
Abstract
The Heartland virus (HRTV) is a tick-borne human pathogenic phlebovirus that primarily causes leukopenia and thrombocytopenia. It is transmitted by Amblyomma americanum type of tick, that is, notable for their aggressive biting behavior, affinity for human hosts, and high prevalence. Developing vaccines or immunizations against HRTV is gaining importance as a public-health preventive strategy. The current study was planned to prioritize a multi-epitope stable mRNA vaccine model against HRTV from lead B-cell and T-cell epitopes (with IC50 < 100 nM) of HRTV proteome following advanced immunoinformatics approaches. Model constructs were designed by linking the most potent, nonallergenic epitopes along with incorporation of human ribosomal protein adjuvant for immune response enhancement. The immunogenic potential of the coding vaccine molecule was examined via molecular docking against toll-like receptors immune receptors followed by normal mode analysis and molecular dynamics simulations-based energy minimization, molecular stability, and flexibility assessments. A robust, stable circular mRNA precursor of multi-epitopes vaccine model was designed by incorporating the Kozak consensus sequence, a start codon, and essential elements such as MHC class I trafficking domain (MITD), tPA, Goblin 5′ and 3′ Untranslated Region (UTRs), and a poly (A) tail. This strategic amalgamation ensures elevated immunogenicity and predicts a promising circular mRNA vaccine model against HRTV. The immune simulation predicted that the designed model vaccine is capable to elicit cell-mediated and humoral immune responses. The predicted circular mRNA vaccine precursor model is promising against HRTV to examine experimentally for its immunogenicity and safety features.
Introduction
Heartland virus (HRTV) is a newly identified phlebovirus RNA virus, emerged in 2009 after being isolated from two northwestern Missouri farmers, exhibiting symptoms such as fever, leukopenia, and thrombocytopenia (McMullan et al., 2012). The HRTV symptoms resemble Ehrlichiosis, encompassing fatigue, fever, leukopenia, and thrombocytopenia (Savage et al., 2013). HRTV-mediated infection frequently leads to hospitalization and results in fatalities (Bosco-Lauth et al., 2015). The Amblyomma americanum tick is a host organism responsible for transmitting HRTV and acts as a potential vector for HRTV-borne infection (Godsey et al., 2016). Serological surveys identified the existence of HRTV-specific antibodies in various mammals, including raccoons and white-tailed deer that may possibly act as HRTV hosts (Bosco-Lauth et al., 2015; Riemersma and Komar, 2015).
HRTV exhibits a notable connection to maintain its distinctiveness from the virus causing severe fever and thrombocytopenia syndrome (SFTSV), a pathogen detected in China, as well as in the surrounding regions (Kim et al., 2013; Takahashi et al., 2014). SFTSV infection manifests with fever, thrombocytopenia, and leukopenia, showcasing severe symptoms such as neurological afflictions and hemorrhaging. Mortality rates vary between 6% and 17% among SFTSV cases, underscoring the critical nature of this emerging health concern (Deng et al., 2013; Gai et al., 2012). A majority of fatalities caused by SFTSV in China have been observed among individuals aged over 70 years (Ding et al., 2013).
The confirmation of transmission of HRTV is a pivotal, revealing instances of vertical infection and cofeeding transmission among A. americanum. Remarkably, a 33% vertical infection rate was observed from adult females to larvae in controlled settings. Intriguingly, only one out of 386 pools of molted nymphs, derived from cofeeding larvae, tested positive for HRTV. This indicates a low maximum-likelihood estimate of infection rate at 0.52 per 1,000 (Muehlenbachs et al., 2014). The significance of these findings was underscored by the documented occurrence of over 35 human HRTV cases within the distribution range of A. americanum. Serological testing of wildlife in regions adjacent to index human cases and widely separated areas in the eastern United States, where A. americanum are present, identified raccoons and white-tailed deer as potential hosts (Pastula et al., 2014). Responding to the urgency of the situation, the Centers for Disease Control and Prevention initiated a national protocol in 2013 to comprehensively evaluate patients across the United States, aiming to unveil evidence of HRTV disease, define the disease geographical distribution, elucidate HRTV epidemiology and clinical characteristics, and devise diagnostic assays for this emerging virus (Brault et al., 2018).
Currently, there is no vaccine for the prevention or treatment of HRTV-mediated infection. As diseases are transmitted through ticks or other insects, safeguarding oneself involves the judicious use of insect repellents, adopting protective clothing such as long sleeves and pants, steering clear of densely vegetated areas, and conscientiously inspecting for ticks post-outdoor activities. It is crucial for health care providers to consider HRTV testing when patients exhibit fever, leukopenia, and thrombocytopenia without a more apparent cause (Bosco-Lauth et al., 2015).
