
Abstract
Abstract
Mechanisms of bone formation and skeletal development have been successfully investigated in zebrafish using a variety of in vivo approaches, but in vitro studies have been hindered due to a lack of homologous cell lines capable of producing an extracellular matrix (ECM) suitable for mineral deposition. Here we describe the development and characterization of a new cell line termed ZFB1, derived from zebrafish calcified tissues. ZFB1 cells have an epithelium-like phenotype, grow at 28°C in a regular L-15 medium supplemented with 15% of fetal bovine serum, and are maintained and manipulated using standard methods (e.g., trypsinization, cryopreservation, and transfection). They can therefore be propagated and maintained easily in most cell culture facilities. ZFB1 cells show aneuploidy with 2n=78 chromosomes, indicative of cell transformation. Furthermore, because DNA can be efficiently delivered into their intracellular space by nucleofection, ZFB1 cells are suitable for gene targeting approaches and for assessing gene promoter activity. ZFB1 cells can also differentiate toward osteoblast or chondroblast lineages, as demonstrated by expression of osteoblast- and chondrocyte-specific markers, they exhibit an alkaline phosphatase activity, a marker of bone formation in vivo, and they can mineralize their ECM. Therefore, they represent a valuable zebrafish-derived in vitro system for investigating bone cell differentiation and extracellular matrix mineralization.
Introduction
Materials and Methods
Zebrafish maintenance
Wild-type zebrafish were maintained in 10-L aquaria with water recirculation (ZebTec housing System; Tecniplast) under the following conditions: temperature, 28°C; photoperiod, 14/10-h light/dark; pH, 7.5; conductivity, 660 μS; density, two fish L−1. Fish were fed twice a day with dry food (Tetramin flake C) and live Artemia nauplii.
Tissue collection and processing
Three-month-old healthy zebrafish ranging from 3 to 4 cm in length were given a lethal anesthesia of 0.015% tricaine (Sigma-Aldrich). Calcified tissues, that is, vertebra, upper and lower jaws, and branchial arches, were collected aseptically from four zebrafish and cleaned from adherent tissues in phosphate-buffered saline (PBS; pH 7.4) supplemented with 500 IU/mL of penicillin and 500 μg/mL of streptomycin (Invitrogen). Tissues were minced into small pieces (∼8 mm3) using sterile instruments and fragments were placed in 25-cm2 cell culture flasks (Sarstedt) containing 3 mL of the Leibovitz's L-15 medium supplemented with 20% fetal bovine serum (FBS), 100 IU/mL of penicillin, 100 μg/mL of streptomycin, and 2.5 μg/mL of fungizone (all from Invitrogen). Cells were then allowed to migrate from fragments and to adhere to the surface of the flask for 2 weeks at 28°C. The culture medium was replaced every 5 days. At confluence, cells were subcultured (1:2) using 0.25% trypsin (Invitrogen) and 0.2% EDTA in PBS.
Chromosome analysis
Cell cultures at passage 43 were exposed for 2 h to colchicine (0.0025% in PBS; Sigma-Aldrich). Cells were detached using trypsin-EDTA and harvested by centrifugation (2000 g for 3 min). Cell pellets were gently resuspended in 0.027 M KCl in PBS and incubated at 25°C for 30 min. Cells were harvested again and fixed in a freshly prepared mixture of cold methanol/acetic acid (3:1) for 15 min. After three washes in fixative, cells were resuspended in a small quantity of fixative, then dropped onto cold glass slides, air-dried, and stained with 5% Giemsa (pH 6.8; Merck Chemicals) for 20 min. Chromosomes were counted from micrographs of 50 metaphase plates.
Cryopreservation
ZFB1 cells from subconfluent cultures were harvested by centrifugation (2000 g for 5 min) and resuspended in an ice-cold medium supplemented with 10% cell culture grade dimethyl sulfoxide (Sigma-Aldrich) at a density of 106 cells/mL. Cell suspensions were dispensed into 2-mL cryotubes (Sarstedt), placed at −80°C in a cell freezing device (Nalgene) overnight, and then transferred into liquid nitrogen. Frozen cells were recovered in 9 mL of the culture medium and placed in a flask at 28°C. The medium was renewed after 8 h and cell viability was determined using the Trypan blue dye exclusion method. 36
Cell proliferation
Cells were seeded at a density of 1.5×103 cells/well in 96-well plates and proliferation was determined at appropriate times using the CellTiter 96 Non-Radioactive Proliferation Assay kit (Promega).
