
Abstract
Abstract
We implemented a noninvasive optical method for the fast control of Cre recombinase in single cells of a live zebrafish embryo. Optical uncaging of the caged precursor of a nonendogeneous steroid by one- or two-photon illumination was used to restore Cre activity of the CreERT2 fusion protein in specific target cells. This method labels single cells irreversibly by inducing recombination in an appropriate reporter transgenic animal and thereby can achieve high spatiotemporal resolution in the control of gene expression. This technique could be used more generally to investigate important physiological processes (e.g., in embryogenesis, organ regeneration, or carcinogenesis) with high spatiotemporal resolution (single cell and 10-min scales).
Introduction

Strategies used to make accurate spatiotemporal regulation of Cre recombination. (
Materials and Methods
Plasmid construction and transgenic line
Plasmid coding the CreERT2 was a kind gift of D. Metzger (Strasbourg) and the plasmid coding the Cre was a kind gift of W. Wurst (Munich). The ef1α:loxP-GFP-loxP-dsRed2 transgenic line was established with standard conditions using a Tol2 transposon system and the pT2K-XIG plasmid backbone. 22 A detailed description of the cloning procedures is available on request.
Microinjection
Tg zebrafish embryos were obtained by natural spawnings according to the zebrafish book, 23 injected at one-cell stage with mRNA synthesized with an in vitro transcription kit (mMessage mMachine; Ambion), and stored at −80°C in small aliquots.
Pharmacological treatments and CreERT2 activation
4-Hydroxy-cyclofen (4-OH cyclofen or Ind) and caged 4-OH cyclofen (cInd) were prepared as previously described. 24 Embryos injected with CreERT2 mRNA were subsequently dechorionated by pronase treatment at dome stage before incubation for at least 90 min in an aqueous solution (embryo medium) of the various substrates (e.g., cInd). Ind was photoreleased at different stages of development. Embryos were observed at 2 days postfertilization (dpf ) after UV illumination. Embryos positive for either GFP or dsRed fluorescence (Cre-induced recombination) were scored under microscope in a double-blind protocol.
Illumination experiments
One-photon excitation experiments were performed at 20°C, with a bench top UV lamp (365 nm, 8 W; Fisher Bioblock) providing a typical 4 × 10−5 Einstein min−1 photon flux 25 (essentially Gaussian spectral dispersion around 350 nm with a 40 nm width at half height). For two-photon excitation, a 40 × 0.8 NA water-immersion objective (Olympus) was used to image the embryos on a CCD camera (Andor Luca) and locate the focal spot of the two-photon excitation. Illumination (200 fs, 76 MHz, 750 nm) was provided by a mode-locked Ti-Sapphire laser (Mira, Coherent). The incident power at the sample (<20 mW) was measured with a NOVA II power meter (Laser Measurement Instruments). The geometrical characteristics of the focal point were determined by fluorescence correlation spectroscopy measurements using a fluorescein solution of known concentration (100 nM in 0.1 M NaOH).
Image acquisition
The fluorescence images of the embryos were acquired using confocal microscopes Leica TCS SP2 AOBS or Zeiss Axiovert 200M LSM510-Meta. In a given series of experiments, all the conditions (EM gain, exposure duration, lamp power, etc.) were identical to allow for a comparison of the observed fluorescence intensities.
Quantitative PCR
Total DNA was extracted from 30 embryos, and 10 ng was used for a quantitative PCR assay. Quantitative PCR was performed using a Roche light cycler 480 detection system and SYBR green labeling system (Qiagen). Details concerning the parameters used are available on request. LoxP recombination was normalized to the number of transgene copies in each sample. Each sample was tested in triplicate. Specific primers were designed by Primer 3 online software (http://frodo.wi.mit.edu/primer3/input.htm): Recombine transgene: forward (5′-AGGTCGACCGTTTAGGGAAC-3′), reverse (5′-ACCTTGAAGCGCATGAACTC-3′); full transgene: forward (5′-CAACGAGGACTACACCATCG-3′), reverse (5′-TCTAGTTGTGGTTTGTCC-3′).
