainly results in genital ulcers (Zhao et al., 2021).
The role of AIM2 and IFI16 in HSV infection
When HSV-1 DNA binds to IFI16 after infection, IFI16 can promote antiviral cytokine production and suppress virus gene expression. During HSV-1 infection, the viral capsid attaches to the nuclear pore complex, allowing the dsDNA genome to be injected into the nucleus, after which IFI16 moves from the nucleolus to the periphery of the nucleus and interacts with vDNA through the HIN domain, resulting in PYD-dependent oligomerization.
The combination of IFI16 and vDNA is required for the production of antiviral cytokines (Howard et al., 2022); in addition, this binding restricts viral gene expression. IFI16 forms a nuclear filament structure on DNA after HSV DNA replication in human cells infected with HSV-1 mutant viruses lacking the ICPO protein (the ICP0 protein promotes the degradation of PML and IFI16 proteins and inhibits their restriction functions). IFI16 filaments prevent RNA polymerase II (Pol II) from binding to viral genes. The IFI16 filaments and related restriction factors create a “restriction body” structure that communicates to other areas of the nucleus where foreign DNA is present, implying that it should be silenced (Merkl and Knipe, 2019).
Johnson et al. found that during the early (2 to 4 h) infection of human foreskin fibroblasts infected with HSV-1, HSV-1 activated the IFI16 inflammasome and triggered IL-1β maturation, which is unrelated to viral gene expression. IFI16 relocalizes and colocalizes with ASC in the cytoplasm after recognizing the HSV-1 genome in the nucleus of infected cells. During late infection, however, HSV-1 preferentially targets IFI16 for fast proteasomal breakdown (Johnson et al., 2013). The envelope protein VP22 of HSV-1 inhibits AIM2-dependent inflammasome activation. VP22 binds to AIM2 and prevents it from oligomerizing, which is the first stage in AIM2 activation. VP22 promotes HSV-1 replication in vivo by inhibiting AIM2-dependent inflammasome activation (Maruzuru et al., 2020; Maruzuru et al., 2018).
Viral hepatitis
Research on viral hepatitis revealed two unique infection modes: infectious hepatitis (Hepatitis A) and serum hepatitis (Hepatitis B). Hepatitis A virus is a positive RNA virus of the Hepaviridae family with four human-specific genotypes (Abutaleb et al., 2020). Hepatitis A is an acute liver infection that is mostly asymptomatic in children and progressively worsens with age (Pintó et al., 2021). Hepatitis B virus (HBV) infection is a leading cause of liver cirrhosis and hepatocellular cancer. Despite the availability of vaccines to prevent HBV infection, ∼350 million people are still infected with HBV. Much evidence suggests that HBV interacts with the innate immune signaling pathway of hepatocytes and inhibits innate immunity (Megahed et al., 2020).
The role of AIM2 and IFI16 in viral hepatitis
To date, the mechanisms underlying chronic development after acute HBV infection remain to be fully elucidated. During early infection, the HBV virus is termed a stealth virus because it generates few host innate immune responses. HBV is a relaxed circular DNA (rcDNA) molecule with two strands. When HBV penetrates hepatocytes, the rcDNA is carried to the nucleus and transforms into covalently closed circular DNA (cccDNA, a determinant of HBV persistence and a critical hurdle to developing a cure for chronic hepatitis B [CHB]), which serves as the virus's transcriptional template. IFI16 can identify and interact with HBV cccDNA in the hepatocyte nucleus, target the ISRE of cccDNA (ISRE is important in IFN-α-mediated epigenetic alteration of cccDNA), and combine innate immune activation and epigenetic control to prevent cccDNA transcription and HBV replication (Yang et al., 2020).
Chen et al. discovered that AIM2 and IFI16 were highly expressed in the blood of patients with CHB and acute hepatitis B (AHB); however, only AHB patients had considerably higher IL-1β and IL-18 levels. In a subsequent investigation, AIM2 and IFI16 mRNA levels in a CHB group were shown to be significantly positively linked with blood HBV DNA load. In contrast, Hepatitis B e antigen (HBeAg) levels were adversely linked with IFI16 mRNA levels. Both acute and chronic HBV infection resulted in the formation of AIM2 and IFI16 inflammasome complexes, and HBeAg inhibited their activation without affecting AIM2, IFI16, or CASP1 mRNA expression (Chen et al., 2018).
