
Abstract
COVID-19, caused by the SARS-CoV-2 virus, can have neurological effects, including cognitive symptoms like brain fog and memory problems. Research on the neurological effects of COVID-19 is ongoing, and factors such as inflammation, disrupted blood flow, and damage to blood vessels may contribute to cognitive symptoms. Notably, some authors and existing evidence suggest that the SARS-CoV-2 virus can enter the central nervous system through different routes, including the olfactory nerve and the bloodstream. COVID-19 infection has been associated with neurological symptoms such as altered consciousness, headaches, dizziness, and mental disorders. The exact mechanisms and impact on memory formation and brain shrinkage are still being studied. This review will focus on pathways such as the olfactory nerve and blood–brain barrier disruption, and it will then highlight the interactions of the virus with different cell types in the brain, namely neurons, astrocytes, oligodendrocytes, and microglia.
Introduction
The coronavirus disease 2019 (COVID-19), which has affected million people worldwide and has posed a serious health threat on a global scale, is caused by the positive-sense, enveloped, single-stranded RNA virus known as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (Esch et al., 2020). The authors' opinions and the published evidence, to date, both indicate that the virus can enter the central nervous system (CNS) through the olfactory nerve, most likely using axonal transport and transsynaptic transport. In addition, the hematogenous pathway and other ways have been identified as other ways by which the virus might enter the brain (Karuppan et al., 2021; Keyhanian et al., 2020; Rhea et al., 2021).
The effects of COVID-19 on the brain are becoming more obvious. According to recent research, the coronavirus may attack specific brain cells directly, decrease blood supply to brain tissue, or cause the creation of immune chemicals that can damage brain cells (Marshall, 2021). Former studies also reported the complications including agitation, impaired consciousness, headache, and dizziness stemmed from COVID-19 infection (Mao et al., 2020). Thereafter recovery, alas, suffering from few adverse effects of this infection, has also been shown, namely fatigue, anxiety, depression, and insomnia (Huang et al., 2021). Accordingly, impediment to memory, concentration, or attention has been reported over the acute phase, accounting for a third of participants (Rogers et al., 2020).
Oxford University's study reveals that COVID-19 patients experience brain shrinkage, reduced gray matter in memory-related areas, and persistent impairments in episodic memory and attention. Despite the unknown mechanisms, most expect a return to normal cognitive function within 6–9 months (Zhao et al., 2022).
This review explores the entry and impact of the SARS-CoV-2 virus on various brain cells, including neurons, astrocytes, oligodendrocytes, and microglia.
General SARS-CoV-2 Pathophysiology Mechanism
SARS-CoV-2's pathophysiology can be summarized as follows: Coughing and sneezing can spread the SARS-CoV-2 virus to other people. Following this, the virus enters the lungs through the respiratory tract and assaults alveolar epithelial type 2 (AT2) cells, which are also in charge of creating the surfactant that lowers the surface tension in alveoli and lowers the pressure that is collapsing. According to reports, the angiotensin-converting enzyme 2 (ACE2) receptors on AT2 cells are engaged by the SARS-CoV-2 spike proteins (Li et al., 2019; Wang et al., 2020c).
The virus uses the ribosome of the host cell to produce polyproteins after entering the host cell and releasing its positive-sense ssRNA. The ssRNA can also replicate its RNA using RNA dependent RNA polymerases. The cell packaging structure can be used to deliver synthesized spike proteins to vesicle carriers. The produced polyproteins of SARS-CoV-2 are broken down by proteinases in the cytoplasm (Sigrist et al., 2020).
Cytokine storm
Cytokine storms occur when the body's immune system reacts excessively, leading to the release of cytokines, including interleukins, interferons, chemokines, and other mediators. Notably, these mediators are crucial for the innate immune response, which has evolved to eliminate infectious agents and initiate subsequent repair processes. Cytokine storms can manifest in different medical conditions, such as severe infections, autoimmune diseases, cancer immunotherapy, and viral infections (Chousterman et al., 2017; Fajgenbaum and June, 2020; Zanza et al., 2022).
The cytokine storm has emerged as an important factor in COVID-19. Patients with COVID-19 often have elevated levels of several proinflammatory cytokines, such as IL-1, IL-2, IL-6, TNF-α, IFN-γ, IP-10, GM-CSF, MCP-1, and IL-10, which are associated with the severity of the disease (Table 1) (Fajgenbaum and June, 2020; Huang et al., 2020; Montazersaheb et al., 2022; Tang et al., 2020).
Cytokines, Including Interleukins, Interferons, Chemokines, and Other Mediators, with Their Sources and Functions
BLC, B-lymphocyte chemoattractant; CCL, chemokine ligand; CRP, C-reactive protein; CXCL, CXC motif chemokine ligand; IL, interleukin; IP-10, IFN-gamma-inducible protein 10; MCP-1, monocyte chemoattractant protein-1; MIG, monokine induced by IFN-gamma; MIP-1, macrophage inflammatory protein 1; NK cell, natural killer cell; Teff cell, effector T cell; TH cell, T helper cell; Treg cell, regulatory T cell.
Headache is a common neurological symptom in COVID-19 patients, with 13.9% experiencing it, followed by dizziness and altered consciousness in 1.17% and 1.17%, respectively (Daou et al., 2020), and as per a meta-analysis study, the incidence of it has been as high as 12% (Auyeung et al., 2005). Headaches have been associated with the cytokine storm, which can be caused by various factors such as the activation of trigeminal nerve endings by proinflammatory cytokines, direct invasion of nerve endings by SARS-CoV-2, or vasculopathy in ACE2-expressing endothelial cells.
For instance, elevated levels of cytokines like IL-1β, TNF-α, IL-1, TGF-β1, MCP-1, and IL-6 have been found in patients with different types of headaches (Bø et al., 2009; Koçer et al., 2010). In a specific case involving the administration of TGN1412, a monoclonal antibody stimulating T cells, a cytokine storm occurred, resulting in symptoms, including headache, myalgias, erythema, vasodilation, and hypotension, with elevated levels of TNF-α, IFN-γ, IL-1β, IL-6, IL-8, and IL-10 (Suntharalingam et al., 2006).
Furthermore, to activate macrophages, the virus releases certain inflammatory mediators that cause the production of cytokines (IL-1, IL-6, and TNF) (Zalpoor et al., 2022) and chemokines (CCL2 and CXCL10) into the bloodstream. Thus, plasma will leak into the interstitial spaces of alveolar cells, noticeably lowering the amounts of surfactant in AT2, and compressing alveoli cells. The implications of releasing such molecules are to promote both vasodilation and capillary permeability (De Wit et al., 2016; Guan et al., 2020; Wu et al., 2020a; Yang et al., 2020; Zaki et al., 2012). Alveolar collapse and reduced gas exchange are the final results of cascade processes.