Results
For optical uncaging, one of the most desirable photophysical properties of the caged inducer is a low photodamage to the released inducer. Although 4-OHT has been used successfully as an inducer to activate CreERT2 recombinase in fish, 20 we observed that 4-OHT was susceptible to UV-induced isomerization and degradation on the typical time scales of uncaging. 24 Therefore, we looked for another inducer, structurally related to 4-OHT, but not subject to isomerization or degradation upon UV illumination. We adopted the core motif of the estrogen cyclofenil. After grafting on one of its phenol groups the basic pendant chain present in 4-OHT, we obtained the nonendogenous and water-soluble 4-OH cyclofen (Ind), which binds to the ERT2 as efficiently as 4-OHT 24 and does not isomerize or degrade upon illumination.
We assayed the toxicity of 4-OH cyclofen in live embryos. The viability test for the ef1α:loxP-GFP-loxP-dsRed2 transgenic zebrafish embryos was performed at various concentrations of the inducer: it was observed that animals develop normally for concentration of 4-OH cyclofen at least as high as 5 μM. These embryos expressed GFP only and no fluorescence of dsRED was observed irrespective of the presence or absence of the inducer in embryo medium (data not shown). It demonstrates as expected that the transgenic zebrafish line has no endogenous recombination. However, injection of 10 pg of Cre mRNA at the one-cell stage causes global recombination in all the cells of the embryo as evidenced by the expression of dsRED in 100% of Cre-injected embryos. Figure 2A shows a representative fluorescence image of dsRED in a Cre-injected embryo observed at 2 dpf. In Figure 2B–D we show that by controlling the amount of injected Cre-ERT2 mRNA and the concentration of inducer, we can tune the recombination efficiency in transgenic zebrafish embryos constitutively expressing a loxP-gfp-loxP-dsRed2 gene construct, and consequently control the activation of dsRED expression. To do this, different amounts of mRNA coding for Cre-ERT2 recombinase were injected in transgenic embryos at the one-cell stage. Figure 2B shows that the injection of 3 pg of CreERT2 mRNA at the one-cell stage causes no recombination. However, upon addition of (3 μM) 4-OH cyclofen in the embryo medium at 50% epiboly (Fig. 2C, D), global recombination (dsRED expression) was observed at 2 dpf. Spontaneous (inducer-independent) recombination after CreERT2 mRNA injection increased with the amount of injected nucleic acid, presumably because the concentration of the cytoplasmic chaperones was insufficient to complex with every CreERT2 protein synthesized. We measured the percentage of embryos that underwent recombination (partial or global) as a function of the amount of injected mRNA, in the presence or absence of Ind (Fig. 2D). Upon addition of 3 μM Ind in the embryo medium, we found a significant increase in the frequency of recombination for embryos injected with less than 5 pg of mRNA (at concentration 50 ng/μL). Thus, by appropriately tuning the amount of injected mRNA, the ratio of induced versus spontaneous recombination can be optimized. Henceforth, we fixed the injection condition at 3 pg of mRNA for all optical uncaging experiments.