HBeAg may suppress the activation of IFI16 and AIM2 inflammasomes by attenuating the phosphorylation of NF-κB and inhibiting the activation of the NF-κB signaling pathway. AIM2 is most abundant in CHB patients during the immunological clearance phase of the illness, when the immune system is aggressively combating infection and immune-mediated liver damage develops. During HBV infection, AIM2 may aid in the immunological clearance of HBV in immune cells. However, AIM2 expression in hepatocytes may be implicated in the inflammatory damage associated with HBV infection (Lozano-Ruiz and González-Navajas, 2020).
Bacterial infections
Periodontitis
Periodontitis is an inflammatory illness caused by an infection that is characterized mostly by gingival inflammation and bone loss. Recent hypotheses ascribe periodontitis development to oral microbiota dysbiosis, in which Porphyromonas gingivalis plays a considerable role by altering host immunological homeostasis. Porphyromonas gingivalis employs virulence factors such as lipopolysaccharides and others to promote bacterial colonization and the expansion of the surrounding microbiota. By altering the host's immunological homeostasis, these virulence agents can evade clearance and induce an inflammatory environment (Xu et al., 2020). Two factors affect the integrity of periodontal support tissues: tooth biofilm and occlusal trauma. Occlusion is a major factor affecting periodontal support tissue, and analyses suggest that traumatic forces may promote the apical spreading of oral biofilms and inflammatory exudates, leading to periodontal pocket deepening, attachment loss, and bone loss (Passanezi and Sant'Ana, 2019).
Periodontitis risk factors include poor nutrition, mental stress, and being overweight. Lifestyle decisions might indirectly affect the immune system and cause inflammation in the periodontal tissues. Ultimately, this makes gingivitis and periodontitis more likely to develop (Haverkort and Laleman, 2021). After cells are triggered by damage-associated molecular patterns (DAMPs) and PAMPs, IL-1β is first generated as inactive pro-IL-1β during the development of periodontal disease. These molecules control the routes for gene expression through the action of PRRs on the cell membrane. By stimulating endothelial cells and enabling eosinophil adhesion, IL-1β heightens the inflammatory response. In addition, IL-1β controls the loss of alveolar bone by fostering the growth and activity of osteoblasts (Aral et al., 2020b).
The role of AIM2 and IFI16 in periodontitis
In 2020, Wen et al. further validated the evidence for the role of AIM2 as a risk factor of periodontitis by integrating the genetic association of large-scale periodontitis genome-wide association scans (GWAS) and expression quantitative trait Loci data. In addition, individuals with periodontitis have markedly higher AIM2 levels in gingival tissue than healthy individuals, and genes that coexpress AIM2 are markedly enriched in pathways that are related to the immune system and inflammation.
This evidence supports the involvement of AIM2 in the formation of periodontitis (Li et al., 2020). According to a study by Aral, patients with chronic periodontitis had considerably lower levels of AIM2 than patients with aggressive periodontitis and healthy individuals. The results suggest that chronic periodontitis may be associated with AIM2 downregulation and that AIM2 downregulation may negatively affect periodontal tissue (Aral et al., 2020a).
However, Park et al. reported that compared with that in the gingival tissue of healthy subjects, patients with chronic periodontitis have considerably higher AIM2 expression in their gingiva (Park et al., 2014). Currently, the specific mechanism of action of AIM2 in periodontitis is still unclear, and further exploration is needed. In a GWAS of 4910 European Americans, Marchesan et al. found that IFI16 and AIM2 single-nucleotide polymorphisms were linked to greater amounts of periodontal bacteria and a large number of clinical markers for periodontal disease (Marchesan et al., 2017). It was discovered that P204, the mouse equivalent of IFI16, has the same domain structure as IFI16; this PYHIN protein similarly has one pyrin and two HIN domains.