Inflammatory cytokine (cytokine storm) production is also visible simultaneously (Palm and Medzhitov, 2007); inflammatory mediators increase the production and recruitment of neutrophils and macrophages, which produce and utilize IL-21, IL-22, and IL-17 (De Wit et al., 2016). All these phases result in coughing, hypoxemia, and breathing difficulties in the later stages of the illness. To clarify, proinflammatory cytokines, such as IL-1, IL-6, and TNF-α, exert their effects on brain cells through several interconnected pathways. These cytokines disrupt the blood–brain barrier (BBB), increasing its permeability and facilitating the entry of immune cells and inflammatory mediators into brain tissue. This inflammation can potentially damage brain cells (Yang et al., 2022a).
In addition, cytokines activate glial cells, initiating neuroinflammation and further amplifying the immune response within the brain (Yang and Zhou, 2019). Moreover, they contribute to oxidative stress by promoting reactive oxygen species (ROS) production, disrupting the delicate balance of neurotransmitters, especially glutamate, and leading to excitotoxicity. In severe cases of COVID-19, cytokines can activate apoptotic pathways, triggering programmed cell death and exacerbating neuronal injury and cell loss (Miller et al., 2013; MohanKumar et al., 2023; Ramos-González et al., 2021; Solleiro-Villavicencio and Rivas-Arancibia, 2018).
In addition, COVID-19 encephalitis/meningitis cases exhibited the presence of SARS-CoV-2 in the cerebrospinal fluid (CSF), along with brain abnormalities and a cytokine storm (Moriguchi et al., 2020). These findings indicate a potential association between the inflammatory response and the development of meningitis, characterized by elevated levels of IL-6 and IFN-γ (Nagafuchi et al., 2006).
Accordingly, a 54-year-old COVID-19 patient with pneumonia and seizures exhibited brain and spine demyelination, as revealed by MRI scans. Multiple nonenhancing demyelinating lesions were observed in these areas (Zanin et al., 2020). The authors of a study suggested that the cytokine storm, particularly elevated levels of IL-1, IL-6, and TNF-α, may trigger the activation of glial cells and subsequent demyelination. In laboratory cultures of cerebellar cells, activated microglia induced by lipopolysaccharides released proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, which led to increased expression of inducible nitric oxide synthase (NOS) and the production of ROS. These processes collectively contributed to the observed demyelination and damage to axons (di Penta et al., 2013). Furthermore, in a mouse model of experimental encephalomyelitis, IL-11 was found to play a role in regulating autoimmune demyelination (Gurfein et al., 2009).
Taken together, cytokines and mediators are key components of the immune response in brain cells during COVID-19, contributing to the inflammatory process. Their dysregulation can influence the progression and severity of neurological manifestations in affected individuals.
Brain cell damage caused by the SARS-CoV-2 virus: mechanisms
To enter into cells, the virus binds through its S1 spike protein to ACE2 (Alagheband Bahrami et al., 2022; Wan et al., 2020). In human, the ACE2 RNA expression, even though shown chiefly in lung tissues (Bao et al., 2021), heart, gastrointestinal system, and kidney (Harmer et al., 2002), has been detected in both brain tissues and CSF (Kehoe et al., 2016; Xu et al., 2017); thus, the nervous system—both the neurons and glial cells of the brain—can be damaged by COVID-19 infection (Xiang et al., 2020). Incidentally, a blood circulatory pathway has been proposed by which this virus—SARS-CoV-2—directly infects the CNS (Koyuncu et al., 2013), thereby escalating the permeability of the BBB. Explicitly, BBB is vital to maintain CNS homeostasis and to prevent the neurons against the penetration of pathogens, such as bacteria and viruses (Rhea and Banks, 2019).
The exact pathophysiological mechanisms by which the SARS-CoV-2 virus leads to the neurological adverse symptoms is still not clear. However, some neurotoxic mechanisms rooted from COVID-19 infection have been stated, most noticeable of which will be cited as follows: The virus is neurotropic and enters into both neurons and glial cells. Also, the virus reaches indirectly the CNS through the BBB and/or directly through olfactory receptor neuron situated in axons thereof, thus causing neuronal dysfunction and damage—neuroinvasion (Alomari et al., 2020; Brann et al., 2020; Briguglio et al., 2020; Butowt and Bilinska, 2020; Meinhardt et al., 2021). The virus affects cerebral blood vessels and causes coagulopathy (Ding et al., 2003; Manousakis et al., 2009; Starke et al., 2013). The virus can cause a huge releasing proinflammatory cytokines so-called “cytokine storm” and peripheral organ dysfunction relating to the brain (Chousterman et al., 2017; Clark and Vissel, 2017). All these mechanisms have remarkable roles to play in the cognitive impairment etiology in COVID-19 survivors (Heneka et al., 2020) (Fig. 1).