Transfection
Transfection efficiency of ZFB1 cells was determined using pEGFP-N1 (Clontech) or pmaxGFP (Lonza) vectors expressing the green fluorescent protein (GFP) under the control of a CMV promoter. ZFB1 cells were seeded at a density of 5×105 cells/well in 6-well plates and cultured until subconfluence. The pEGFP-N1 vector (1 μg) was delivered into the cells using FuGENE6 or FuGENE-HD (both from Roche) according to the manufacturer's instructions, through calcium phosphate coprecipitation according to Pfitzner et al. 37 or using polyethylenimine (PEI) according to Braga et al. 38 The pmaxGFP vector was delivered into the cells through nucleofection. In this case, 6×105 cells were resuspended in 100 μL of buffer L or V (Nucleofection optimization kit; Lonza) and nucleofected with 2 μg of the pmaxGFP vector using the Amaxa Nucleofector (Lonza). Cells were seeded into 6-well plates and incubated for 6 h at 28°C. The culture medium was changed 48 h after transfection and 6 h after nucleofection, and efficiency of DNA delivery was determined by flow cytometry (BD Biosciences).
ECM mineralization
Cells were seeded at a density of 105 cells/well in 6-well plates and grown to confluence. ECM mineralization was induced by supplementing the culture medium with a mineralogenic cocktail composed of 10 mM β-glycerophosphate, 4 mM calcium chloride, and 50 μg/mL of L-ascorbic acid. 39 The medium was changed twice a week. After 3 weeks of treatment, levels of mineralization were assessed through Alizarin Red S (AR-S, 40 mM at pH 4.2; Sigma-Aldrich) and von Kossa's (VK) stainings. 40
Proteoglycan production
Cells were seeded at a density of 105 cells/well in 6-well plates and grown to confluence. Proteoglycan production was induced by supplementing the culture medium with 6.25 μg/mL of human insulin, 50 nM ascorbate 2-phosphate, and 10 ng/mL of human transforming growth factor-β1 (all from Sigma-Aldrich). After 3 weeks of treatment, the presence of proteoglycans was assessed through Alcian Blue 8GS staining (0.5% [w/v] in 0.1 N HCl at pH 1.0; Sigma-Aldrich).
Alkaline phosphatase activity
Cells were seeded at a density of 3×105 cells/well in 6-well plates and grown under mineralizing or normal conditions. At appropriate times, cells were washed with PBS, fixed in a 1% (v/v) glutaraldehyde solution (prepared in PBS) for 10 min, washed twice with PBS, and then incubated (in the dark) in a solution containing 50 mg/mL of bromo-chloro-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT; Sigma-Aldrich). The development of a blue signal indicative of an alkaline phosphatase (ALP) activity was observed by microscopy. At appropriate times, the ALP activity was also measured in cell extracts by spectrophotometry according to the method described by Tiago et al. 41
Immunofluorescence staining
For immunophenotyping, cells were grown on 13-mm glass coverslips (VWR), fixed with 3.7% (v/v) formalin (prepared in PBS) for 10 min at 4°C, washed with PBS, permeabilized with 0.1% Triton X-100 for 5 min, and blocked in PBS containing 1% bovine serum albumin (BSA). Mouse monoclonal antibodies (Developmental Studies Hybridoma Bank) against Spp1/osteopontin (MPIIIB10-1), type II collagen (II-II6B3), type X collagen (X-AC9), and chondroitin sulfate proteoglycans (9BA12), were diluted 1:10 in PBS with 1% BSA and directly added to the fixed cells at room temperature for 2 h. Cells were washed with 1:10 dilution of a blocking buffer in PBS and incubated with a Alexa Fluor 594-linked secondary antibody (Invitrogen) for 45 min at room temperature in the dark. Cells were washed several times with PBS, mounted in Mowiol (Sigma-Aldrich), and observed using an Olympus IX81 motorized fluorescence microscope equipped with an F-View camera (Olympus).
Immunohistochemistry
The suitability of mouse monoclonal antibodies against type II collagen and chondroitin sulfate proteoglycans for detecting zebrafish proteins was determined through whole-mount immunohistochemistry and staining of 6- to 7-cm-long zebrafish larvae, according to the procedure described by Verstraeten et al. 42 Control fish were treated with a secondary antibody alone.
Western blot analysis
The suitability of mouse monoclonal antibodies against type X collagen and osteopontin/Spp1 for detecting zebrafish proteins was determined through western blot analysis of proteins prepared from zebrafish vertebrae and mouse femur. Protein extracts were fractionated on 4%–12% acrylamide NuPAGE Novex Bis-Tris gels (Invitrogen) and transferred onto PVDF membranes (Millipore) using the XCell SureLock blot module (Invitrogen). Chemiluminescence of the anti-mouse IgG-peroxidase conjugate (Sigma-Aldrich; 1:30,000 dilution) was detected using the Western Lightning ECL kit (Perkin Elmer) and Hyperfilm ECL (Amersham, GE Healthcare).