In addition, a Basic Local Alignment Search Tool comparison of the murine PYHIN protein with human IFI16 indicated that p204 shares the highest amino acid similarity with IFI16 (37%), and both P204 and IFI16 have a similar DNA-binding mechanism (Fan et al., 2021; Unterholzner et al., 2010). In a recent study, a ligation-induced periodontitis model was established in IFI204 knockout mice and AIM2 knockout mice, and it was found that IFI204 knockout mice had more loss of alveolar bone than did the healthy controls and that the deletion of IfI204 resulted in an increase in the expression of osteoclast-activating factor IL-1β in gingival tissue, thereby increasing the number of osteoclasts in the periodontium (Swanson et al., 2022).
The expression of IFI16 was also dramatically higher early during the experiment after the authors induced experimental periapical periodontitis in mice; the control group showed the lowest expression. In contrast, IFN-α/β receptors were strongly expressed during later phases of experimental development, and there were more TRAP-positive cells (Pucinelli et al., 2022). The above results further support the role of IFI16 as a novel regulator of periodontal tissue that prevents bone loss.
The relationship between periodontitis and aseptic inflammation
Periodontitis is chronic inflammation caused by an imbalance of oral microflora and is a risk factor for aseptic inflammation. Through overflowing periodontal pockets and blood-borne diffusion, periodontal bacteria can produce systemic inflammation. Moreover, they can impact the metabolic state by pushing the threshold for systemic inflammation (Hajishengallis and Chavakis, 2021). The two primary pathogenic bacteria that cause periodontitis are Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans.
According to research, Pseudomonas gingivalis makes a specific peptidyl arginine deaminase that citrullinates proteins and has the potential to raise the likelihood of anti-cyclic citrullinated peptide formation. It may operate as a potent antigen and break host immune tolerance as a bacterial self-citrullinated protein to cause rheumatoid arthritis (RA) clinical symptoms. Similarly, Aggregatibacter actinomycetemcomitans can cause host neutrophils to become hypercitrullinated, which can cause the development of antibodies against citrullinated proteins, membrane damage in neutrophils, and unregulated citrullinase activation, all of which can culminate in the development of RA (González-Febles and Sanz, 2021). Studies have shown that through small fimbriae, Porphyromonas gingivalis targets and enters dendritic cells derived from monocytes and spreads to atherosclerotic plaques and other distant sites with dendritic cells, thereby triggering inflammation (El-Awady et al., 2022).
Another study found that periodontitis can trigger atherosclerosis (AS) and vascular inflammation by activating monocytes/macrophages to induce NF-κB activation and vascular cell adhesion molecule 1 (VCAM-1) expression (Miyajima et al., 2014). Furthermore, effective periodontal therapy reduces systemic inflammatory markers and reduces blood glucose and glycated hemoglobin in diabetic patients, while improving vascular and renal functions (D'Aiuto et al., 2018).
The Role of AIM2 and IFI16 in Aseptic Inflammation
DNA often resides in nuclei and mitochondria. Inflammation can result from the persistence of factors produced by the host when extracellular DNA is cleared and degraded or apoptotic bodies show abnormalities or faults in apoptosis. Other cell death processes result in the release of host DNA into the extracellular environment, where it can be ingested and detected by endosome or cytoplasmic nucleic acid sensors. Nuclear chemicals also function as inflammatory stimuli outside cells by obliterating the membranes of nearby cells (Lupfer et al., 2017).
IFI16 is a multifunctional protein that serves as a nuclear pathogenic DNA detector that is activated by a variety of proinflammatory cytokines. In addition, autoantibodies are drawn to it. Numerous data have demonstrated disease-specific mislocalization of nuclear IFI16 in inflammatory environments, where IFI16 dissociates from the nucleus to the cytoplasm after transfection with virus-derived DNA or after treatment with UV light, and is then finally released into the extracellular environment.
When extracellular IFI16 binds to particular cell surface receptors, it produces a series of effects. This action is blocked when the PYD domain on IFI16 is covered by an anti-ifi 16-N-terminal antibody. Due to its function, IFI16 is a potentially harmful endogenous dislocation molecule, much as DAMPs, which is crucial for the emergence of chronic inflammation and autoimmune disease (Bawadekar et al., 2015; Gugliesi et al., 2013;). Similarly, a major contributor to aseptic inflammation and autoimmune disorders is the abnormal activation of AIM2 in self-DNA. According to studies, self-DNA buildup causes skin cells to produce IL-1β in an AIM2-dependent manner, which causes psoriasis (Dombrowski et al., 2011; Man et al., 2016).
