
Abstract
Hepatitis C virus (HCV), despite the availability of effective direct-acting antivirals (DAAs) that clear the virus from >95% of individuals treated, continues to cause significant health care burden due to disease progression that can lead to fibrosis, cirrhosis, and/or hepatocellular carcinoma. The fact that some people who are treated with DAAs still go on to develop worsening liver disease warrants further study into the immunopathogenesis of HCV. Many viral infections, including HCV, have been associated with activation of the inflammasome/pyroptosis pathway. This inflammatory cell death pathway ultimately results in cell lysis and release of inflammatory cytokines, IL-18 and IL-1β. This review will report on studies that investigated HCV and inflammasome activation/pyroptosis. This includes clinical in vivo data showing elevated pyroptosis-associated cytokines in the blood of individuals living with HCV, studies of genetic associations of pyroptosis-related genes and development of liver disease, and in vitro studies aimed at understanding the mechanism of pyroptosis induced by HCV. Finally, we discuss major gaps in understanding and outstanding questions that remain in the field of HCV-induced pyroptosis.
Introduction
Hepatitis C virus
Chronic hepatitis C virus (HCV) infection is associated with the development of liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC), with ∼58 million individuals currently infected worldwide (World Health Organization, 2023). HCV is a small, enveloped, icosahedral, single-stranded RNA virus in the hepacivirus genus of the family Flaviviridae (Lindenbach and Rice, 2001). If left untreated, ∼80% of individuals who are infected will develop chronic infection along with increased risk of liver disease, including cirrhosis and HCC (WHO, 2017). HCV is, in some respects, a scientific success story as it is currently the only chronic viral infection that can be completely cured using direct-acting antivirals (DAAs; WHO, 2017). Due to the success of DAAs, the WHO has launched an ambitious goal of HCV elimination by 2030 (WHO, 2017). While theoretically possible to treat every infected individual, success is dependent on the ability to locate and treat every individual living with HCV, including those who are currently unaware of their infection status. The goal of elimination is challenged by three factors. (1) The scientific community has been unable to produce an HCV vaccine offering effective protection against infection (approaches and difficulties in developing an HCV vaccine are discussed in Bailey et al., 2019). (2) Individuals who are infected and subsequently treated with DAAs or those who spontaneously clear the virus have no detectable immune memory response, resulting in the possibility of repeat infections (Grebely et al., 2012; Shoukry, 2018). (3) Some individuals who achieve sustained virological response continue to develop worsening liver disease despite virus elimination (Abe et al., 2020; D'Ambrosio et al., 2021; Montaldo et al., 2021; Welsch et al., 2017). With these considerations in mind, understanding how HCV causes disease, how the immune system responds to this virus, and the role of infection-induced immune system dysfunction, are vital if 2030 elimination targets are to be met.
Inflammasome activation and pyroptosis
Pyroptosis is an inflammatory form of programmed cell death that has implications for the innate immune responses to stimuli such as viruses as well as contributing to the pathogenesis associated with many infectious diseases (de Castro-Jorge et al., 2019; de Sousa et al., 2018; Han et al., 2015; Lê et al., 2019; Li et al., 2022; Lien et al., 2021; Pan et al., 2019; Platnich et al., 2018). The pyroptosis pathway can be triggered by numerous stimuli, including some yet to be identified. Known and described triggers of the pyroptosis pathway include, but are not limited to, viral RNA (Allen et al., 2009; Chen et al., 2014; Gao et al., 2020), cytoplasmic mitochondrial DNA (mtDNA; Shimada et al., 2012; Wu et al., 2019), reactive oxygen species (ROS; Qiu et al., 2019; Wang et al., 2019), viral proteins (Chen et al., 2019; Ding et al., 2019; McAuley et al., 2013; Shrivastava et al., 2020; Siu et al., 2019; Xie et al., 2020), ion concentration changes (Di et al., 2018; Muñoz-Planillo et al., 2013), cell swelling (Compan et al., 2012), and ATP (Duncan et al., 2007; Ye et al., 2021). The classic pathway begins when a receptor, such as nucleotide oligomerization domain, leucine-rich repeat protein 3 (NLRP3; Ting et al., 2008), absent in melanoma 2 (AIM2), NLRC5, or IFI16, among others, is triggered, leading to the recruitment and oligomerization of ASC (apoptosis-associated speck-like protein containing a CARD [caspase recruitment domain]). Pro-caspase-1 is then recruited and undergoes self-proteolysis whereby caspase-1 is cleaved into its active form. The inflammasome (the effector protein complex for the pyroptosis pathway) contains the receptor, ASC (if the receptor does not contain its own CARD domain), and effector molecule, caspase-1. The inflammasome then cleaves gasdermin-D (GSDM-D) into the C- and N-terminus. The N-terminus subsequently localizes to the plasma membrane and forms pores. This pore formation will ultimately lead to cell lysis following overwhelming pore numbers when compared with the ability of membrane repair complexes to fix these pores (Evavold and Kagan, 2019; Evavold et al., 2021; Liu et al., 2016; Rühl et al., 2018). The inflammasome complex also mediates cleavage of pro-IL-1β and pro-IL-18 into their mature forms, which are subsequently released from cells, utilizing GSDM-D pores for their escape (Evavold et al., 2018; Monteleone et al., 2018; Schneider et al., 2017; Tsuchiya et al., 2021). The pyroptosis pathway is extensively reviewed in Galluzzi et al., 2018 and HCV-induced pyroptosis is summarized in Fig. 1.