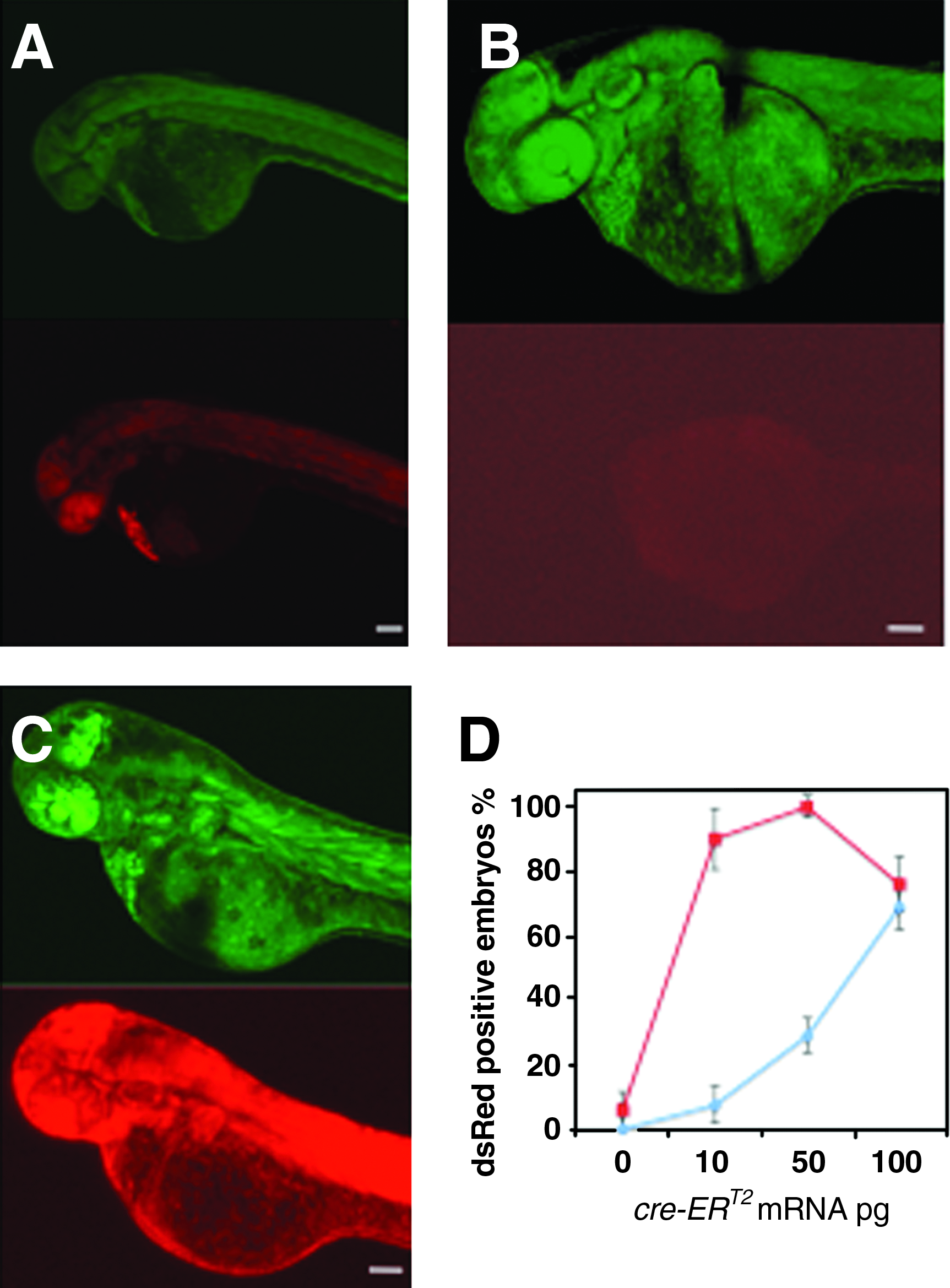
Cre-mediated recombination in the green-to-red reporter line. In Tg (ef1a: loxP-GFP-loxP-dsRed2) embryos injected with 3 pg of Cre mRNA at one-cell stage (
To test the ability of our method to locally or globally photocontrol the activity of CreERT2, we investigated the activation of the Cre-ERT2 recombinase via uncaging of caged cyclofen (cInd) in the transgenic zebrafish line mentioned earlier. For these experiments, embryos were injected with CreERT2 mRNA at the one-cell stage and incubated in embryo medium supplemented with 3 μM of cInd. In the absence of UV illumination, when observed with epifluorescence microscopy at 2 dpf, embryos showed ubiquitous expression of GFP but no recombination (dsRed expression) as shown in Figure 3A, implying that the caged ligand is indeed inactive. Upon uncaging of cInd with UV illumination at one somite, the embryos ubiquitously expressed dsRed at 2 dpf (Fig. 3B). We also checked that when the embryos were illuminated during early development (50% epiboly to six somites) for up to 4 min with the UV lamp (8 W, 365/40 nm), the recombined embryos developed normally proving that neither UV illumination nor the side product resulting from uncaging (i.e., the caging group) was detrimental to the embryo development (data not shown). One of the limits of the system can be the delay between cInd releasing and Cre recombination. We then analyzed the recombination by the CreERT2 upon cInd uncaging. Recombination was measured by quantitative PCR. Figure 3C shows that recombination occurred on the 10-min scale; it can be detected as soon as 3 min after uncaging and 13 min after uncaging 30% of the floxed sequences have been recombined. This time scale is perfectly relevant for zebrafish rapid development.