Diabetes
Diabetes is a major worldwide health problem. High blood sugar levels brought on by inadequate insulin synthesis by the pancreas define type 2 diabetes mellitus (T2DM), a metabolic condition. Insulin resistance, which is defined as a state of reduced responsiveness of insulin-targeting tissues to physiological levels of insulin, is a key pathogenic component of many metabolic diseases, including T2DM. Because some types of tissues cannot respond to normal insulin levels, higher than normal levels of insulin are needed to maintain normal functions of insulin (Lee et al., 2022). The macrophages, elevated blood glucose levels, and inflammatory mediators produced by adipose tissue all play a role in the inflammatory response. Due to the pancreatic beta cells being damaged by this low-grade, ongoing inflammation, there is inadequate insulin production, which causes hyperglycemia (Berbudi et al., 2020).
The role of AIM2 and IFI16 in diabetes and its complications
One of the main risks for T2DM is being overweight. Mandy et al. found that mice overexpressing IFI202b developed significantly increased body weight and showed liver tissue did not contain as much IFI202b as adipose tissue did. IFI202b appears to decrease insulin sensitivity since it increases glucose absorption in adipose tissue, while insulin inadequately inhibits lipolysis stimulation in mouse explants.
Like IFI202b in mice, IFI16 has been shown to be involved in human adipogenesis and adipose tissue inflammation. The results indicated that IFI16 could function as a diabetes gene (Stadion et al., 2018). Diabetic cardiomyopathy is also one of the complications of diabetes. Inflammasome AIM2 and IL-1β levels were shown to be considerably higher in the cardiac tissue of diabetic rats, according to Wang et al. Cardiac function in diabetic rats improved when AIM2 expression was suppressed, indicating that AIM2 may be triggered in diabetic rats to hasten the development of diabetic cardiomyopathy (Wang et al., 2019).
Rheumatoid arthritis
RA is a complex autoimmune disease that first affects the joints, but has an unclear cause. The synovium is invaded by proliferating synovial fibroblasts, neutrophils, monocytes, and lymphocytes, which results in chronic joint inflammation and joint degeneration. RA is thought to be primarily caused by genetic and environmental factors; however, the exact pathophysiology of RA is currently unknown (Edilova et al., 2021). Emerging research over the past two decades has demonstrated that the innate immune system is crucial to the onset and development of RA. In RA patients, innate immune cells are engaged in the inflammatory response and induce adaptive immune system activation.
The role of AIM2 and IFI16 in RA
Chen et al. found that AIM2 in fibroblast-like synovial (FLS) cells was upregulated in RA and that inhibiting its expression inhibited FLS cell proliferation, suggesting that the AIM2 inflammasome pathway may promote the occurrence of RA (Chen et al., 2020). Alunno et al. discovered that IFI16 protein and anti-IFI16 antibody concentrations in RA patients' synovial fluid were greater than in control arthritis patients. Serum IFI16 and anti-IFI16 antibody levels were greater in RA patients than in healthy controls, and there was a clear link between IFI16 concentration and anti-IFI6 antibody titer. RA patients exhibited greater levels of circulating IFI16 protein than healthy controls, and they also tested positive for rheumatoid factor/anti-cyclic citrullinated peptide antibody. These findings imply that IFI16 may be involved in the pathophysiology of RA (Alunno et al., 2016).
Atherosclerosis
AS, the common pathological basis of many cardiovascular diseases, is characterized by the accumulation of lipid-rich, immune cell plaques in large- and medium-sized arteries, termed atherosclerotic aneurysm. AS occurrence begins with endothelial dysfunction in arterial susceptible areas and is characterized by blood flow disturbances, where chemokine signaling and the expression of adhesion molecules trigger a cascade of signaling within immune cells that lead to the nucleation of lesions and thus the formation of atherosclerotic plaques (Roy et al., 2022).