Summary of HCV-induced pyroptosis. HCV activates NLRP3 (and/or another yet-unidentified sensor), which subsequently oligomerizes with ASC. Caspase-1 is then recruited as the executioner caspase. This protein complex is dubbed the inflammasome. Caspase-1 within this complex then cleaves GSDM-D into its pore-inducing form, which migrates to the cell membrane to create pores. Caspase-1 also cleaves pro-IL-1β and pro-IL-18 into their mature forms for subsequent release from cells through GSDM-D pores. This figure was created using
Pyroptosis has been dubbed a “doubled-edged sword” as it is thought to contribute to innate immune responses by acting as an alarm system to recruit other immune cells and contribute to an effective adaptive immune response (Deets and Vance, 2021; Hachim et al., 2020; Ichinohe et al., 2009; Niu et al., 2019). However, this can also be detrimental to the host, as the pyroptosis pathway can become overactivated, leading to uncontrollable inflammation. The overactivation of the inflammasome is thought to contribute to the pathogenesis of many viral infections, including, but not limited to, influenza A virus (Bauer et al., 2012; Chen et al., 2018; Cilloniz et al., 2010; Corry et al., 2022; McAuley et al., 2013; Ren et al., 2017; Zhang et al., 2017), SARS-CoV-2 (Pan et al., 2021; Rodrigues et al., 2021; Vora et al., 2021), HIV (Ahmad et al., 2018; Doitsh et al., 2014) and, as discussed in detail below, HCV.
It is important to note that most publications regarding HCV-induced inflammasome activation/pyroptosis pay particular interest to the NLRP3 inflammasome. There seems to be a bias in the field, suggesting that RNA viruses only activate NLRP3 despite extensive reports of other RNA viruses also activating traditionally “DNA-associated” sensors (extensively reviewed in Wallace and Russell, 2022). Investigation into whether HCV can activate multiple inflammasomes, including those traditionally associated with DNA sensors, is lacking. To fully understand the effect of HCV activation of inflammasomes and pyroptosis, investigation of other sensors must be expanded.
For many years, it was unclear whether inflammasome activation always led to pyroptosis (Martín-Sánchez et al., 2016). However, more recent studies have shown that this varies substantially and that not only does inflammasome activation not always lead to lytic cell death (Rühl et al., 2018), but that there are alternative pathways and crosstalk between various cell death pathways that can lead to inflammasome activation (Conos et al., 2017; Denes et al., 2015; Heilig et al., 2020; Rogers et al., 2019; Wallace et al., 2022; Xu et al., 2022), and that IL-1β can be secreted without GSDM-D pore formation and cell lysis (Karmakar et al., 2020). Therefore, this review will consider publications documenting both inflammasome activation as well as pyroptosis induction by HCV (Green, 2019).
Is there a role for the inflammasome in HCV-induced disease even after cure?
Pathogenesis of HCV has been well studied, but has never been completely understood. Although DAA treatment results in curative rates above 90% (WHO, 2017), and is associated with decreased serum levels of many inflammatory markers, such as CXCL10, RIG-I, STAT1, and IRF7, levels of IL-1β and IL-18 in serum, even after cure, remain elevated (Burchill et al., 2017). Some work from the post-DAA introduction era has documented 10% of individuals with ongoing increased levels of alanine transaminase (ALT), whereas another 25% have ALT levels above the “healthy range” (Welsch et al., 2017).
A 2021 study detailed the importance of ongoing follow-up with individuals who have achieved sustained virologic response (SVR), since HCC was the most widely reported complication, including among individuals who had achieved SVR (D'Ambrosio et al., 2021). Another 2021 study supports the idea that there is ongoing inflammation, even following achievement of SVR. Montaldo et al. isolated extracellular vesicles (EVs) from healthy controls and from individuals with HCV before and after achieving SVR. Upon exposure of cells in culture to EVs from individuals with HCV (even following SVR), analysis showed markers of fibrosis (both mRNA and protein), indicating that profibrotic signals persist even after cure (Montaldo et al., 2021). Given accumulating evidence suggests that some individuals who undergo curative therapy still develop worsening liver disease in the absence of productive infection (Abe et al., 2020; D'Ambrosio et al., 2021; Montaldo et al., 2021; Welsch et al., 2017), additional research is needed to understand HCV-induced inflammation and pyroptosis.
What do we know about HCV-induced inflammasome activation/pyroptosis?