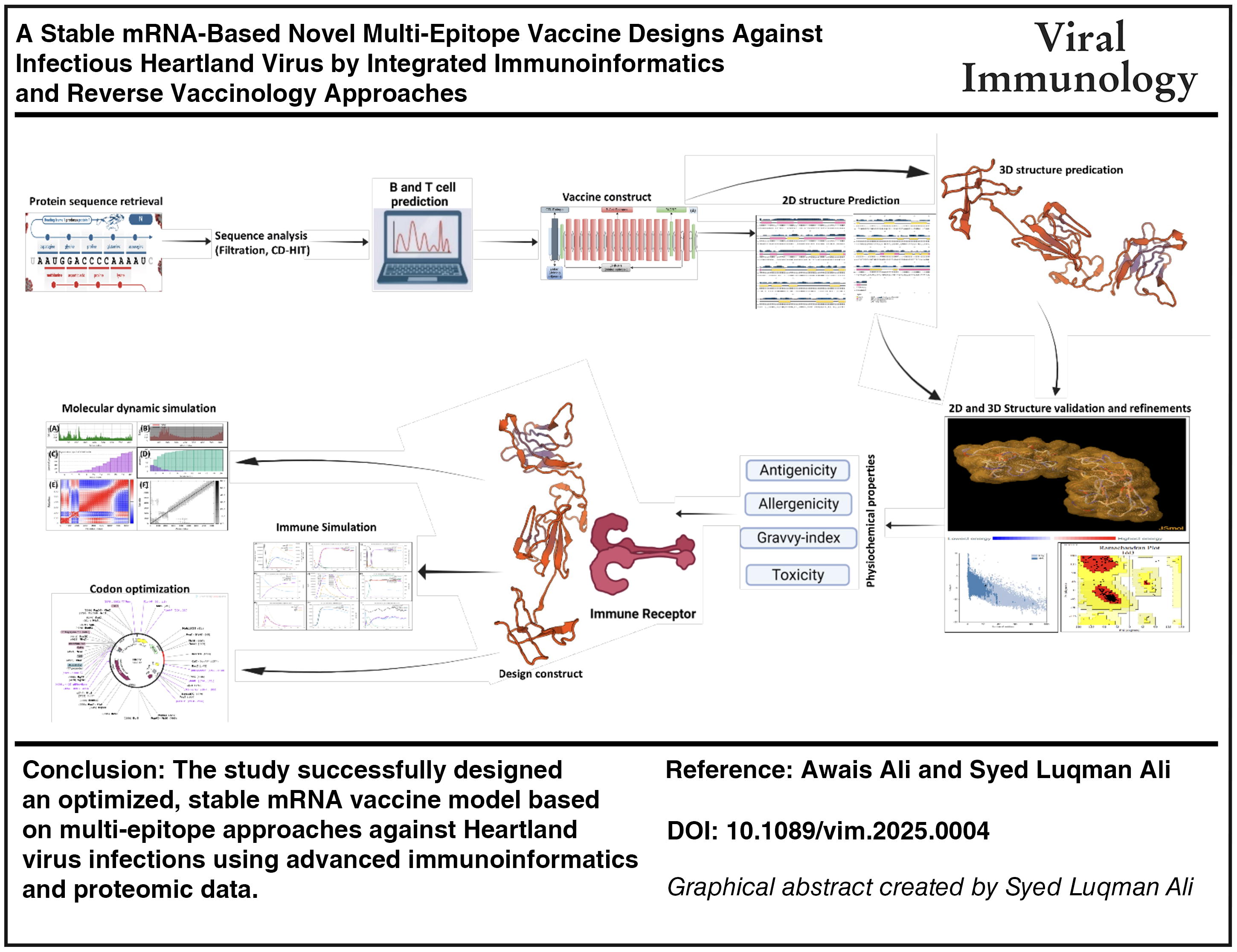
Circular RNA (circRNA) vaccines are closed loops, ultra-stable for easy storage and transport, even at room temperature (Kim et al., 2023). CircRNA vaccines are reported to lead stronger, longer-lasting immunity with limited or no side effects. In addition to combat viral infection, the circRNA vaccines are being tested against cancer and autoimmune diseases (Ali et al., 2023a; Borah et al., 2021). CircRNA vaccines hold immense promise for revolutionizing health care, though still the circRNA vaccines-based therapies are in early stages (Bai et al., 2022). The current study aimed to design an effective circRNA vaccine against HRTV by analyzing the viral complete proteome data. The top-ranked immunogenic, host non-cross reactive T-cell and B-cell epitopes from HRTV vaccine candidate proteins were utilized in designing chimeric vaccine constructs. Several models of chimeric vaccine constructs were assessed, and the lead construct sequence was subjected to circRNA vaccine designing by utilizing additional helper sequences. The validity of the model was finally evaluated.
Methodology
Vaccine candidate proteins prioritization
The National Center of Biotechnology Information (NCBI) facilitates the acquisition of full proteome sequences of HRTV (Taxonomy ID: 1216928). The data were accessed on June 6, 2023, for the analysis being pursued in the current study. The sequences redundancy was eliminated to yield a distinct, nonparalogous protein sequences set by employing the CD-hit suite with an 80% sequence similarity threshold (Huang et al., 2010). Homology of pathogen sequences against human host proteome was examined via BLASTp following the parameters as according to Aiman et al. (2023). The immunogenic and toxicity potential of HRTV proteins was assessed by following the VaxiJen v 2.0 and ToxinPred platform as according to (Ali et al., 2023c, 2023d). Additionally, the AllerTOP2.0 web platform was utilized for allergenicity assessment (Dimitrov et al., 2013).
Multi-epitope vaccine construct designing, structural modeling, and evaluation
The HRTV vaccine candidate proteins predicted in the above steps were prioritized for promising immunogenic T-cells epitopes prediction via the Immune Epitope Database (IEBD) server as according to the IC50 threshold criteria (Atukpa et al., 2024; Zhuang et al., 2024a). The IEBD prediction is based on seven distinct sets of Human Leukocyte Antigens (HLAs) alleles to predict strong HLA binder’s epitopes via following the Stabilized Matrix Method (SMM) method (Nielsen et al., 2007). ABCPred web server was used to predict the pivotal B-cell epitopes (Aziz et al., Zhuang et al., 2024b). The predicted epitopes were further examined for antigenic potential as antigens and adverse effect of allergenicity following the VaxiJen and Allertop 2.0 tools. The epitopes exhibited highest antigenic reactivity while maintaining nonallergenic attributes were prioritized for multi-epitope vaccine (MEV) designing. A potent immune-boosting adjuvants were utilized in MEV constructs to elevate the immune response effectiveness and to trigger enduring innate and adaptive immune reactions (Ali et al., 2024b; Nezafat et al., 2016). The Heparin-Binding Hemagglutinin (HBHA) protein, β-defensin, 50S ribosomal protein L7/L12 adjuvants, and HBHA conserved peptide sequences were used in MEV construct as adjuvants (Ali et al., 2024c). The EAAAK linkers and the adjuvant sequences were combined at the N-terminus of the periodized epitopes. The selected epitopes are suitable to align via HEYGAEALERAG and GGGS linkers application (Sette and Rappuoli, 2010). By utilizing innovative linker strategy, we harnessed the potential of the EAAAK linker as a rigid spacer, deftly anchoring the adjuvant’s N-terminus to the epitope (Ali et al., 2025). These linkers were properly arranged in model vaccine construct in order to enhance expression and bolster bioactivity, as well as to orchestrate a symphony of heightened immunogenic responses (Solanki and Tiwari, 2018). Subsequently, the MEV constructs underwent a comprehensive evaluation of their antigenic and allergenic characteristics by leveraging the VaxiJen (Doytchinova and Flower, 2007) and AllerTop v.20 resources as according to the criteria of Aiman et al. (2023). Moreover, the ProtParam web-based application was employed to assess an array of pivotal physicochemical attributes, including molecular weight, theoretical isoelectric point (pI), hydrophobicity (GRAVY), aliphatic index, instability, and grand average of hydropathicity of tailored MEV constructs (Ali et al., 2023c). The secondary and tertiary structures of the MEV constructs were modeled via PSIPRED (McGuffin et al., 2000) and SWISS-MODEL (Biasini et al., 2014) resources. The structural integrity and accuracy of the model structures were assessed via RAMPAGE (Lovell et al., 2003) and ProSA-web (Wiederstein and Sippl, 2007).