RNA preparation and gene expression analysis
Total RNA was extracted from cell cultures according to Chomczynski and Sacchi. 43 RNA integrity was assessed through 1% (w/v) agarose/formaldehyde gel electrophoresis and RNA quantity was determined through spectrophotometry (NanoDrop 1000; Thermo Scientific). Total RNA (1 μg) was treated with RNase-free DNase I (Promega) for 30 min at 37°C and reverse transcribed at 37°C for 1 h using the Moloney-murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen), RNase Out (Invitrogen), and oligo-d(T) [5′-ACGCGTCGACCTCGAGATCGATG(T)13-3′]. Levels of gene expression were determined through real-time PCR amplification carried out using gene-specific primers (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/zeb) and normalized using the eef1a1l1 housekeeping gene. A reaction mixture containing 1×Sso Fast EvaGreen supermix (Bio-Rad), 10 μM of forward and reverse primers, and 1:10 dilution of reverse-transcribed RNA was submitted to the following PCR conditions: 2 min at 95°C and 50× (20 s at 95°C, 45 s at 68°C). Amplifications were performed by using the StepOnePlus Real-Time PCR system (Applied Biosystems).
Results and Discussion
Establishment of ZFB1 cells
Several primary cell cultures derived from zebrafish-calcified tissue explants were developed as described in the Materials and Methods section and cultured at 28°C in the L-15 medium supplemented with 20% (until passage 30) and then 15% FBS. One culture—named ZFB1—was selected based on morphological criteria and mineralogenic potential and further characterized. After few passages, ZFB1 cells exhibited a polygonal shape and an epithelial-like phenotype (Fig. 1A) and growth appeared to be contact inhibited; this phenotype was maintained for more than 70 passages. The cell monolayer was routinely dissociated every 3–5 days using trypsin-EDTA and divided 1:2. The L-15 medium/FBS appeared to be adequate to sustain the growth of ZFB1 cells without the need for adjustments (e.g., osmolality and pH) or further supplements (e.g., fish serum). Whereas previous reports on the development of mineralogenic cell lines from marine teleosts 39 mentioned the use of an enzymatic cocktail to digest the bone collagenous matrix (collagenase) and cleave proteins anchoring bone cells to the matrix (trypsin), thus facilitating migration of bone cells from explants, ZFB1 cell culture was developed without any enzymatic treatment of bone fragments. The only apparent consequence was a slight delay in the initial cell migration from tissue fragments.
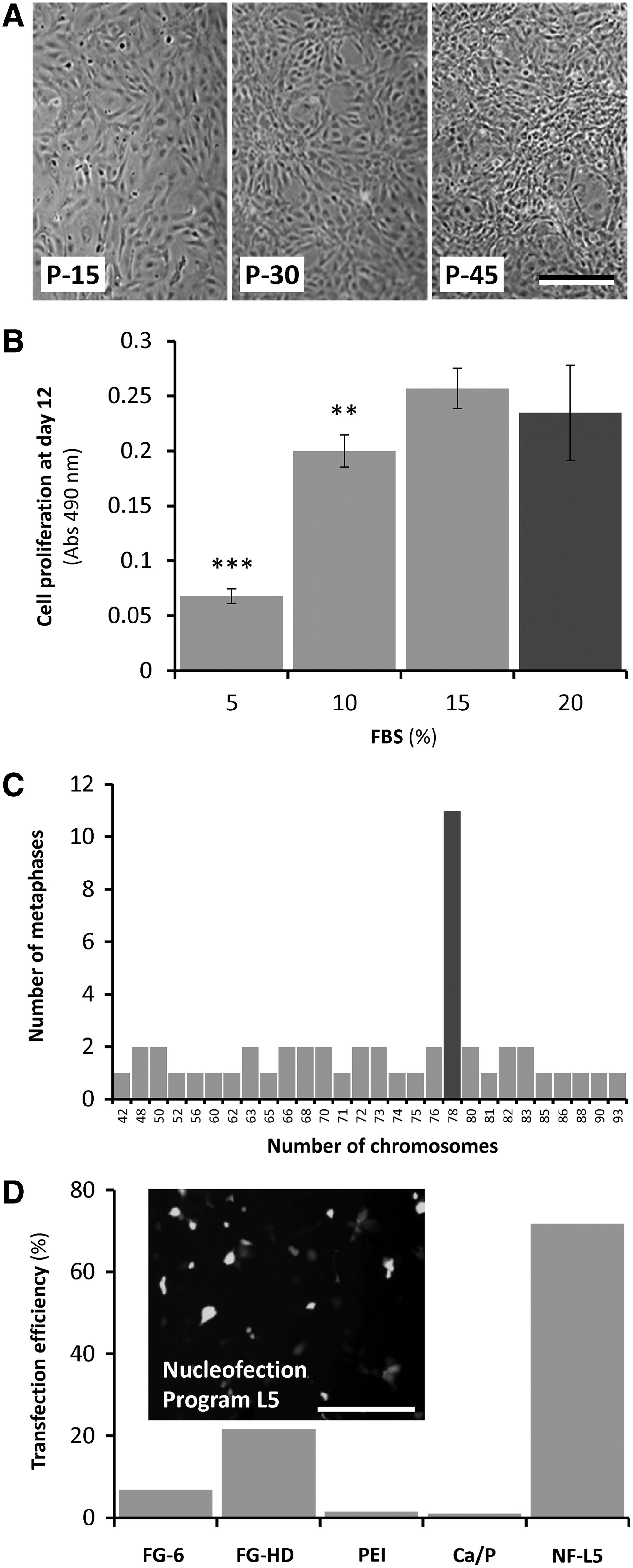
Characterization of ZFB1 cells.