Evidence for pyroptosis in vivo
Many early studies of HCV-induced inflammasome activation/pyroptosis did not set out to consider this pathway but noticed elevated levels of what are known now to be pyroptosis-associated inflammatory cytokines. The earliest known report (2002) was published when the link between IL-1β/IL-18 and inflammasome activation/pyroptosis was still being teased out and long before development and implementation of curative DAAs. This early publication compared serum levels of a variety of inflammatory cytokines in individuals living with HCV who were either undergoing hemodialysis or were not. While the authors did not report many stark differences in cytokine levels, they did note that individuals undergoing hemodialysis had significantly lower serum levels of IL-1β compared with counterparts who were not regularly undergoing hemodialysis. The authors suggested that hemodialysis may aid in elimination of inflammatory triggers from the blood of individuals living with HCV (Spanakis et al., 2002; Fig. 2).
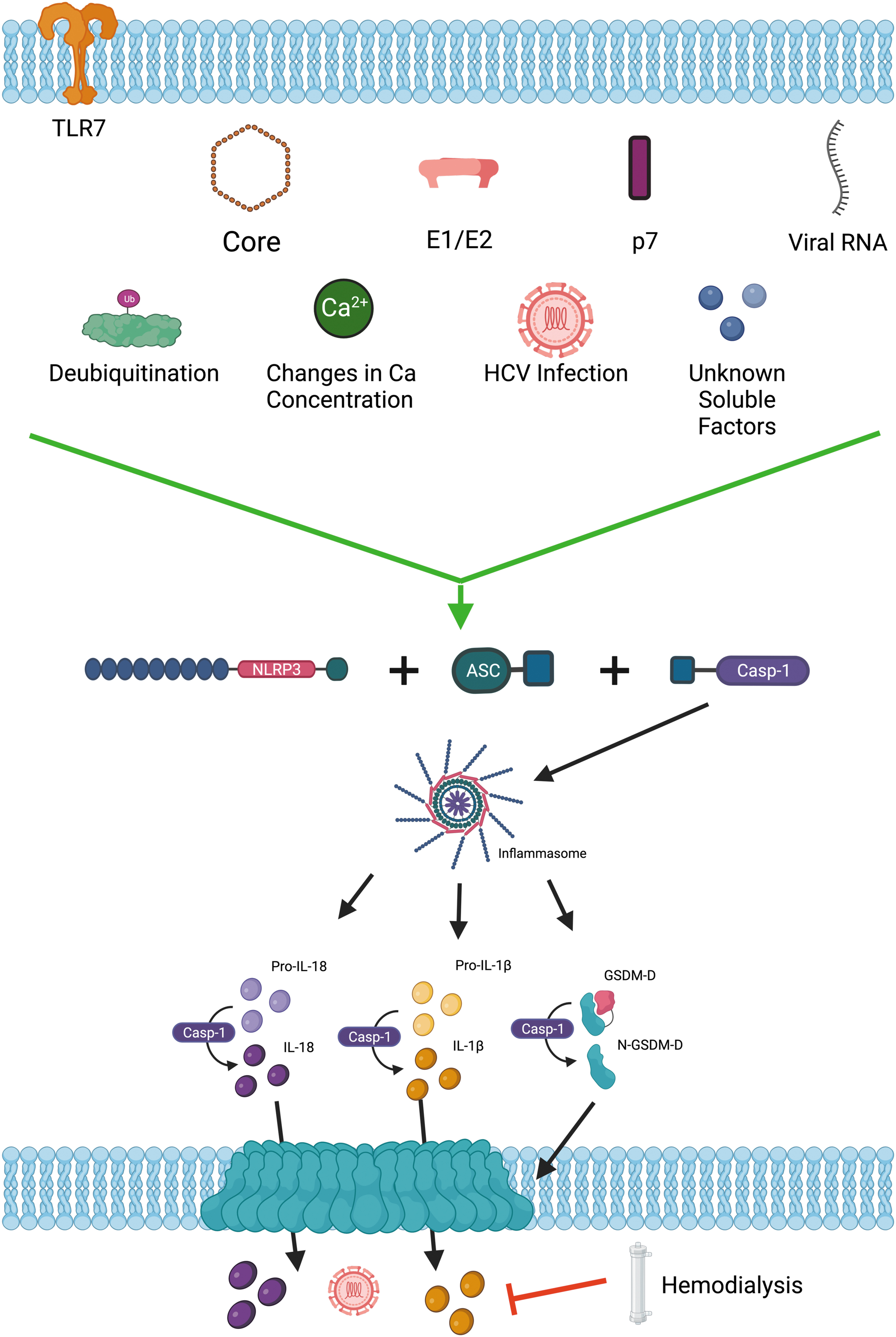
Summary of published findings on HCV-induced pyroptosis/inflammasome activation triggers and the one published way of inhibiting this process. This figure should serve as a visual representation of the summary of in vitro findings included in Table 2. This figure was created using
A 2006 study noted elevated levels of plasma IL-6 and IL-18 in individuals living with either HCV or Hepatitis B virus (HBV) when compared with healthy control groups. This work also indicated that IL-18 was positively correlated with serum ALT activity and serum aspartate transaminase activity, both of which are associated with liver damage. The authors of this study proposed that IL-6 and/or IL-18 may have important roles as markers of inflammation and hepatic injury, particularly during the course of HCV infection (Falasca et al., 2006).