The BBB is a protective barrier that separates the bloodstream from the brain tissue and plays a crucial role in maintaining the brain's homeostasis. The disruption of the BBB can lead to increased permeability, allowing various substances, including drugs, to enter the brain more easily. In the case of SARS-CoV-2, the virus responsible for COVID-19, several mechanisms contribute to the disruption of the BBB and increased permeability to drugs. SARS-CoV-2 interacts with the BBB by binding to specific receptors present on the vascular endothelium, particularly the ACE2 receptors. The viral spike (S) protein plays a key role in this interaction. Other proteins, including NRP1, TMPRSS2, BSG, and cathepsin L, are also involved in facilitating viral entry into the host cells. The proteolytic activation of the S protein is an essential step in SARS-CoV-2 infection. It is initially cleaved by the furin protease at the S1/S2 site, and further cleavage at the S2’ site by TMPRSS2 allows the virus to fuse with the host cell membrane. Once infection of the BBB endothelial cells occurs, viral replication takes place, leading to the activation of inflammatory mechanisms. Inflammatory responses triggered by the infection promote the release of proinflammatory cytokines, such as IFN-γ and TNF-α. These cytokines activate immune cells, ECs, astrocytes, and microglia, which are specialized cells in the brain. Activated microglia release cytokines like IL-1, IL-6, and TNF-α, which further activate astrocytes. Activated astrocytes release various mediators, including TNF, prostaglandins, glutamate, ROS, and nitric oxide. These mediators can induce neurotoxicity and contribute to neuroinflammation. IL-6, in particular, can cause excessive activation of the complement system and coagulation cascades, leading to a harmful cycle of cytokine storm. This cytokine storm can destabilize tight junctions and damage BBB endothelial cells, increasing vascular permeability. The infection of endothelial cells by the virus can also lead to their lysis, which further alters BBB permeability. In addition, SARS-CoV-2 infection promotes the upregulation of MMP9, an enzyme responsible for degrading collagen IV, an essential component of the basal membrane of the BBB. The increased expression of MMP9 can result in the rupture of the BBB. Moreover, the S protein of SARS-CoV-2 can activate RhoA, a protein involved in cytoskeletal restructuring. Activation of RhoA leads to the disassembly of tight junctions, further compromising the integrity of the BBB and increasing its permeability. The cytokine storm, neuroinflammation, and disruption of the BBB can have detrimental effects on various brain cells, including neurons, astrocytes, and microglia. These effects can contribute to neuronal cell death and the development of neurological disorders. BBB, blood brain barrier; BSG, basigin; MMP9, matrix metalloproteinase 9; NRP1, neuropilin-1; ROS, reactive oxygen species; TMPRSS2, transmembrane protease serine 2.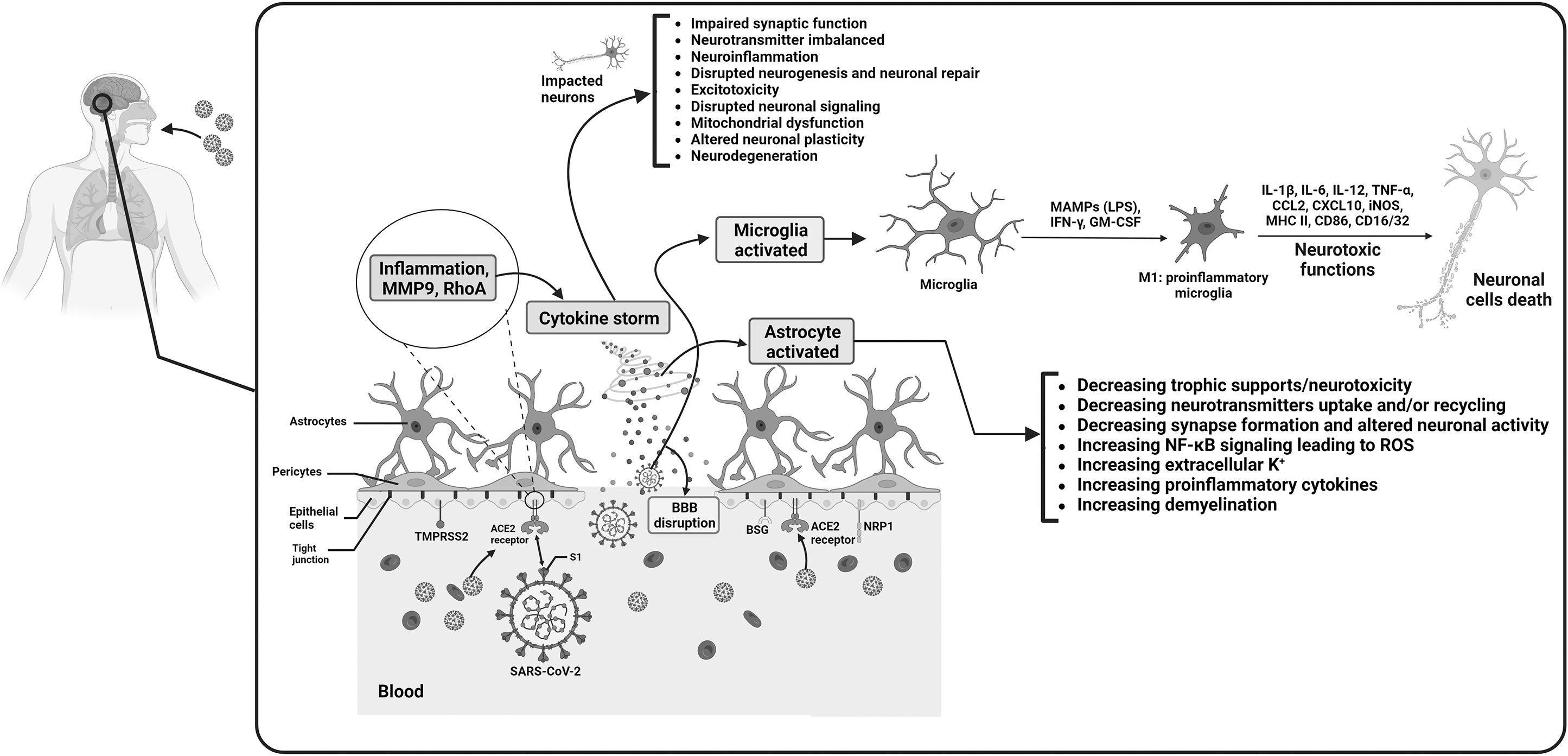
As per the available information and the authors' perspectives, the virus can enter the CNS through the olfactory nerve, most likely using transsynaptic and likely axonal transport. In addition, the hematogenous route has been identified as the second way the virus might enter the brain (Karuppan et al., 2021; Keyhanian et al., 2020; Rhea et al., 2021).
According to fierce evidence, damaging the zones of the brain relating to memory by SARS-CoV-2 virus elevates proinflammatory cytokines such as IL-6, TNFα (Poduri et al., 2020), and IL-1β (Ye et al., 2020a). The implication of this event is to inactivate the synthesis of all forms of NOSs, which in turn gives rise to reducing the generation of NO that acts as one of the key mediators of local inflammation. NO is important for regulating blood flow, modulating immune responses, and supporting synaptic plasticity in the brain (Azargoonjahromi, 2023). The decrease in NO levels caused by COVID-19-related inflammation can disrupt blood flow, impair immune regulation, and affect synaptic plasticity. These processes contribute to localized inflammation and damage in the brain, particularly in memory-related regions. Consequently, cognitive impairments, including memory deficits, can occur (Azargoonjahromi, 2023; Colombo et al., 2002; Lee et al., 2017; Madhu et al., 2016).
Noteworthy, hippocampus zone that has a unique role in memory is shrunk as a result of exceed release of cytokines, culminating in hippocampal atrophy (Lindlau et al., 2015). To clarify, in individuals infected with SARS-CoV-2, the excessive release of proinflammatory cytokines can lead to hippocampal atrophy, which is the shrinking of the hippocampus, a brain region crucial for memory. The mechanisms underlying hippocampal atrophy involve chronic inflammation, oxidative stress, excitotoxicity, disrupted neurotrophic factors, and impaired neurogenesis (Chesnokova et al., 2016; Juszczyk et al., 2021). Activation of glial cells and the release of proinflammatory molecules further contribute to damage in the hippocampus (Sajja et al., 2016). Hippocampal atrophy can result in memory deficits and cognitive impairments, which are commonly observed in individuals recovering from severe COVID-19 (Hashimoto et al., 2017). However, further research is needed to fully understand the complex relationship between SARS-CoV-2 infection, inflammation, and hippocampal atrophy.
In addition, the ACE2 receptor is a protein found on the surface of cells, including brain cells (Alagheband Bahrami et al., 2022; Kuhn et al., 2006). It acts as the entry point for the SARS-CoV-2 virus, allowing the virus to enter and infect cells. However, apart from its role in viral entry, ACE2 also has other functions in the body. One of these functions involves ACE2's interaction with brain-derived neurotrophic factor (BDNF) (Zheng et al., 2014). BDNF is a protein that plays a crucial role in promoting the growth, development, and survival of neurons (nerve cells) in the brain.