The role of AIM2 and IFI16 in AS
AS has been shown to result from chronic inflammation caused by the constitutive activation of PRRs. Using small interfering RNAs targeting IFI16, Hao et al. found that IFI16 expression was upregulated in senescent primary human aortic smooth muscle cells (HASMCs) and that the proliferation and migration of HASMCs decreased. Moreover, metformin was able to activate IFI16, thereby inhibiting the proliferation and migration of HASMCs. When IFI16 was knocked out, metformin could not inhibit the proliferation and migration of HASMCs (Hao et al., 2018). It has been shown that IFI16 effectively inhibits the proliferation and migration of HASMCs, thereby inhibiting the development of AS.
Lüsebrink et al. found that the activation of AIM2 in mice induced a systemic inflammatory response, interfered with endothelial cell regeneration after injury, increased plaque formation, and led to the accumulation of macrophages in aortic plaques. The findings revealed that AIM2 expression aided in the formation of AS (Lüsebrink et al., 2020). Pan et al. found that the area of aortic atherosclerotic lesions increased when AIM2 was overexpressed in mice and that the number of smooth muscle cells increased with increasing AIM2 levels. In vitro studies have found that AIM2 mediates the migration of vascular smooth muscle cells (VSMCs) (Pan et al., 2018). In summary, AIM2 can mediate the migration of VSMCs, leading to plaque formation and AS and inhibits endothelial cell regeneration after injury.
Summary and Outlook
The literature supports that PRRs play a considerable role in the body's innate immune system, and therefore, manipulation of its components has proven to be therapeutically beneficial. Kim et al. discovered that Aggregatibacter actinomycetemcomitans stimulates IL-1β release through the AIM2 inflammasome and that xylitol inhibits this activity. This finding shows that xylitol might be utilized to treat aggressive periodontitis in the future (Kim et al., 2016). Zhang et al. established a periodontitis model in mice and locally injected super activated platelet lysate (SPL). SPL-injected animals had much lower AIM2 expression than the control group, and the level of alveolar bone loss in these animals was dramatically lower. The findings revealed that SPL controlled the expression of inflammasomes and cytokines and had a clear anti-inflammatory impact on rats with experimental periodontitis (Zhang et al., 2020).
Recent research has revealed that IFI204 can prevent alveolar bone loss in periodontitis mice, suggesting that it could be an option for the creation of novel periodontal diagnostics and treatment medicines. According to research by Tao et al., tissue-resident memory CD8 T cells (CD8TRM) detect viral antigen and secrete IFN-γ, which causes the production of a number of interferon-stimulating genes in the cells around them, including IFI16. IFI16 can both inhibit the reproduction of the wild-type HSV-1 virus and the HSV-1 mutant ICPO-virus at the overexpressed level and at the basic level. This connection between innate and adaptive immunity may be a crucial immunological pathway to regulate immunity and herpes (Peng et al., 2021). A unique method of treating AS is made possible by the ability of metformin to activate IFI16 and prevent the onset of AS.
Based on the IFI16 protein's capacity to detect cytosolic DNA, innate immune responses are either triggered to create IFN-β (through the STING-TBK1-IRF3 axis) or to generate the IFI16 inflammasome. In the future, it will be important to determine whether we can control the activation of AIM2 or IFI16 to regulate the other process to develop a new anti-inflammatory treatment strategy for diseases. Because these two approaches will have different effects on diseases and because AIM2 and IFI16 are negative regulators of one another, this will be important. Future research will need to clarify the inflammasome pathway's function as a target for disease therapy.
Footnotes
Acknowledgments
The authors would like to acknowledge the support of the School of Stomatology, Hainan Medical University, and Affiliated Hainan Hospital, Hainan Medical University.
Authors' Contributions
Z.F. contributed to analysis and interpretation of data and the writing, reviewing, and finalizing of the article. R.C., F.X., W.Y., and X.X. contributed to analysis and interpretation of data. Z.F. is responsible for preparing figures. S.W. and C.H. contributed to critical revision of all versions of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by the Research Foundation of Hainan Medical University (No: XRC2022008), Hainan Province Science and Technology Special Fund (ZDYF2022SHFZ017), and Key Research and Development Project of Hainan (ZDYF2019216).