Several years later, elevated levels of another inflammasome-associated cytokine, IL-18, in plasma, were documented. In a 2011 longitudinal study, the authors tested plasma from individuals living with HCV (with known date of virus acquisition) for changes over time in levels of inflammatory markers, including IL-1β, IL-6, IL-8, IL-12, IL-18, IFN-γ, TNF-α, and ALT. Interestingly, the only significant changes detected were in the levels of IL-18, which increased in each individual with initial detection of HCV RNA. For spontaneous clearers (individuals who cleared the virus without curative drug therapy), IL-18 plasma levels decreased to preinfection baseline following infection resolution. In contrast, individuals who developed persistent HCV infections were found to have decreased plasma IL-18 following the acute phase of infection, but levels remained above baseline preinfection levels and did not correlate with HCV RNA or ALT levels. These findings suggest IL-18 may play a role in the modulation of the innate immune response to persistent infection (Chattergoon et al., 2011).
More recently, particularly following the finding that pyroptosis of CD4+ T cells is involved in the pathogenesis of HIV infection (Ahmad et al., 2018; Doitsh et al., 2014), publications once again focused on pyroptosis-associated cytokines. A 2016 study by Veenhuis et al. documented systemic elevation of IL-18 in individuals living with HIV/HCV coinfection versus individuals living with monoinfection of HIV or HCV. In individuals living with HIV, IL-18 serum levels correlated with detectable HIV viremia and were inversely correlated with CD4+ T cell counts (Veenhuis et al., 2016).
Genetic associations of HCV and inflammasome-related genes
Following early findings of elevated levels of IL-1β and IL-18 in serum of individuals living with HCV, there was interest in determining whether any genetic polymorphisms were associated with these genes and whether that could influence the response to HCV and the potential for viral clearance. Previous studies had suggested that IL-18 plays a role in viral clearance and fibrosis development, and identified polymorphisms at −607 and −137 of the IL-18 promoter sequence that were associated with susceptibility to HBV infection (Kimura et al., 2002; Kitasato et al., 2004).
Manohar et al. investigated the role of these SNPs in HCV infection. More than 200 individuals living with HCV as well as 350 healthy controls were enrolled in this study. The authors found no significant difference with regard to the frequency of the −607 and −137 alleles between those living with HCV versus healthy controls. However, when individuals living with HCV were divided into groups based on mild or severe liver disease, there were significantly more individuals in the mild disease group with the −607 A/A allele than in the group with severe liver disease. Therefore, the authors suggested that the −607 allele may have a protective effect as it was associated with more mild disease (Manohar et al., 2009).
In 2019, a group from Egypt was interested in understanding the potential associations of nine SNPs in four inflammasome-related genes (nlrp3, card8, IL-1β, and IL-18) with susceptibility to HCV infection as well as outcomes for individuals treated with interferon. This study included 201 individuals with chronic HCV and 95 healthy controls. Genotyping was performed and the CC genotype of NLRP3-re1539019 was found to be correlated with decreased risk of developing chronic HCV. The same association was found for the CA genotype of the A allele of NLRP3-rs35829419, as well as the GG genotype of the G allele of IL-18-rs1946518.
Interestingly, the AA genotype of IL-1β-rs1143629 was found significantly more frequently in individuals living with HCV than in healthy controls. The frequencies of the AA genotype of NLRP3-rs1539019, CC genotype and C allele of NLRP3-rs35829419, and the TT genotype and T allele of CARD8-rs2043211 were all found more frequently in individuals who did not respond to treatment with interferon. Important to note, SNPs in the genes encoding IL-1β and IL-18 did not show any significant correlation with response to treatment with interferon. Altogether, the authors suggested that there are genetic variants associated with both susceptibility to HCV infection as well as response to interferon treatment (Estfanous et al., 2019).
Following up on these earlier studies, a 2021 study in Brazil showed polymorphisms in inflammasome-associated genes in 151 individuals living with HCV (subdivided into mild or advanced fibrosis) and compared them with 206 healthy controls. The genes encoding IL-1β, IL-18, NLRP3, CARD8, CTSB, and AIM2 were genotyped for all individuals. Among the healthy controls, the NLRP3-rs10754558 C/C genotype was associated with higher serum IL-1β compared with the G/G genotype. A similar pattern was observed in individuals living with HCV. This suggests that changes to genes coding for proteins upstream of pro-IL-1β processing may have effects on serum levels. This is supported by the fact that serum IL-1β from individuals living with HCV was higher than that from healthy controls.