It also has anti-inflammatory properties (Joosten and Houweling, 2004) and helps regulate the activity of microglia (Wu et al., 2020b), which are immune cells in the brain. When ACE2 is activated by the virus, it can influence the activity of BDNF. However, COVID-19 infection has been associated with lower levels of BDNF in the brain. This decrease in BDNF levels may contribute to cognitive impairment observed in individuals affected by COVID-19. In the context of COVID-19, cognitive impairment can manifest as brain fog, difficulties with concentration, or memory problems (de Pins et al., 2019; Ng et al., 2019; Wu et al., 2020b).
The exact mechanisms by which decreased BDNF levels lead to cognitive impairments in COVID-19 are not yet fully understood. However, BDNF's role in reducing neuronal inflammation and modulating microglial activation suggests that its deficiency may contribute to neuroinflammation and altered immune responses in the brain, which can subsequently affect cognitive function (Joosten and Houweling, 2004; Wu et al., 2020b).
Both IL-6 and TNFα, furthermore, pass the BBB and then activate microglia (Akrout et al., 2009). This activation leads to releasing IL-1β, the receptors for which are specifically concentrated in the postsynaptic sections of hippocampal neurons (Gardoni et al., 2011). This event makes the hippocampus susceptible to IL-1β, which has been shown to interfere with LTP and memory (Prieto et al., 2019).
SARS-CoV-2 Virus Impact on Neurons
Neuronal dysfunction resulting from SARS-CoV-2 infection can lead to a range of neurological symptoms and disorders. The neurologic manifestations of COVID-19 vary extensively from patient to patient, and involve both the CNS and PNS. These effects include cognitive impairments, altered sensation (such as tingling or loss of taste/smell), headaches, dizziness, autonomic dysfunction, mood and behavioral changes, and, in rare cases, seizures and encephalopathy. The mechanisms underlying these effects are complex and involve neuroinflammation, disrupted blood flow, and direct viral invasion of neurons (Carod-Artal, 2020; Filatov et al., 2020; Guadarrama-Ortiz et al., 2020; Mao et al., 2020; Wang et al., 2020a).
Seizures, strokes, and encephalopathy are neurological complications that can arise from SARS-CoV-2 infection. Seizures are believed to be triggered by hyperinflammation caused by the virus, which creates an environment in the brain that promotes seizure activity (Myint et al., 2006; Najjar et al., 2020; Wu et al., 2020c).
Strokes occur due to the virus-induced inflammation and vascular dysfunction, leading to an increased risk of blood clot formation in the cerebral vasculature, which can result in ischemic or hemorrhagic strokes (Guadarrama-Ortiz et al., 2020; Levi et al., 2020; Myint et al., 2006). Encephalopathy, characterized by impaired attention, confusion, delirium, lethargy, or coma, is thought to be a consequence of various factors, including direct viral invasion, neuroinflammation, and systemic effects of the infection. While these complications are relatively rare, ongoing research is needed to uncover the precise underlying mechanisms and identify risk factors associated with these neurological manifestations in COVID-19 patients (Islam et al., 2022; Pennisi et al., 2020; Wu et al., 2020c; Zubair et al., 2020).
Mitochondria play a vital role in maintaining neuronal health by providing energy for their functions, including electrical signaling and synaptic transmission. Through oxidative phosphorylation, mitochondria generate ATP, which is essential for the demanding energy requirements of neurons with their dense axonal networks and synaptic connections. However, neurons are susceptible to degeneration due to the intricate nature of their structure and the stress placed on their mitochondria. This mitochondrial stress can lead to neuronal dysfunction and degeneration (Murali Mahadevan et al., 2021; Sheng and Cai, 2012).
In the context of the COVID-19, these neuronal vulnerabilities are exacerbated. The systemic inflammation and lack of oxygen associated with COVID-19 can further disrupt the health and function of mitochondria, and consequently, patients may experience neurological complications such as encephalopathy, cognitive impairment, and long-term neurological effects (Nunn et al., 2022). The substantial stress placed on mitochondria in neurons renders them vulnerable to degeneration. The result of substantial mitochondrial stress in neurons is an increased risk of neuronal dysfunction, degeneration, and potentially the development of neurological disorders, including Parkinson's disease, multiple sclerosis, Alzheimer's disease, depression, and epilepsy (Alshial et al., 2023; Norat et al., 2020).
Individuals with declined mitochondrial functions and/or mitochondrial stress may be more susceptible to SARS-CoV-2 infection and experience severe COVID-19 complications, potentially leading to increased mortality (Yao and Lawrence, 2021). This may be because the CoV open reading frame 9 (ORF-9) protein interacts with mitochondrial outer membrane receptors, such as mitochondrial antiviral signaling systems (MAVS) (Shi et al., 2014) and the translocase of the outer mitochondrial membrane complex (Miserey-Lenkei et al., 2021), functioning as viral recognition receptors during viral replication (Malavolta et al., 2020). This interaction leads to the degradation of MAVS and Drp1 (Shi et al., 2014), causing mitochondrial hyperelongation. In addition, CoV ORF-9 is suggested to induce the release of mitochondrial DNA (mtDNA) and activate mtDNA-induced inflammasome, limiting the host cell's innate immune response (Shi et al., 2014; Singh et al., 2020).
SARS-CoV-2 Virus Impact on Oligodendrocytes
Increasing evidence indicates that SARS-CoV-2 virus infection is a definite risk factor for demyelination in both the peripheral and CNSs. This notion is supported by the description of a COVID-19 patient who was hospitalized for interstitial pneumonia and seizures and then described by Zanin et al. (2020) Damage to the demyelin sheath was detected recently, according to a brain MRI. However, high-dose steroid therapy triggered repair in the neurological and respiratory systems. They suggested that a SARS-CoV-2-induced delayed immune response was what led to the degeneration of the myelin sheath (Zanin et al., 2020).
Mehta et al. (2020) discovered demyelinating lesions linked to neurological impairment in a COVID-19 case in a different study. The patient's brain and spine MRIs revealed multiple demyelinating lesions that had recently developed, but were not enhancing. They postulated that glial cell activation and subsequent demyelination during SARS-CoV-2 infection would be due to the proinflammatory environment caused by the cytokine storm (Mehta et al., 2020). Likewise, encephalomyelitis was reported by Zoghi et al. (2020) in a male patient, age 21, who had upper respiratory symptoms 2 weeks before to this presentation and intermittent vomiting for 4 days. Brain MRI revealed bilateral posterior internal capsule lesions extending to the ventral part of the pons as well as a marbled splenium hyperintensity pattern. A substantial transverse myelitis was discovered throughout their research using thoracic and cervical MRI (Zoghi et al., 2020).
Brun et al. (2020) reported a 54-year-old woman who had been exposed to SARS-CoV-2 and had brain lesions that indicated acute demyelination. The pallidum and supratentorial white matter on both sides of the patient's brain were afflicted by hypodense lesions, according to the patient's brain CT scan. Confined diffusion lesions were seen on an MRI of the brain, but there was no bleeding or amplification. Unaffected areas included the striatum, posterior fossa, and thalamus.