Immunogenic inspection of MEV via molecular docking against immune-receptor molecules
Toll-like receptors (TLRs) are indispensable orchestrators of the inflammatory pathway and play vital roles in shaping immunogenic responses against infections. These essential regulators finely tune the immune reactions, ensuring optimal defense mechanisms against various pathogens (Thompson and Iwasaki, 2008). The innate immune system of the host recognizes these intruders, launching a tailored response through TLR recognition. In particular, TLRs assume a critical function in pinpointing distinct components of viruses, such as nucleic acids and envelope glycoproteins, inciting a series of cascades encompassing the generation of Interferon (IFN), inflammatory cytokines, and chemokines. Moreover, TLRs orchestrate the maturation of dendritic cells, triggering adaptive immune reactions (Ali A, Ali SL, Alamri A, et al., 2024). Therefore, gauging the interplay between TLRs and tailored vaccines becomes imperative for anticipating the intricacies of the ensuing immune response (Lawrence and Escudero-Pérez, 2022). The TLR3 (PDB ID: 2a0z), TLR4 (PDB ID: 3FXI), and TLR8 (PDB ID: 3w3m) molecules were selected in the current study to evaluate the molecular binding affinity potential of the designed MEVs via molecular docking inspection. The HDOCK server is a suite of integrated tools for rapid and precise protein–protein docking (Yan et al., 2020). We harnessed the capabilities of the HDOCK server to evaluate the binding interaction of MEV models, derived from the HRTV proteome, with the crucial human TLRs, specifically TLR4. The HDOCK server aptly uncovers protein–protein interactions by employing a unique hybrid algorithm amalgamating template-based and template-free approaches. The analysis was fortified by harnessing the prowess of the HawKDock web server for the precise estimation of binding free energies for the vaccine-TLR complex by employing the sophisticated MM/GBSA methodology (Weng et al., 2019).
Normal mode analysis and molecular dynamics simulation inspection
A comprehensive molecular dynamics (MD) simulation analysis was pursued for the prioritized vaccine-TLR complex obtained from docking analysis. The MD simulations encompassed energy minimization and the assessment of protein flexibility by employing the iMODS web server. The normal mode analysis (NMA) via iMODS adeptly replicates the coordinated functional motions intrinsic to biological macromolecules through internal (dihedral) coordinates. iMODS constructs tangible transition pathways, linking homologous macromolecular structures, uncovering latent conformational changes, and gauging elastic network potentials. With a plethora of coarse-grained atomic representations, it refines resolution and augments the depiction of complex domain dynamics using an affine-model-based arrow representation. Notably, iMOD explores the structural dynamics of proteins and their interactions, yielding insights into deformability, eigenvalues, variance, mobility profiles (B-factors), covariance maps, and elastic network data via NMA. In the current study, the docked PDB file of the prioritized complex underwent meticulous analysis through the iMODS server, yielding results that aligned with default parameters across all facets.
In addition, the GROMACS simulations emerge as an invaluable computational ally, delving into the intricacies of vaccine constructs at the atomic level. With a meticulous initiation involving force field parameter definition and simulation conditions, GROMACS orchestrates the dance of atomic motions and forces, allowing a panoramic exploration of the construct’s behavior across diverse environments. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), and GROMACS energies unravel nuanced insights into structural stability, flexibility, and thermodynamics. The Radius of Gyration evaluates compactness throughout the simulation. GROMACS MD simulations contribute in exploration of molecular interactions and structural dynamics, pivotal in fine-tuning the optimization of vaccine candidates. In addition to NMA, the GROMACS-based MD simulation was pursued to culminate the revelation of the most energetically favorable HRTV vaccine complex.
Immune response simulation of designed MEV
The tailored MEV model immune potential was further examined via a provoke immune response calculation model implemented in C-ImmSim web service (Rapin et al., 2010). The resource dissects the diverse facets of the immune system’s response to the administered vaccine by employing a PSSM model. This encompassed the dynamic reactions of pivotal components, including fluctuations in antibodies, cytokines, and interferons levels, along with the changes in T-cells and B-cells populations.