Brazilian scientists also performed some gene–gene interaction studies, which revealed that individuals who were heterozygotes for both CARD8-rs2009373 and IL-1β-rs16944, heterozygotes for CTSB-rs1692816 and AIM2-rs1103577, or heterozygotes for IL18-rs187238 and NLRP3-rs10754558 were less likely to develop chronic HCV. This again supports the idea that inflammasome-related genes have impacts on outcomes and infection with HCV (Toro et al., 2021).
A summary of the genetic associations of HCV and inflammasome-related genes can be found in Table 1.
Summary of Genetic Associations of Hepatitis C Virus and Inflammasome-Related Genes
Mechanistic studies in vitro
By 2013, inflammation and associated increases of blood IL-1β and IL-18 had been well documented in individuals living with HCV, but the mechanism, other than some genetic associations, had not been discerned.
One of the early studies (2013) that attempted to determine the mechanism by which HCV causes inflammation pointed out that the mechanism of hepatic inflammation was unknown, and that the mechanism by which HCV triggers this inflammation was unknown at that time. Attempting to understand these mechanisms and knowing that macrophages had been studied extensively in terms of their ability to undergo pyroptosis and secrete IL-1β and IL-18, the authors searched for evidence that hepatic macrophages play a role in HCV-associated inflammation. Data suggested macrophages taken from both peripheral blood mononuclear cells (PBMCs) as well as liver-resident Kupffer cells produced IL-1β following HCV exposure. They investigated this further using THP-1 cells, where they were able to document a rapid, although transient, increase in caspase-1 activation, which subsequently led to secretion of IL-1β. Although HCV can enter macrophages by CD81-independent phagocyte-mediated uptake, productive HCV infection has not been documented in macrophages, suggesting virion production is not necessary to trigger the pyroptosis pathway in these cells. To follow up, the authors showed that HCV RNA can trigger TLR7-mediated signaling, which leads to an increase in IL-1β mRNA levels. Taken together, the authors concluded that IL-1β production and secretion may contribute to HCV-associated hepatitis (Negash et al., 2013). Another study was also published in 2013 with similar findings. The authors also exposed THP-1 cells, macrophages from PBMCs, or Kupffer cells to cell culture-derived HCV, which led to increased activation of caspase-1 in an NF-κB-dependent manner. Because some literature has suggested that viral proteins with viroporin function can trigger the inflammasome pathway (Chen et al., 2019; Ichinohe et al., 2010; Nieto-Torres et al., 2015; Triantafilou et al., 2013; Zhi et al., 2020), the authors investigated whether RNA coding for HCV p7 could trigger pyroptosis. They documented exposure of macrophages to RNA encoding p7 was, indeed, sufficient to trigger IL-1β secretion. The authors concluded, similarly to Negash et al. that circulatory and/or resident macrophages may be involved with HCV-induced liver inflammation (Shrivastava et al., 2013).
HCV had been shown previously to trigger a type I IFN response in hepatocytes through TLR3 activation. Chattergoon et al. were interested in the differences in inflammatory pathway induction in response to HCV in various cell types. They demonstrated that HCV can activate the inflammasome, but not type I IFN responses, in monocytes and macrophages. They further determined that this was infection and TLR3 independent, but was dependent on endosomal TLR7 activation. This was an interesting finding, as TLR7 sensing of HCV in monocytes does not lead to downstream IFN production. Altogether, the authors reported infection-independent mechanisms of inflammation with TLR utilization that was cell type-dependent and suggested this could play a role in chronic inflammation associated with HCV (Chattergoon et al., 2014).
In contrast to some earlier studies discussed above, a 2014 publication by Chen et al. did not confirm that HCV exposure alone was sufficient to trigger inflammasome activation in THP-1 cells. However, if THP-1 cells were transfected with HCV viral RNA, there was a clear dose-dependent increase in the secretion of IL-1β. It was also noted that HCV RNA transfection resulted in ASC oligomerization and cleavage of pro-caspase-1 into its active form. Initiation of the inflammasome pathway was also determined to be dependent on the production of ROS, but was independent of RIG-I signaling (Chen et al., 2014).
Before 2014, work had already been completed that indicated chronic HCV infection leads to decreased IFN-γ production by natural killer (NK) cells. In a 2014 publication, Serti et al. aimed to further understand the mechanism by which this occurs. The authors used PBMCs from individuals living with HCV or healthy controls, and NK cells were isolated or eliminated. The cells were then cocultured with Huh7 cells that were left untreated or were expressing an HCV subgenomic replicon before examining antiviral activity, cytotoxicity, and cytokine production.