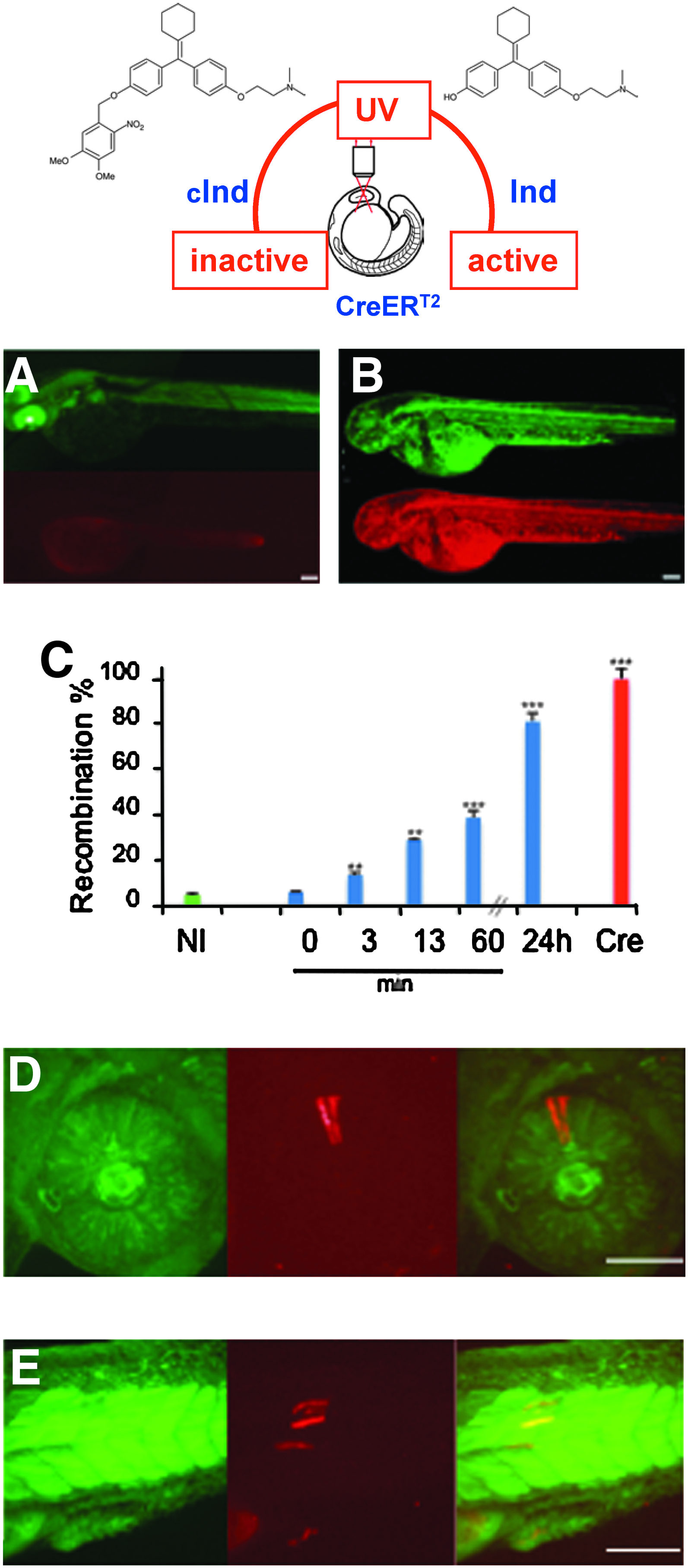
Photocontrol of CreERT2 recombinase activity in Tg reporter line. Transgenic (ef1α:loxP-GFP-loxP-dsRed2) embryos were injected with 3 pg of CreERT2 mRNA at the one-cell stage and further incubated with 3 μM of caged inducer (cInd). Photoactivation of cInd at the 3/6-somite stage resulted in dsRed expression observed here at 2 dpf. In epifluorescence microscopy, the non-UV illuminated embryos (
Finally, to target the activation in a single cell, we used two-photon illumination to release cInd in a specific cell (in a single cell of the forming retina [Fig. 3D] or in a newly formed somite [Fig. 3E]) at three- to four-somite stage of a live zebrafish embryo. This cell's progeny could be observed at 2 dpf to exhibit the characteristic red fluorescence of dsRed in an embryo that is otherwise fluoresced in green (Fig. 3D, E).
Discussion
We used two-photon excitation to quickly and noninvasively achieve spatiotemporal control of CreERT2 activity and gene expression at the single-cell level in a live animal. We show that photoreleasing 4-OH-cyclofen from a caged precursor on a second time scale is an efficient strategy to restore the function of Cre fused to the ERT2 ligand-binding domain in a live animal down to the single-cell level. This method offers a great versatility: virtually any cell or any cell combinations can be chosen. Complex temporal inductions within the same lineage are also feasible, potentially allowing a dynamic dissection of fate commitment events. Moreover, there is no longer the need for a complicated promoter engineering to achieve the envisioned Cre expression.