Construction of circular mRNA precursor, mRNA folding, stability, and secondary structure prediction
In pursuit of unraveling the intricate mRNA vaccine constructs, we strategically utilized two distinct servers, that is, Transcription Mfold v2.3 (Zuker, 2003) and Translation RNAfold (Gruber et al., 2008), specialized in predicting the secondary structure, thereby delving into the molecular intricacies embedded in the genetic code of the vaccine construct. The investigation based on calculation of the minimum free energy, expressed in Kcal/mol. The reduced values in this metric signify a heightened stability in the mRNA folding structure, offering profound insights into the structural robustness and resilience of the mRNA. This scrutinizes the mRNA stability and deciphers the nuanced intricacies of its folding dynamics. The optimization of the vaccine DNA involved strategic modifications at both the 30 and 50 ends. To enhance mRNA translation in eukaryotic cells, a Kozak motif was incorporated at the 30 end, while the 50 end underwent a refinement that included the addition of a stop codon. Recognizing the pivotal role of the internal ribosome entry site (IRES) in translating circular mRNAs, we chose the IRES from the CBV3 strain, leading to a transformative alteration at the 30 end of the DNA sequence. Inspired by proven success with group I catalytic introns in RNA circularization, the intron sourced from the T4 phage thymidylate synthase (Td) gene was symmetrically inserted on both sides of the DNA sequence. By integrating the homology arms and spacers into the vaccine DNA sequence, the RNA circularization efficiency was elevated. The designing of the circular mRNA precursor employed a complementary base pairing approach, culminating in a constructed RNA vaccine. In addition to MEV construct sequence, the final circRNA vaccine molecule was engineered to encompasses homology arms at both ends to enhance splicing efficiency, Group I permuted intron-exon sequences, T1 and T2 from the Td gene of T4 phage (Ford and Ares, 1994), Coxsackievirus B3 (CVB3) IRES sequence, a spacer sequence for improved circulation efficiency (Ali et al., 2025; Wesselhoeft et al., 2018), a Kozak sequence for enhanced RNA stability and translation efficiency (Kim et al., 2022), tissue plasminogen activator (tPA) sequence, MITD, and a TAA stop codon (Fig. 1). The secondary structure of the mRNA vaccine was predicted using the RNAfold web server. Employing McCaskill’s algorithm, the server calculated the minimal free energy (MFE) of the RNA secondary structure, providing invaluable insights into the MFE and the secondary structure, as well as the centroid secondary structure (Zhuang et al., 2024a, 2024b).
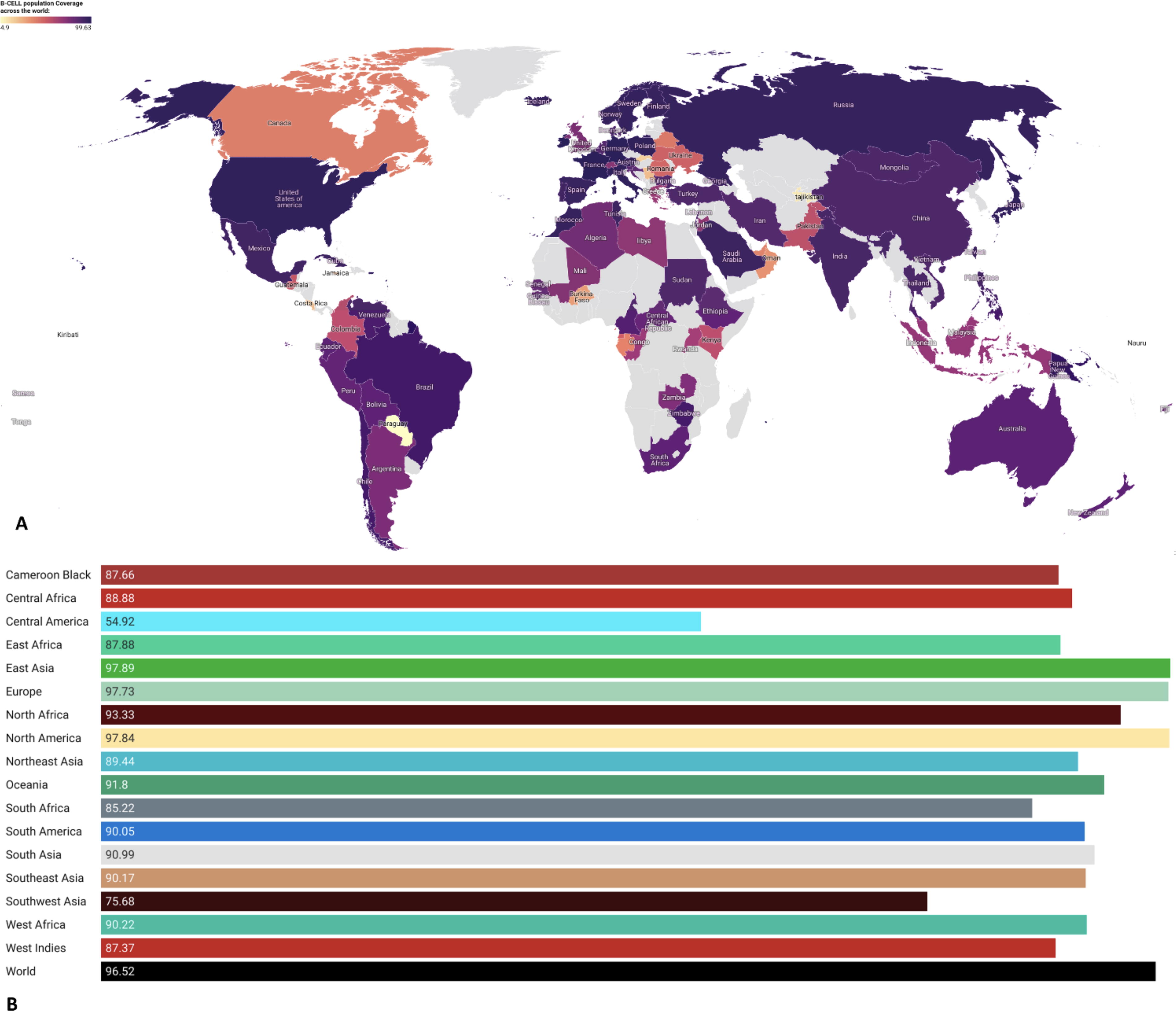
Population coverage inferences of the lead epitopes.