NK cells displayed greater amounts of IFN-γ when PBMCs were cocultured with Huh7 cells expressing the replicon than when cocultured with untreated Huh7 cells. Interestingly, NK cell production of IFN-γ was reduced when NK cells were isolated from PBMCs and then the NK fraction was cocultured with Huh7 cells. The same result of decreased IFN-γ production by NK cells was shown when PBMCs were depleted of monocytes, leaving all other immune cells, and then cocultured with Huh7 cells. This decrease in IFN-γ was not seen when all PBMCs were cocultured with Huh7 cells, suggesting that monocytes play an important role in IFN-γ production by NK cells in this context. The authors noted that knockdown of the inflammasome pathway, or IL-18 neutralization, both had the same effect as depleting monocytes, suggesting that the pyroptosis pathway in monocytes is necessary for stimulating antiviral effects of NK cells during HCV infection. Since individuals living with HCV are known to have decreased monocyte function, this could explain why NK cell antiviral activity is also attenuated during infection with HCV (Serti et al., 2014). Although the authors did not aim to study the effects of inflammasome activation/pyroptosis, a 2013 study by Holder et al. also showed NK cell dysfunction when NK cells were cocultured with HCV-infected Huh-7.5 cells, further supporting the idea that HCV can negatively affect NK cell function (Holder et al., 2013).
Early literature relating to the pyroptosis pathway focused mostly on its occurrence in immune cells, with general consensus that other cell types did not undergo this process (Brough et al., 2009; Duncan et al., 2007; Fernandes-Alnemri et al., 2009; Ivan et al., 2013; Liu et al., 2004; Poeck et al., 2010; Thomas et al., 2009). Our laboratory first identified pyroptosis of Huh-7.5 cells infected with HCV (JFH-1T) in 2016. Kofahi et al. identified not only pyroptosis and apoptosis of infected cells, but also pyroptosis and apoptosis of uninfected “bystander” cells. This was the first report of bystander pyroptosis induced by HCV in vitro and is thought to potentially contribute to the pathogenesis associated with chronic infection (Kofahi et al., 2016).
Many viruses encode for viroporins, which have been suggested to induce activation of the inflammasome in response to changes in ion concentrations within the cell (Chen et al., 2019; Ichinohe et al., 2010; Nieto-Torres et al., 2015; Shrivastava et al., 2020; Triantafilou et al., 2013; Zhi et al., 2020). The exact function of the HCV p7 is not fully understood but one of its proposed functions is to act as a viroporin, as mentioned above. To study this, the authors of a 2017 study recorded that IL-1β secretion differed between HCV genotypes despite TNF secretion remaining consistent across the various genotypes. The authors pointed out that it had already been reported that HCV infection can activate the NLRP3 inflammasome, but the mechanism had yet to be confirmed. The authors proposed that p7, as a viroporin, was the trigger of inflammasome activation.
The full function of p7 has never been elucidated as generating an antibody against it has been problematic and adding a FLAG tag to either the C- or N-termini leads to different cellular localizations of p7 (Griffin et al., 2005) (reviewed in Atoom et al., 2014). The data showed that when pH decreased, the activity of p7 increased. Overall, the authors concluded that their data matched that of others who had proposed that viroporins trigger the activation of inflammasomes. There are some inherent limitations to this study as HEK293T cells were used, which differ substantially from hepatocytes (Farag et al., 2017).
After Negash et al. (2013) previously published their findings of NLRP3 inflammasome activation triggered by HCV in hepatic macrophages, they published a follow-up study to better understand this cascade of events (Negash et al., 2019). In this study, the authors identified HCV core protein as a viroporin that can trigger inflammasome activation. It was determined that macrophages exposed to either HCV or soluble core protein alone was both necessary and sufficient to trigger IL-1β production. To further understand the mechanism behind this process, the authors investigated the role of each of the structural proteins and p7. They observed that either p7 or HCV core was able to trigger activation of the inflammasome and that this process was dependent on calcium mobilization. Overall, this adds to our understanding of innate immune responses to HCV (Negash et al., 2019).
It is well established that cells infected with HCV display substantial rearrangement of organelles (Egger et al., 2002; El-Hage and Luo, 2003; Gosert et al., 2003; Mottola et al., 2002; Shi et al., 2003). The authors of a recent article were interested to learn whether the pyroptosis pathway was involved, specifically with regard to Golgi fragmentation. At homeostasis, it was determined that ASC resides at the Golgi with Golgi-resident protein immunity-related GTPase M (IRGM). Following HCV infection, ASC disassociated from the IRGM and the Golgi and, instead, associated with HCV-triggered pyroptosis sensor NLRP3. Inhibition of NLRP3 through siRNA knockdown led to decreased Golgi fragmentation, while ASC inhibition, also by siRNA knockdown, was associated with changes to Golgi structure in both control and infected cells, accompanied by decreased IRGM localized within the Golgi. Altogether, these results suggest a role of ASC upstream of the pyroptosis pathway where it may regulate IRGM and control of the Golgi. To determine whether this phenomenon is HCV specific, the authors repeated experiments using either related Zika virus or the common NLRP3-specific positive control, lipopolysaccharide (LPS) with nigericin stimulation. Results obtained when LPS/nigericin was used were similar to those acquired when cells were infected with HCV. Interestingly, the results were not similar when cells were infected with Zika virus. Overall, these findings suggest additional functions of inflammasome components during cell organelle restructuring, at least within the context of specific stimuli (Daussy et al., 2021). Other research on Zika virus-induced cell death has demonstrated activation of both NLRP3 and AIM2 in the brains of fatal cases of Zika virus-induced microcephaly (de Sousa et al., 2018). Perhaps the results reported by Daussy et al. (2021) are a finding specific to NLRP3, which could account for their negative results with Zika virus. These observations could also be a cell type-dependent phenomenon, since a hepatoma cell line was used for the HCV experiments while a macrophage cell line was used for the Zika experiments. While both hepatoma-like and macrophage-like cell lines have been reported to undergo inflammasome activation/pyroptosis in the context of viral infections, it is unclear whether the details regarding this pathway are exactly the same in both cell types for a given virus or family of viruses (Kofahi et al., 2016; Negash et al., 2019; Negash et al., 2013; Wallace et al., 2022).