A follow-up MRI revealed no new abnormality, and the spinal cord MRI was normal (Brun et al., 2020). Furthermore, two other studies (Agarwal et al., 2020; Karapanayiotides et al., 2020) presented demyelination in patients afflicted with COVID-19; MRI on both studies presented that the corpus callosum and the pericallosal white matter were where the majority of the myelin destruction foci and a concentric demyelination pattern and the presence of hemosiderin deposit together. Given such evidence, SARS-CoV-2 virus is able to cause demyelination in sundry ways.
SARS-CoV-2 Virus Impact on Astrocytes
SARS-CoV-2 targets astrocytes as its primary target in the brain. It has been proven that SARS-CoV-2 preferentially infects astrocytes over neurons in primary and organoid cortical cultures, causing astrocyte reactivation and non-cell autonomous neuronal death. Furthermore, it is found that basigin (BSG), also known as extracellular matrix metalloproteinase inducer or cluster of differentiation 147 (CD147), and dipeptidyl-peptidase 4 are significant SARS-CoV-2 infection-related molecular actors in cortical astrocytes (Huang and Fishell, 2022).
As per a study carried out by Andrews et al. (2022), infected astrocytes exhibit heightened reactivity and cellular stress. In addition, SARS-CoV-2-infected cultures show non-cell autonomous inflammatory effects such as an increase in reactive microglia and a general loss of neurons due to apoptosis. According to research, astrocytes may have a significant supporting function in the regulation of brain energy, metabolism, and the microenvironment (Beard et al., 2022). It is interesting to note that BSG/CD147 is important for the astrocyte metabolic pathways that support the energy requirements of neurons (Chao et al., 2019).
Kriegstein and his colleagues also showed in a preprint published that SARS-CoV-2 preferentially infects astrocytes over other types of brain cells, leading to fatigue, depression and “brain fog.” The virus was introduced to brain organoids, which are tiny, laboratory-grown structures that resemble the brain. Over all other cells present, astrocytes were almost completely infected by SARS-CoV-2 (Andrews et al., 2021).
Most importantly, astrocytes play a vital role in the maintenance and regulation of the BBB by means of their end-feet structures. These structures not only establish a physical barrier but also govern the movement of molecules (Abbott, 2002). Nonetheless, in the presence of pathological conditions or brain injury, astrocytes can become activated, resulting in astrogliosis. This activation disrupts the tight junctions and enhances BBB permeability, potentially giving rise to neuroinflammation, neuronal damage, and various neurological disorders (Azargoonjahromi, 2023; Yue and Hoi, 2023). Moreover, astrocytes release signaling molecules such as cytokines, growth factors, and enzymes to modulate the tight junctions between endothelial cells. Astrocytes can interact with some proteins, such as NRP1, TMPRSS2, furin, BSG, and cathepsin L; these proteins are involved in tight junctions and are essential for maintaining the selective permeability of the BBB (Bai et al., 2021; Burks et al., 2021).
SARS-CoV-2 engages with the BBB by attaching its viral S protein to ACE2 receptors found on the endothelial cells of blood vessels. Other proteins, including NRP1, TMPRSS2, BSG, and cathepsin L, are also involved in this interaction. Activation of the S protein by the furin protease at the S1/S2 site is a crucial step for the virus to establish infection. TMPRSS2 further cleaves the S protein at the S2’ site, facilitating the fusion of the virus with the host cell membrane.
Once inside the BBB endothelial cells, the virus replicates and triggers an inflammatory response. Proinflammatory cytokines like IFN-γ and TNF-α are released, activating immune cells, endothelial cells, astrocytes, and microglia. Activated microglia release cytokines such as IL-1, IL-6, and TNF-α, which further stimulate astrocytes. These activated astrocytes, in turn, release substances like TNF, prostaglandins, glutamate, ROS, and nitric oxide, which contribute to neurotoxicity associated with neuroinflammation. Excessive activation of IL-6 can initiate a detrimental cytokine storm, triggering the complement system and coagulation cascades.
This storm disrupts the tight junctions between endothelial cells, causing damage to BBB integrity and increased permeability. In addition, the lysis of infected endothelial cells further compromises the BBB. The virus also induces the upregulation of MMP9, an enzyme responsible for breaking down collagen IV, a key component of the BBB basement membrane. This process ultimately leads to the rupture of the BBB. Furthermore, the S protein activates RhoA, a protein involved in cellular signaling, which leads to reorganization of the cytoskeleton and disassembly of tight junctions. These changes further disrupt the permeability of the BBB (Bai et al., 2021; Hernández-Parra et al., 2023; Tavčar et al., 2021; Wan et al., 2021).
What is notable is that SARS-CoV-2 can affect brain endothelial cells (Yang et al., 2022b) and pericytes (McQuaid and Montagne, 2022), resulting in disruptions to the BBB and brain health. Infected cells exhibit dysfunction and increased permeability, and release proinflammatory cytokines and chemokines, leading to neuroinflammation and neuronal damage (Tohidpour et al., 2017). Simultaneously, dysfunctional pericytes contribute to increased permeability and an inflammatory response. These effects can manifest as neurological symptoms such as headaches, confusion, dizziness, and loss of taste or smell. In severe cases, they can even lead to encephalopathy or stroke (Uemura et al., 2020).
In healthy tissue, pericytes play a crucial role in maintaining vascular functions by closely interacting with endothelial cells and the extracellular matrix (Bergers and Song, 2005). However, in the case of SARS-CoV-2 infection, the viral spike protein (SARS-CoV-2 Sp) binds to the ACE2 receptors found on pericytes. This interaction leads to the detachment and loss of pericytes from the blood vessels. The loss of pericytes disrupts the normal crosstalk between pericytes and endothelial cells (McQuaid and Montagne, 2022). Consequently, endothelial cells become activated and exhibit increased expression of ICAM1, a cell adhesion molecule involved in immune cell recruitment. This increased expression of ICAM1 promotes immune cell infiltration into the affected area (Bui et al., 2020; Tong et al., 2020a).
In addition, the disruption of pericyte-endothelial cell interactions results in decreased levels of VE-Cadherin, a protein involved in maintaining endothelial cell junctions. The decrease in VE-Cadherin contributes to vascular breakdown and compromised integrity of the blood vessels (Bouillet et al., 2023; Dejana et al., 2009; Harris and Nelson, 2010). Overall, the binding of SARS-CoV-2 spike protein to ACE2 receptors on pericytes disrupts their interaction with endothelial cells, leading to vascular breakdown and potential compromise of the BBB, exacerbating neurological implications in COVID-19.