Results
Epitopes prediction, MEVs designing, and validation
A dataset of all available 124 HRTVs’ protein sequences was compiled from the GenBank, NCBI database. Afterward, 22 nonparalogous sequences were obtained by filtering redundant entries via 80% sequence similarity threshold using CD-Hit resource. The exclusion of human-homologous sequences, a refined collection of eight HRTV sequences was remained. Five among these proteins were found nonallergenic and demonstrated pronounced antigenicity surpassing the VaxiJen 2.0 threshold of 0.4 scores. Notably, these identified vaccine candidates exhibit characteristics of highly antigenicity, nonallergenicity, and nontoxicity (Supplementary Table S1), signifying a targeted immune response toward the virus rather than the host, as indicated by rigorous prediction analysis (Gupta et al., 2013). The subsequent analysis was pursued to identify prime immunogenic epitopes from these five HRTV vaccine candidate proteins and to formulate potent chimeric vaccine blueprints against HRTV. The T-cell epitopes (both MHC-I and MHC-II) were predicted from IEDB server calculations based on a stringent IC50 threshold of <100 nm (Supplementary Table S2). Complementary prediction for overlapped B-cell epitopes accompanied with ABCpred scores surpassing 0.8 with a specificity rating of 75%. This identified three intersecting lead epitopes for each key protein. The prioritization of 15 epitopes was based on their remarkable antigenicity, positive IFN-impact, negligible toxicity, and nonallergenic potential (Table 1).
Physiochemical Properties of the Prime Prioritized and Overlapped Cells and T-Cells Epitopes
The primary goal was to identify key epitopes that possess the potential to trigger both robust humoral and cell-mediated immune reactions, as well as host interferon responses. The selected epitopes’ consistency across multiple HRTV strains was validated. Notably, the selected epitopes exhibit a comprehensive global coverage of 100% across diverse populations (Supplementary Table S2). The IEDB analysis notably underscored substantial population coverage of the predicted epitopes particularly in the regions and countries reported with HRTV infection (Fig. 1).
MEV construction, validation, and physiochemical properties
A combination of carefully selected lead epitopes forms the basis for a novel chimeric vaccine construct linking via GGGS and HEYGAEALERAG linkers (Fig. 2). These linkers develop the backbone of stability and independent functionality for each epitope in the construct. Four distinct adjuvants as shown in the Supplementary Data (Fig S1A, S2A and S3A), that is, HBHA protein, β-defensin, 50S ribosomal protein L7/L12, and HBHA conserved peptide sequences were incorporated in the constructs at the N-terminal using EAAAK linkers to invigorate immunogenic responses. The PADRE peptide sequences were strategically incorporated to mitigate HLA-DR variation issues across populations to improve global adaptability. Previous studies underscored the augmented immune protection and robust cytotoxic T lymphocyte (CTL) responses inherent in PADRE-infused vaccine constructs. Comprehensive immunological analysis unveiled the nonallergenic, nontoxic features for all four constructs. The Antigenpro’s antigenicity scores surpassing 0.9 underpin their robust antigenic nature, derived from meticulous peptide sequence cross-validation and protective features identification. The VaxiJen 2.0 scores, ranging from 0.5397 to 0.5834, exhibit sufficient antigenicity potential of the constructs. The molecular weight, hydrophilic tendency, and theoretical pI values of the constructs were in ideal ranges (Table 2). Aliphatic index scores of 45.00–58.11 underscore thermos stability of MEV constructs. The predicted instability index scores, that is, varying from 15.28 to 25.33, predicting construct stability across varied temperature ranges. The designed vaccine constructs demonstrated diverse secondary structure composition, encompassing α-helices, β-strands, random coils, and extended strains. Albeit with differing adjuvants, the constructs were predicted to radiate robust immunogenic response initiation within the human host. These models are promising, and worthy to pursue for experimental validation that remains as an imperative step in affirming these results’ accuracy.
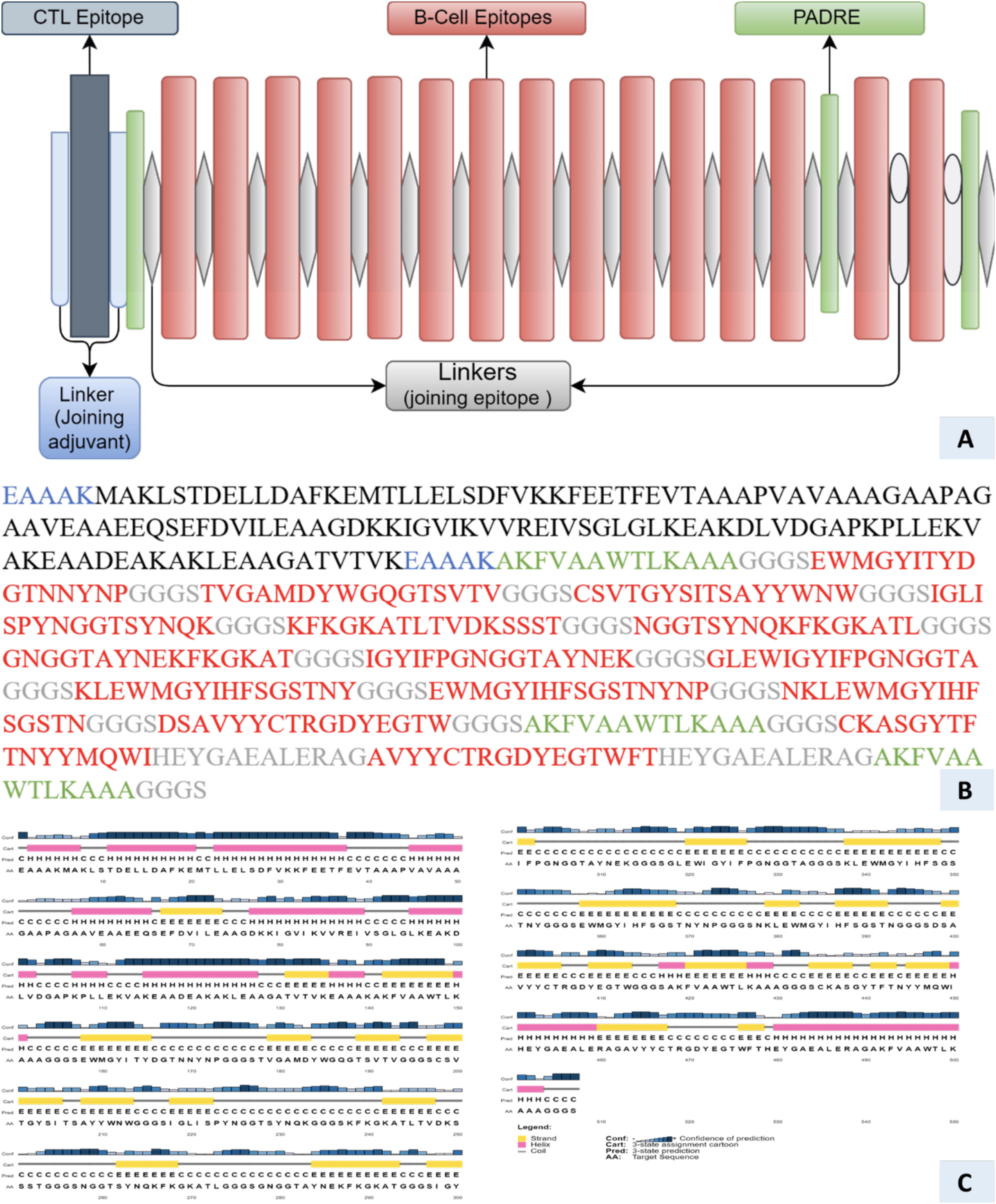
Physiochemical Properties of the Vaccine Constructs Using ProtParam and JCAT Servers
Positive GRAVY value indicated that a protein is hydrophobic, while negative value indicates that it is hydrophilic.