Previous literature has documented activation of NLRP3 by HCV in hepatocytes. Ramachandran et al., in their 2021 article discuss that, in hepatocytes, NLRP3 inflammasome activation is regulated by posttranslational modifications and is specifically modified by lysine-63 ubiquitination chains, as well as the fact that NLRP3 is specifically deubiquitinated during HCV infection. Chemical or siRNA inhibition/knockdown of deubiquitinases blocked inflammasome assembly and activation, which was accompanied by decreased IL-1β maturation, reduced HCV protein expression, and lower levels of HCV virion release from cells. Altogether, these findings suggest that NLRP3 inflammasome activation is necessary for the HCV life cycle, as well as suggest that deubiquitinases are one of the main regulators of the inflammasome during HCV infection (Ramachandran et al., 2021).
An ongoing theme in the virus-induced pyroptosis literature is the attempt and desire to determine which viral proteins specifically trigger pyroptosis. As viral glycoproteins are often a major antigen during infection, a 2021 study compared the glycoproteins from several different viruses to evaluate their ability to trigger inflammasome activation in THP-1 cells. Like many of the other viral glycoproteins tested (SARS-CoV-1/2 and HCMV), the HCV glycoproteins (E1/E2) triggered the NLRP3 inflammasome, indicated by ASC oligomerization, GSDM-D cleavage, and IL-1β secretion. This result was confirmed by using CRISPR-Cas9 knockout of NLRP3 and GSDM-D, which abrogated the above result, confirming their findings (Eisfeld et al., 2021).
The most recent work from our own laboratory focused on crosstalk between apoptosis and pyroptosis, but in this study, we will discuss the findings related to inflammasomes and pyroptosis. We found that NLRP3 inhibition by CRISPR-Cas9 knockdown did not completely eliminate caspase-1 activation, suggesting that multiple inflammasomes may be involved in HCV-induced pyroptosis. We also found that when GSDM-D was knocked out, there was a shift toward apoptosis, suggesting substantial crosstalk between these two cell death pathways. We also showed for the first time, to our knowledge, the simultaneous activation of both the apoptosis and pyroptosis pathways within the same HCV-infected cell. Additionally, we also showed, similarly to the findings by Ramachandran et al. (2021), that inhibition of the pyroptosis pathway by CRISPR-Cas9 knockdown of NLRP3 or GSDM-D led to both a decrease in extracellular titer (or virion release) as well as a switch in the ratio of intra- to extracellular titer. Traditionally, extracellular titer is ∼1 log higher than intracellular, but in the knockdown cells, the intracellular titer was significantly higher, suggesting HCV utilizes cell death pathways as a mechanism of pathogenesis (Wallace et al., 2022), further supporting earlier findings by Ramachandran et al. (2021).
A summary of the main findings from mechanistic studies of HCV-induced inflammasome activation/pyroptosis in vitro can be found in Table 2 and a visual summary can be found in Figure 2.
Summary of In Vitro Findings Related to the Trigger and General Understanding of Hepatitis C Virus-Induced Pyroptosis, Separated by Study
What are some of the main unknowns?
There is still much to be learned in the field of HCV-induced pyroptosis, including both basic and applied science studies. In our opinion, the main questions remaining can be grouped together into three main categories.
Mechanistically, which part of HCV or its life cycle triggers pyroptosis?
There is some evidence, as mentioned above, that p7, core, and E1/E2 contribute to HCV-induced pyroptosis, but data are not conclusive at the present time and warrants further research.
What impact does inflammasome activation have on postcure liver disease? We know that there are signs of ongoing liver inflammation post HCV cure, but the extent to which this inflammation is driven by the inflammasome pathway remains unknown.