SARS-CoV-2 Virus Impact on Microglia
Microglia, which resemble macrophages and seem to be CNS immune cells, play a crucial role in preserving brain homeostasis and in the quick response to damage and inflammation (Block et al., 2007). Microglia can be activated in response to immunological stimuli, thereby shifting from ramified to amoeboid phenotype and secreting TNF-α, IL-1, and IL-6 (Fetler and Amigorena, 2005). Activated microglia are included in both the neurotoxic M1 phenotype, which contributes to neuroinflammation, and the neuroprotective M2 phenotype. Based on an increasing number of studies, the neurological system may suffer irreparable harm from dysregulation and overactivation of microglia (Block et al., 2007; Kim and Joh, 2006; Polazzi and Contestabile, 2002; Zecca et al., 2006).
A patient may be more likely to experience neurological and mental issues if their microglia are activated even after they have fully recovered clinically from the infection (Tremblay, 2020). Furthermore, poor or aberrant microglial function may substantially impair cognitive abilities like judgment, decision-making, learning, and memory. Accordingly, proinflammatory activation brought on by SARS-CoV-2 infection of microglia may significantly affect the short-, moderate-, or long-term neurological and psychiatric effects of infection with SARS-CoV-2 (Tay et al., 2017).
There is evidence that SARS-CoV-2 can infect a human microglial cell line directly. The RNA-seq research findings showed that a viral infection resulted in ER stress, immunological reactions, and apoptosis in the late phase. In addition, SARS-CoV-2 infection caused human microglia to undergo apoptosis by activating both intrinsic and extrinsic pathways. In short, in lung tissues and cell lines, SARS-CoV-2 has been demonstrated to induce intrinsic and extrinsic apoptosis (Jeong et al., 2022). In a preprint study also, transgenic mice were found to have microglia that were infected with SARS-CoV-2, which resulted in the ongoing death of microglia and the release of proinflammatory cytokines (Jeong et al., 2022).
Postmortem Brain Evidence from COVID-19 Patients
As per some studies using postmortem brain samples from COVID-19 patients, researchers found common neuropathological findings in COVID-19 patients' postmortem brain samples, suggesting neurological symptoms may be related to other mechanisms beyond direct virus infection. For instance, in a study, the researchers employed various methods to investigate the neuropathological findings and presence of the SARS-CoV-2 virus in postmortem brain samples from COVID-19 patients.
They conducted microscopic examination, immunohistochemistry (IHC) using two different antibodies, and PCR-based techniques. Their cohort of 18 cases revealed common neuropathological findings such as macrophages containing iron deposits around blood vessels and neuronal changes associated with the lack of oxygen. Only one brain tissue sample showed viral protein expression, while all samples tested positive for viral RNA. A co-localization IHC study indicated that SARS-CoV-2 antigens were located in brain perivascular macrophages. These findings suggest that neurological symptoms in COVID-19 patients may be due to other mechanisms than direct virus infection, with few studies confirming SARS-CoV-2 antigen presence in brain tissue samples (Lebrun et al., 2023).
Likewise, a study conducted autopsies on 44 patients who died from COVID-19 and extensively examined the CNS in 11 of these patients. The researchers aimed to understand the distribution, replication, and cell-type specificity of the virus throughout the body, including the brain, from the acute stage of infection to more than 7 months after the onset of symptoms.
The findings revealed that SARS-CoV-2 was widely distributed in multiple tissues, including the brain, especially in patients with severe COVID-19. The virus was present early in the infection, and SARS-CoV-2 RNA could persist in various anatomical sites, including the brain, for an extended period, such as up to 230 days after symptom onset in one case. However, despite the widespread presence of the virus, there was little evidence of inflammation or direct viral damage outside the respiratory tract. The study suggests that SARS-CoV-2 can cause systemic infection and persist in the body for months in some patients (Stein et al., 2022).
Another study employed minimally invasive autopsy techniques to examine brain changes in deceased individuals. They used brain imaging and tissue sampling in seven COVID-19 patients and found abnormalities such as infarcts, hemorrhages, and signal abnormalities in various brain regions. Histological analysis confirmed reactive gliosis, congestion, and axonal disruption, and SARS-CoV-2 RNA was detected in brain tissue samples (Martin et al., 2022). Moreover, the other study focused on microglial and macrophage activation in the brains of deceased COVID-19 patients. They analyzed brain tissue samples from 17 individuals who died from COVID-19.
The study found pronounced activation of microglia and macrophages in the white matter, brain stem, and cerebellar areas. However, there was no correlation between disease severity and neuropathological changes. Macrophage clusters were not associated with disease severity, but represented a more advanced stage of microglial and macrophage activation. The study also noted that the reaction of microglia and macrophages was less pronounced in post-COVID-19 patients. The findings suggest that the activation of microglia and macrophages in the brains of deceased COVID-19 patients is not directly associated with disease severity and may be more prominent during the acute phase of the illness (Stein et al., 2023).
In addition, a study examined neuropathological findings in COVID-19 patients who died after being hospitalized in an intensive care unit. The study included 20 decedents who underwent brain autopsy. Acute vascular changes, hypoxic/ischemic injury, hemorrhages, and microglial activation were common findings in the brains. Encephalitis-like changes, neurodegenerative diseases, and inflammatory responses were also observed. Notably, there was no evidence of viral RNA or protein in the CSF or brain samples. Older subjects showed age-related brain pathologies even without known neurological diseases (Agrawal et al., 2022).
Taken together, these studies indicate that COVID-19 can lead to various brain abnormalities, including vascular changes, hypoxic injury, inflammation, and activation of microglia and macrophages. The presence of SARS-CoV-2 RNA in brain tissue samples suggests possible viral invasion, although it is not consistently detected. The findings highlight the need to consider both direct and indirect effects of the virus on the brain. In addition, pre-existing brain diseases and age-related pathologies may contribute to the observed neurological abnormalities in COVID-19 patients. Further research is needed to fully understand the mechanisms underlying brain involvement in COVID-19 and its long-term implications.
Long Covid and Brain Fog
“Long COVID,” also known as postacute sequelae of SARS-CoV-2 infection (PASC), is a term used to describe a condition where individuals continue to experience persistent symptoms even after recovering from the acute phase of COVID-19 infection. These symptoms may last for an extended period, beyond what would typically be expected for recovery. One of the most commonly reported symptoms among individuals with long COVID is cognitive impairment, specifically difficulties with concentration (Raveendran et al., 2021).
Several factors may contribute to long COVID, including viral persistence, immune dysregulation, organ damage, neurological involvement, blood clotting and vascular issues, autoimmune responses, psychological factors, pre-existing health conditions, and individual factors like age and genetics. These factors can interact and lead to ongoing inflammation, tissue damage, and psychological distress, prolonging the duration of symptoms (Astin et al., 2023; Davis et al., 2023; Turner et al., 2023).
Patients with long COVID, who experienced mild acute COVID-19, often exhibit immune dysregulation characterized by various immune cell alterations. These alterations include exhausted T cells (Jon et al., 2022), reduced CD4+ and CD8+ effector memory cell numbers, and elevated PD1 expression on central memory cells, persisting for at least 13 months (Glynne et al., 2022; Jon et al., 2022). In addition, there is a depletion of naive T and B cells and an upregulation of interferon expression (Phetsouphanh et al., 2022).