Subsequently, the constructs were subjected to comprehensive protein 3D structure models designing (Supplementary Fig. S1B, S2B and S3B). The Ramachandran plot analysis and ProSA-web showcasing the highest amino acid residues proportion of the designed 3D models within the favorable zone with least outlier percentage (Supplementary Fig. S1C, S2C and S3C). Concurrently, ProSA-web diligently assessed model quality, and Z-scores adhere to the acceptable range for protein structures of comparable dimensions (Supplementary Fig. S1D, S2D, and S3D) (Table 3).
3D Structural Validation of Vaccine Constructs via ERRAT, PROCHECK, and ProSA-Web Servers
HRTV, Heartland virus.
Molecular binding assessment of HRTV constructs against immune receptors
Molecular docking analysis is effective for discerning the optimal interactions between the designed vaccine models with TLR3, TLR4, and TLR8 immune receptor molecules. The HDock server utilizes a blind docking technique and facilitates the assessment of binding affinities of the vaccine molecule with human TLRs immune receptors. To enhance precision, the top ten HDock outcomes were subjected to further refinement via the HawkDock web server, thereby augmenting binding energy. Although the binding scores for all the vaccine-receptor complexes were somehow parity; however, the V4 construct remarkably displayed the most favorable binding energy with the abovementioned TLR receptors (Fig. 3, Table 4).

The interaction between the vaccine candidates and immune-receptor molecule.
Binding Energies and Docking Scores of Designed Vaccine Constructs
HRTV, Heartland virus; RMSD, root mean square deviation.
Employing the iMODs webserver, a comprehensive NMA was conducted to evaluate the molecular stability and dynamic motions of the top-ranked V4-TLR4 complex (Fig. 4). This analysis revealed pivotal peaks on the deformability graph, pinpointing regions of significant main chain deformation within the V4-TLR4 complex, effectively identifying potential “hinges” or “linkers” (Fig. 4A). The experimental B-factor plot seamlessly demonstrates the correlation between NMA mobility and 3D structure model of the V4-TLR4 docked complex, offering insight into the average RMSD values of the complex (Fig. 6B). The calculated eigenvalue of 6.635862e-06 for the V4-TLR4 complex conveys the intrinsic stiffness inherent in each normal mode (Fig. 4C). Further insights emerge as each normal mode, depicted in both individual (purple) and cumulative (green) variance bars, reveals an intriguing negative correlation between variance and eigenvalue (Fig. 4D). The interactional dynamics between the complex’s constituents find expression in a covariance map, with interrelated motions depicted through correlated (red), uncorrelated (white), and anticorrelated (blue) atomic motions within the HRTV-V4-TLR4 complex (Fig. 4E). The intricate electric network map unfurls showcasing pairs of interconnected atoms held by springs in the V4-TLR4 complex. Colored dots, varying in darkness, ingeniously symbolize the assembly between sizable molecules and their respective stiffness, with deeper grays denoting sturdier springs (Fig. 4F). The overarching revelations of the NMA elegantly indicate steadfast interactions in the docked complex of TLR4 and the prioritized vaccine HRTV-V4.