Inflammasome-component inhibitors have been approved by the FDA for treatment of other inflammatory conditions, generally those associated with genetic conditions that result in overactivation of the inflammasome pathway. Do those inhibitors have any effect on clearance/further development of liver disease? Currently, this question remains unanswered. Drugs already approved by the FDA include anti-IL-1β blocking treatments (ex Canakinumab), IL-1Ra agents (IL-1 antagonist; ex Anakinra and Rilonacept), as well as disulfiram, which blocks GSDM-D pore formation (Hu et al., 2020). There are also a number of drugs currently in clinical trials that work against some part of the inflammasome pathway, including several that inhibit NLRP3. However, extensive testing of these drugs with regard to their effect on clearance of viral infection or decreasing virus-induced inflammation has not yet been performed on a wide scale. Patients with severe coronavirus disease 2019 (COVID-19) are known to have increased levels of inflammatory cytokines in their blood (Blanco-Melo et al., 2020; Jalloh et al., 2022; Rodrigues et al., 2021; Sefik et al., 2022; Vora et al., 2021) and SARS-CoV-2 infection activates the inflammasome in abortively infected human monocytes (Sefik et al., 2022). One IL-1 antagonist (Anakinra) was given emergency use authorization in 2022 for the treatment of hospitalized COVID-19 patients who required supplemental oxygen or who were likely to develop respiratory failure (U.S. FDA, 2022), but these results have not yet been published. Optimistically, this will set a precedent for these compounds to be tested in clinical trials for other viral infections associated with substantial inflammation, including HCV.
Conclusions
It is apparent that our knowledge of HCV-induced inflammasome activation/pyroptosis has expanded greatly in the last 20 years. We have learned that individuals living with HCV have increased levels of pyroptosis-associated inflammatory cytokines, IL-18 and IL-1β, in their serum (Chattergoon et al., 2011; Falasca et al., 2006; Spanakis et al., 2002; Veenhuis et al., 2016), including in individuals who have reached viral cure.
On a genetic level, we now know that many inflammasome-related genes are correlated with various outcomes of infection with HCV. Research has shown various SNPs in inflammasome-related genes to be associated with numerous outcomes to HCV infection: SNPs within the gene coding for IL-18 have been associated with more mild liver disease, SNPs in the genes coding for NLRP3 and IL-18 have been associated with decreased risk of developing chronic HCV, SNPs in the gene encoding IL-1β are found more frequently in individuals living with HCV, and some SNPs are more frequently found in individuals who did not respond to HCV treatment (Estfanous et al., 2019; Manohar et al., 2009; Toro et al., 2021). From a mechanistic, or in vitro perspective, while acknowledging the significant developments and discoveries, we have also learned that there remain many unanswered questions. Some of the earliest work suggested that inflammasome activation in monocytes/macrophages by HCV was triggered by HCV exposure or HCV RNA exposure, and that this may contribute to liver inflammation (Chen et al., 2014; Negash et al., 2013; Shrivastava et al., 2013). We learned that inflammasome activation in monocytes is necessary for NK cell activation during HCV infection (Serti et al., 2014).
Work from our own laboratory has shown activation of both pyroptosis and apoptosis in both infected cells and uninfected bystander cells, and that pyroptosis is, at least in part, dependent on NLRP3 (Kofahi et al., 2016; Wallace et al., 2022). Our work, and that of Ramachandran et al., also showed that inflammasome activation is necessary for the HCV viral life cycle, including progeny virus release (Ramachandran et al., 2021; Wallace et al., 2022). More recent work has shown that p7, core protein, and envelope proteins E1/E2 can act as viroporins and may be involved in initial stimulation of the inflammasome pathway.
Overall, the HCV-induced inflammasome activation/pyroptosis field is in its infancy. Significant progress has been accomplished but more work remains to determine the effect that this pathway has on HCV infection, clearance, and ongoing liver inflammation and disease after curative therapy with DAAs.
Footnotes
Acknowledgments
The authors would like to acknowledge the members of the Russell Laboratory for their support; the scientists around the world who have worked to understand HCV; and the individuals who live or lived with HCV who donated their time, lived experience, and samples who made the work presented in this review possible.
Authors' Contributions
H.L.W. wrote the original draft. H.L.W. and R.S.R. were both involved with conceptualization, review, and editing. R.S.R. supervised and obtained funding. All authors have read and agreed to the published version of the article.
Disclaimer
The funders had no role in review design or topic, data interpretation, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
H.L.W. received the Memorial University School of Graduate Studies Aldrich Award, a Memorial University Faculty of Medicine Dean's Fellowship (MSc and PhD), a Canadian Institutes of Health Research Banting and Best Canada Graduate Scholarship—Masters, and a Canadian Network on Hepatitis C Virus Doctoral Fellowship. CanHepC is funded by a joint initiative of the Canadian Institutes of Health Research (Grant No. NPC-178912) and the Public Health Agency of Canada. Research in this field of virus-induced pyroptosis is supported by a Canadian Institutes of Health Research grant to R.S.R (Grant No. FRN#PJT-159675) as well as by the Medical Research Foundation, Faculty of Medicine, Memorial University.