A comparative study involving individuals with long COVID, uninfected individuals, and those with acute COVID-19 revealed higher numbers of monocytes, activated B cells, and CD4+ T cells secreting IL-4 and IL-6 in long COVID patients. Conversely, conventional dendritic cells and exhausted T cells were found to be decreased (Jon et al., 2022). Notably, elevated levels of cytokines such as IL-1β, IL-6, TNF, and IP10 have been linked to cognitive dysfunction in long COVID patients (Peluso et al., 2021; Schultheiß et al., 2021).
Delayed detection and subsequent delayed initiation of appropriate medical care can impact the overall management of COVID-19 and potentially influence the development of long-term symptoms (Gertz et al., 2022). However, the potential impact of delayed diagnosis on the development of long COVID symptoms is not yet fully understood. A study utilizing a dynamic model has revealed that improving the timeline of diagnosis can play a crucial role in reducing the reproduction rate of the virus.
However, effectively controlling the spread requires a comprehensive approach (Rong et al., 2020). On the whole, long COVID symptoms, including fatigue, shortness of breath, cognitive difficulties, and joint pain, are not definitively linked to delayed detection of COVID-19. The exact causes are unknown, but they are believed to be a complex interplay of viral persistence, immune dysregulation, and other factors (Peluso et al., 2022b; Tsampasian et al., 2023). Research into long COVID is ongoing, and our understanding of its causes and risk factors is evolving.
Reactivation is the process in which a previously dormant virus within a host cell transitions into an active phase, known as the lytic stage. During reactivation, the virus undergoes productive replication, leading to its spread within the host and potentially causing new infections (Traylen et al., 2011). It is important to note that the establishment of latency or dormancy varies among different viruses. While certain viruses, like herpesviruses, have the ability to enter a latent phase, the capacity of SARS-CoV-2 to establish latency is not yet well-established.
Notably, it was determined that the average time between COVID-19 infection and the onset of symptoms (latent period) is estimated to be 5.5 days (with a 95% confidence interval of 5.1–5.9 days), which is shorter than the average time between infection and the detection of the virus (incubation period) at 6.9 days. This suggests that utilizing laboratory testing could potentially lead to shorter quarantine periods since 95% of COVID-19 cases were found to shed the virus within 10.6 days (with a 95% confidence interval of 9.6–11.6 days) after being infected (Xin et al., 2022).
Accordingly, there have been reports of individuals experiencing recurrent or prolonged symptoms resembling COVID-19 after initial recovery, suggesting the possibility of viral reactivation in long COVID. Factors such as immunosuppression, immune dysregulation, tissue damage, inflammation, and the existence of viral reservoirs may influence viral reactivation (Altable Pérez and De la Serna, 2020; Andrea et al., 2022; Opsteen et al., 2023). However, the exact mechanisms and extent of viral reactivation in long COVID are still being investigated.
Noteworthy, reactivation of viruses such as Epstein-Barr virus (EBV), cytomegalovirus, herpes simplex virus 1, human herpesvirus 7, and human herpesvirus 6 has been observed in long COVID patients, similar to those with myalgic encephalomyelitis/chronic fatigue syndrome (Drago et al., 2021; Hashimoto, 2023; Jon et al., 2022; Lehner et al., 2020; Matthew et al., 2022; Shikova et al., 2020; Su et al., 2022; Xu et al., 2020; Zubchenko et al., 2022). This viral reactivation can lead to mitochondrial fragmentation, impairing energy metabolism and contributing to fatigue (Schreiner et al., 2020). In addition, reactivated viruses can trigger immune responses, resulting in inflammation and potential neurocognitive dysfunction. Recent research has found a correlation between EBV reactivation and fatigue and neurocognitive symptoms in long COVID patients (Matthew et al., 2022).
Therefore, the full spectrum of viral species and nonviral pathogens that can be reactivated in long COVID has not been fully characterized, although some herpesviruses and other viruses have been implicated. Plasma DNA PCR screening or RNA sequencing in samples from individuals with long COVID can help identify the specific pathogens reactivated and their timing relative to the onset of symptoms, providing valuable insights into the underlying mechanisms of the condition (Iosef et al., 2023; Jon et al., 2022).
Cognitive symptoms associated with long COVID can manifest as problems with attention, focus, memory, and mental clarity. Some individuals may struggle to concentrate on tasks, experience brain fog, have difficulty retaining information, or find it challenging to organize their thoughts. These cognitive impairments can significantly impact daily functioning, work performance, and overall quality of life (Li et al., 2023).
The exact mechanisms underlying cognitive symptoms in long COVID are still being studied, and multiple factors may contribute to their development. It is believed that the persistent inflammation and immune dysregulation associated with long COVID, even after the acute infection has resolved, may play a role in these cognitive impairments. Inflammatory processes in the brain, disruption of neural networks, and changes in neurotransmitter levels could all contribute to cognitive dysfunction.
In addition, other factors such as psychological distress, fatigue, and the indirect effects of the virus on other organs and systems (such as respiratory or cardiovascular) can also contribute to cognitive symptoms in long COVID. The prevalence and severity of cognitive symptoms can vary among individuals with long COVID. Some individuals may experience mild cognitive difficulties, while others may have more significant impairments. The duration of these cognitive symptoms can also vary, with some individuals experiencing improvements over time, while others may have symptoms that persist for months (Baig, 2020; Becker et al., 2021; Callard and Perego, 2021; Perego et al., 2020).
Likewise, a research showed that almost 80% of subjects discharged from hospitals complained of at least one of the following symptoms: fatigue, myalgia, dizziness, muscle weakness, headache, sleep disturbances, cognitive impairment, and brain fog (Al-Aly et al., 2022; Nehme et al., 2021; Zeng et al., 2023).
In long COVID, there is evidence of activation in the kynurenine pathway, characterized by increased levels of metabolites such as quinolinic acid, 3-hydroxyanthranilic acid, and kynurenine. This activation has been linked to cognitive impairment (Stone and Darlington, 2013), suggesting a potential role of the kynurenine pathway dysregulation in the cognitive symptoms experienced by long COVID patients. The presence of these metabolites, particularly quinolinic acid, is associated with neuroinflammation and neurotoxicity, which could contribute to cognitive dysfunction (Cysique et al., 2022). Notably, long COVID can lead to various neuropathologies, including neuroinflammation, blood vessel damage, and neuronal injury (Spudich and Nath, 2022).