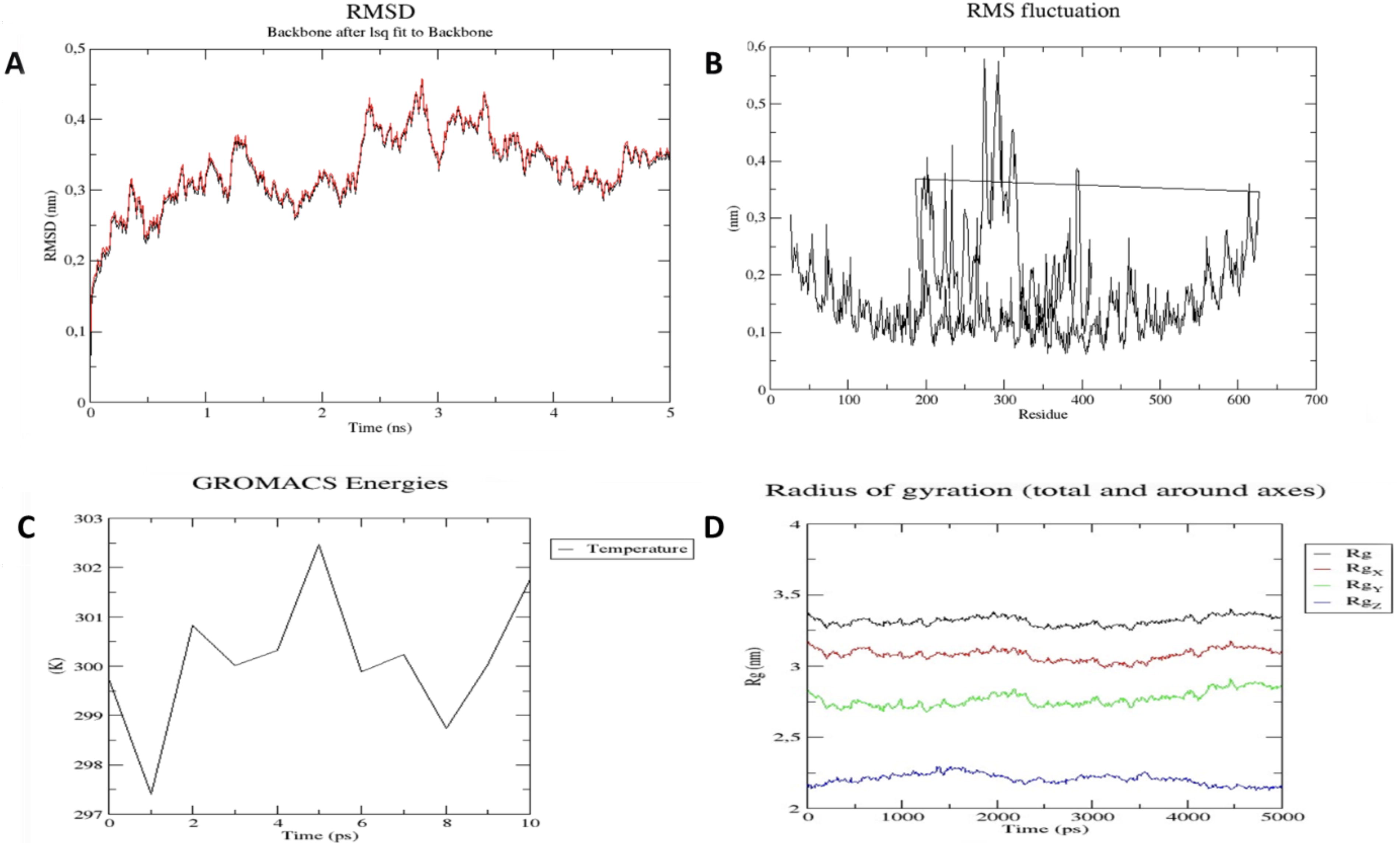
MD simulation of the designed vaccine construct with TLR4.
Additional analysis of MD simulation inferred the intricate dynamics of the HRTV-TLR4-V4 complex. Employing RMSD, RMSF, and Rg analyses, we gained nuanced insights into the construct’s innate flexibility and robust stability. RMSD plot (Fig. 5A) demonstrates the vaccine–receptor complex’s resilience, revealing initial deviations that gracefully stabilized post 5 ns, ensuring a consistently stable structure with deviations below 0.2 nm. The RMSF plot (Fig. 5B) portrayed minimal fluctuations in atomic positions, emphasizing the steadfastness of amino acid residues throughout the simulation. Delving into GROMACS energies, a comprehensive view of the system’s thermodynamic stability and intricate intermolecular interactions was depicted (Fig. 5C). The Rg plot (Fig. 5D) unfolded a compelling narrative, showcasing minimal deviation and a flat curve, affirming the compactness and resilience of the complex structure. In essence, these holistic MD simulations underscored the structural flexibility and stability of the vaccine-receptor complex, that is, promising to elicit an effective immune response.
Immune simulation
The immune simulation predicted a remarkable increase in both primary and secondary immune reactions against the administration of HRTV-V4 proposed vaccine model (Fig. 6). A notable flow was predicted in IgG1 + IgG2, IgM, and IgM + IgG antibody levels, showcasing the robust amplification of immune responses (Fig. 6A). Multiple exposure of the HRTV-V4 model vaccine led to a distinct expansion of B-cell populations, resulting in the establishment of enduring humoral immune memory (Fig. 6B and C). Secondary and tertiary immune responses were marked by an escalated emergence of cytotoxic and helper T cells, accompanied by a significant decline in the antigen population (Fig. 6D and E). Furthermore, the production of natural killer cells, dendritic cells, and macrophages was predicted to exhibit continuous growth during each immunization (Fig. 6F–H). Impressively heightened levels of cytokines and interleukins were also observed to be sustained during the entire immunization period, indicating enduring immune stimulation due to persistent antigen exposure (Fig. 6I). The comprehensive immune simulation predicted that the prioritized vaccine model is immense promising in eliciting robust innate and adaptive immune responses within the human immune system against HRTV.

Construction of stable circular mRNA vaccine precursors and stability assessment
The prioritized HRTV-V4 vaccine construct was prioritized for the construction of a stable circular mRNA vaccine, featuring a CAI of 1.94% and a GC content of 47.09%. These metrics indicate alignment with the ideal codon, boasting high density and thermos-stability. The final circular mRNA vaccine precursor, meticulously crafted from the 5′ to 3′ terminal, incorporates 50 homology arms, a T1 region, CBV3 IRES, a Kozak sequence, signal peptide (tPA), EAAAK linker, β-defensin adjuvant, top prioritized prime epitopes of HRTV, PADRE peptide, MITD sequence, and a 3′ homology arm. The optimal secondary structure of this vaccine precursor mRNA was assessed by the RNAfold server. This analysis demonstrated a notable minimum free energy of −518.60 kcal/mol for the vaccine construct mRNA molecule (Fig. 7A), and the centroid secondary structure displayed a slightly lower minimum free energy of −442.37 kcal/mol (Fig. 7B). Additionally, the minimum free energy of the most favorable secondary structure of the vaccine mRNA was calculated as −541.61 kcal/mol using the mFold v2.3 server. This analysis indicates that the vaccine mRNA secondary structure possesses significant stability characteristics and enhance structural viability upon in vivo expression.