The inactivation of a protein fused to the ER domain has been described for various proteins (e.g., Engrailed, Otx2, Gal4, p53, kinases such as Raf-1, Cre, and Flp recombinases). 26 The strategy described here is thus compatible with the photoactivation of a wide variety of proteins. This noninvasive optical method opens opportunities for the local spatiotemporal investigation of developmental pathways, the identification of stem cells, and the study of cancer and neuronal (e.g., memory) pathways at the single-cell level in a live organism. In particular, it could be used to label individual cells and track their lineage as well as to interfere with morphogen gradients and signaling pathways with unprecedented spatiotemporal resolution. Ultimately, this strategy may provide crucial information about the regulation networks involved in development and differentiation/dedifferentiation: their timing (i.e., when are the genes/proteins activated in the cell), their dynamics (how fast are they activated), and their spread (how far is their influence felt).
Footnotes
Acknowledgments
This work has been supported by the Association pour la Recherche sur le Cancer (ARC, contract no. 3787), the ANR (PCV 2008, Proteophane), the NABI CNRS-Weizmann Institute Program (for a fellowship to D.K.S.), and the Ministère de la Recherche (for fellowships to N.G. and P.N.). P.N. is supported in part by the National Science Foundation under grant no. PHY05-51164. D.B. acknowledges partial support of a PUF ENS-UCLA grant. Work in L.B.-C. laboratory was supported by the Volkswagen Association, the EU 6th Framework Program (ZF-Models IP, contract no. LSHC-CT-2003-503466), and the Center for Protein Science-Munich. The authors thank P. Leclerc and B. Matthieu for their kind help with confocal imaging, and D. Metzger for providing the plasmids coding the Cre-ERT.
Disclosure Statement
No competing financial interests exist.