A study conducted using the UK Biobank dataset discovered several concerning findings in patients who had recovered from COVID-19 compared to control subjects. These included a reduction in gray matter thickness, a decrease in brain size, and cognitive decline (Douaud et al., 2022). Additionally, abnormalities were observed in long COVID patients, such as abnormal levels of mitochondrial proteins, the presence of SARS-CoV-2 spike and nucleocapsid proteins (Peluso et al., 2022a), deficiencies in tetrahydrobiopterin, and increased oxidative stress (Villaume, 2022). These findings highlight the complex and diverse neurological effects of long COVID, shedding light on potential underlying mechanisms and providing avenues for further research and targeted interventions.
Most importantly, COVID-19 is not solely limited to the respiratory tract and can potentially affect cells in other parts of the brain, leading to potential neuropsychiatric complications. Individuals who have survived COVID-19 may be at risk of developing conditions such as anxiety, depression, and post-traumatic stress disorder (PTSD). The development of these symptoms is influenced by a combination of environmental, psychological, and biological factors. Certain risk factors, such as being female and having a history of psychiatric disorders, can increase the likelihood of experiencing these psychiatric complications among COVID-19 patients (Thye et al., 2022). In a systematic review and meta-analysis encompassing 51 studies, the prevalence of neuropsychiatric symptoms in COVID-19 survivors was examined. The analysis revealed that the most prevalent symptoms among survivors included sleep disturbance, fatigue, objective cognitive impairment, anxiety, and PTSD.
The mean duration of follow-up across the studies was 77 days, and the overall quality of the included studies was deemed moderate. Interestingly, the review found limited evidence to suggest differential symptom prevalence based on factors such as hospitalization status, disease severity, or duration of follow-up. These findings highlight the common and persistent nature of neuropsychiatric symptoms following recovery from COVID-19. Notably, the first 6 months following infection were characterized by a particularly high prevalence of insomnia, fatigue, cognitive impairment, and anxiety disorders among survivors (Badenoch et al., 2022).
Brain fog can cause confusion, forgetfulness, and a loss of focus and mental clarity. This can be exacerbated by overwork, inadequate sleep, stress, and excessive internet use. High levels of cellular inflammation and changes in the hormones that regulate your mood, energy, and focus are known to have a role in brain fog. The hormone levels are out of balance, which throws off the entire system. In addition, brain fog syndrome may lead to the development of additional conditions like obesity, irregular menstruation, and diabetes mellitus (Theoharides et al., 2021).
Current Therapeutics
A COVID-19 patient should prioritize resting in bed and staying hydrated to conserve energy and prevent dehydration. If the patient experiences high fever (>38.5°C), they are advised to use antipyretic medications like ibuprofen or acetaminophen, as necessary. The National Health Commission of China (NHC) has introduced several antiviral medications for COVID-19 prevention, diagnosis, and treatment. These include drugs like IFN-α, lopinavir/ritonavir, chloroquine phosphate, ribavirin, and arbidol, as outlined in the subsequent sections (Alagheband Bahrami et al., 2022; Gwenzi et al., 2022) (Table 2).
Medications Employed for Managing COVID-19 Along with Relevant Details
Lopinavir/Ritonavir is known for their ability to inhibit an enzyme called cytochrome P450. They are typically used to enhance the effectiveness of antiretroviral treatments. In a study involving 47 patients, combining these drugs with medications used for pneumonia resulted in positive outcomes, such as reduced body temperature and improved physiological functions, without causing any adverse effect (Ye et al., 2020b).
Chloroquine phosphate and hydroxychloroquine, which are commonly used to treat malaria and rheumatic diseases, have shown to demonstrate broad-spectrum antiviral effects by interfering with the processes within the cell nucleus and hindering RNA transcription. Although hydroxychloroquine shows potential for COVID-19 treatment, its effectiveness against SARS-CoV-2, the virus causing COVID-19, lacks convincing evidence when tested in living organisms (Jin et al., 2020). Furthermore, ribavirin is a type of nucleoside analog that targets a specific enzyme called RNA-dependent RNA polymerase (RdRp) in SARS-CoV-2. Arbidol, another antiviral drug, works differently, inhibiting SARS-CoV-2 without being a nucleoside analog. It is administered orally multiple times a day (Jin et al., 2020).
Remdesivir, ciclesonide, and favipiravir also show promise in treating COVID-19. Remdesivir, known for its tolerability without causing significant harm to the kidneys or liver, targets specific enzymes involved in viral replication. Favipiravir acts similarly by inhibiting RdRp, providing an additional avenue for treatment (Arab-Zozani et al., 2020; Lu, 2020; Young et al., 2021).
Most importantly, Pfizer Inc., (
Molnupiravir, under the brand Lagevrio/Molulife, is an antiviral drug that can inhibit RNA virus replication, diminishing the risk of hospital admission and death by ∼50% among nonhospitalized adults with mild to moderate COVID-19 infection (Toots et al., 2020; Toots et al., 2019). However, according to
Notably, to the best of my knowledge, specific treatments for brain fog and cognitive decline caused by long COVID are still under investigation, and no standardized or universally approved medication existed specifically for these symptoms. However, health care providers used various strategies and therapies aimed at managing and improving cognitive symptoms. For better cognitive function, physical exercise, sleep management, and cognitive rehabilitation programs are necessary. Physical exercise increases blood flow and cell growth, and reduces inflammation (Mandolesi et al., 2018; Scheffer and Latini, 2020); cognitive rehabilitation improves memory and concentration (das Nair et al., 2016); and sleep management is critical for brain cell functions and mental health (Scott et al., 2021).
In addition to the aforementioned, a balanced diet and adequate hydration are crucial for overall health, including brain function. Nutrients like omega-3 fatty acids (Dighriri et al., 2022) and antioxidants like resveratrol and curcumin are associated with cognitive improvement (Azargoonjahromi and Abutalebian, 2023; Cicero et al., 2019; Kuszewski et al., 2018). While specific medications exclusively targeting brain fog in long COVID are unavailable, health care providers may prescribe drugs to alleviate related symptoms. For instance, antidepressants may be utilized to manage mood disturbances, potentially enhancing overall cognitive function (Rosenblat et al., 2015; Schulkens et al., 2022).
Taken together, these approaches aim to ameliorate cognitive function and alleviate brain fog by addressing various contributing factors. However, the effectiveness of each method varies among individuals, prompting health care providers to tailor treatment plans to specific symptoms and individual needs.
Conclusion
COVID-19 can have neurological effects, with potential routes of entry to the CNS, including the olfactory nerve and the BBB. Neurological symptoms can range from loss of taste and smell to more severe complications such as stroke or encephalitis. Neuroinflammation and persistent symptoms in postacute COVID-19 cases are also observed. Ongoing research aims to understand these effects better and develop effective treatments.
Footnotes
Acknowledgment
I would like to express gratitude to researchers and scholars who have conducted previous studies that have contributed to our current knowledge across various fields, regardless of the specific topic.
Authors' Contribution
This article was authored solely by A.A., who conceptualized and completed all aspects of the research.
Author Confirmation
Mr. Azargoonjahromi is from Shiraz University of Medical Sciences (Shiraz, Iran), where education and research are the primary functions.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
No funding was received for this article.