A strategic layout of the designing of HRTV chimeric vaccine precursor circular mRNA model and its molecular stability assessment.
Discussion
In recent years, a series of viral epidemics have silently emerged, casting serious health and economic burden in human societies. Cutting-edge breakthroughs in immunoinformatics, bioinformatics, and structural vaccinomics have ushered in a new era of antigen exploration, revolutionizing the landscape of vaccine development to combat a spectrum of pathogenic threats (Gupta et al., 2013). Prior investigations have illuminated the superior efficacy of multi-epitope peptide vaccines compared to single-epitope counterparts, stimulating defenses against infectious agents (Bourdette et al., 2005; López et al., 2001). The current study delved into the designing an effective stable circular mRNA vaccine model against HRTV with an assembly of potent immunogenic epitopic signatures from pathogenic proteins, orchestrating a precise humoral immune response toward targeted antigenic epitopes, culminating in an enhance and safer shield of immunity (Manzoor et al., 2023; Tahir ul Qamar et al., 2020). The HRTV proteome data from Genbank, NCBI were utilized, and the physicochemical attributes of the viral proteins were scrutinized via the ProtParam server to glean their inherent characteristics (Das et al., 2015). The VaxiJen platform extensively evaluated the compiled protein sequences to identify the most efficacious antigenic proteins that possesses the capacity to induce robust immunity. This resulted in identification of five viral proteins, that is, QKP69782.1, QKP69786.1, QKP69780.1, QKP69788.1, and QKP69790.1, which were identified to play a vital role in their interaction with the host cell (Russell et al., 2008). Besides, the VaxiJen resource assessed each procured protein sequence and exhumed the most formidable antigenic candidate with the potential to bestow immunity (Ali et al., 2023b, 2023c).
The designing of a multi-epitope peptide-based vaccine involved characterizing B-cells and T-cells epitopes followed by identifying common antigenic epitopes to induce a strong immune response against HRTV. The top-ranked vaccine construct against HRTV identified in the current study, encompassing 15 epitopes with 507 amino acids, a PADRE sequence, CTL adjuvant, and a necessary peptide linker, strategically merged the adjuvants and EAAAK peptide linker at the N-terminal end. Additionally, the PADRE sequence, acting as a helper T-cell epitope, was coupled with the EAAAK linker to enhance CTL response across diverse antigens. Computational tools and servers verified the vaccine’s acidic feature compatible to the human physiological pH range and its thermostability based on the aliphatic index score (Ikai, 1980). The positive GRAVY value underscored the vaccine model hydrophobic nature without substantial interaction with water (Ali et al., 2017). The reduced scores for the minimum free energy in the secondary structure of the vaccine mRNA imply its molecular stability. The HRTV whole protein-derived peptide vaccine’s 3D structure was designed using the SWISS MODEL server and validated by ProSA and PROCHECK web servers. ProSA’s negative “Z” score (−4.02) affirmed its structural quality (Bhattacharya et al., 2022). The downstream molecular docking inferences ensured strong interaction of the prioritized model with TLR4, a key immune receptor, and the TLR4/V4 and vaccine candidate docked complex exhibited a significant binding affinity of −45.82 kcal/mol. The NMA and molecular dynamic simulations highlighted the construct’s superior flexibility, while immune simulation profiling showcased its potential for both humoral and cellular immune responses with the secondary response surpassing the primary one. The stringent in silico evaluation indicated that the model anti-HRTV vaccine constructs exhibit ideal structural and functional attribution, enabling these models as promising next-generation vaccine against HRTV. Although the finding of the current study paves a new way to design candidate vaccine against HRTV-mediated infection, however, there is a need to validate the model’s efficacy via experimental and clinical follow-up.
Conclusion
The study embarked on designing a multi-epitope-based vaccine models against emerging HRTV using proteo-genomic data of the virus and employing the cutting-edge immunoinformatics approaches. The scientific exploration commenced by extracting the complete viral proteome, which paved the way for the prediction of potent B-cell and T-cell immunogenic epitopes, critical for instigating robust immunity. To elevate the immunogenic potential of these promising epitopes, they were deftly intertwined with tailor-made linkers and potent immunogenic adjuvants. Subsequently, we pursued comprehensive evaluations, including binding affinities assessment against immune-receptors, molecular stability analyses of the receptor-model-vaccine complexes, as well as allergenicity, antigenicity, and physicochemical attributes. Although the multifaceted approaches followed in the current study culminated with designing of an optimized multi-epitopes chimeric, stable circular mRNA vaccine models based on multi-epitopes against HRTV infections, still it is imperative to corroborate the designed models through rigorous experimental validation.
RAMPAGE servers (http://mordred.bioc.cam.ac.uk/rapper/rampage.php)
ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php)
HDOCK server, http://hdock.phys.hust.edu.cn/
SWISS-MODEL web-server (https://swissmodel.expasy.org/)
AllerTop v.20 (https://www.ddg-pharmfac.net/AllerTOP/)
ProtParam, web-based (https://web.expasy.org/protparam/)
ABCPred online web-server (http://crdd.osdd.net/raghava/abcpred/)
HawKDock web server (http://cadd.zju.edu.cn/hawkdock/)
C-ImmSim web-service http://www.cbs.dtu.dk/services/CImmSim-10.1/
SnapGene tool (https://www.snapgene.com/)
Translation Tool http://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm
Mfold v2.3 (http://www.unafold.org/mfold/applications/rna-folding-form-v2.php)
RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)
Footnotes
Acknowledgment
The authors are very thankful to Abdul Wali Khan University Mardan for providing the best environment and facilities for this research.
Authors’ Contributions
A.A.: Methodology development, software validation, formal analysis, investigation, resource management, data curation, and the drafting of the original article. S.L.A.: Conceptualization of the study, supervised the research, managed project administration, and contributed to visualization, review, and article editing.
Data Availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Author Disclosure Statement
The corresponding authors, on behalf of all the authors of this submission, disclose any potential competing interests that might influence their work. The authors confirm that there are no competing interests to declare.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.