Proliferation of ZFB1 cells was assessed in the presence of four different FBS concentrations. While increasing FBS concentration to 20% (concentration used for the explant culture and the initial steps of the primary cell culture) did not improve the proliferation rate of ZFB1 cells, decreasing it to 10% and 5% significantly reduced cell growth rate by 15% and 75%, respectively (Fig. 1B). It is noteworthy that although 10% of serum will not bring optimal culture conditions, it will sustain cell proliferation to levels suitable for most in vitro experimentation.
Cryopreservation of ZFB1 cells at passage 45 was successfully achieved using methods developed for mammalian cells and applied previously in our laboratory to marine fish cell lines. 44 Approximately 80% of ZFB1 cells survived cryopreservation in 10% DMSO and actively divided thereafter (results not shown).
Ploidy of ZFB1 cells was determined at passage 43 from chromosome counting in 50 metaphase plates. While diploid number of chromosome is 2n=50 in zebrafish, a modal number of 78 chromosomes was observed in ZFB1 cells (Fig. 1C), indicating aneuploidy. Occurrence of abnormal number of chromosomes is frequent in cell lines, particularly in fish, 45 and is considered to be a sign of cell transformation. 46
DNA delivery into ZFB1 cells at passage 40 was assayed through lipofection, nucleofection or using polymers, and was evaluated through GFP fluorescence. While lipofection and polymer-assisted transfection resulted in few GFP-positive cells (efficiency ranged from 1% using calcium phosphate coprecipitation to 22% using FuGENE-HD), nucleofection proved to be an efficient method for delivering DNA into ZFB1 cells (efficiency was up to 72%; Fig. 1D). In combination with nucleofection, ZFB1 cells represent a valid tool for successful gene or mRNA targeting approaches and for assessing gene promoter activity.
ECM mineralization and production of sulfated proteoglycans
Mineralization of ZFB1 ECM was induced by supplementing the culture medium with a mineralogenic cocktail and evidenced through the presence of numerous calcium phosphate deposits of various sizes, found to be positive for VK and AR-S staining (Fig. 2A, D, respectively). No clear mineral deposits were observed in cultures left untreated (Fig. 2B, E, respectively) indicating the requirement for an osteogenic stimulus to trigger cell differentiation, and none was observed in the absence of cells (results not shown) indicating a cell-mediated process. ECM mineralization was further quantified through densitometry (VK; Fig. 2C) and spectrophotometry (AR-S; Fig. 2F) analysis. These results demonstrated the ability of ZFB1 cells in differentiating towards an osteoblastic phenotype and in producing a matrix compatible with in vitro mineralization, as reported previously for marine fish cell lines.39,47 ZFB1 cell cultures exposed for 1 week to the mineralogenic cocktail exhibited a strong ALP activity (Fig. 2G) further confirmed through enzymatic assays of ALP activity (Fig. 2I). However, it was not significantly different from that observed in untreated cells (Fig. 2H) suggesting an acquired capacity to express high levels of ALP affecting positively the predisposition of these cells to differentiate towards an osteoblastic-like phenotype.

Extracellular matrix (ECM) mineralization
While mineralization of osteoblast matrix in rat, mouse and human only requires medium supplementation with ascorbic acid and β-glycerophosphate,48–53 fish osteoblast cells need a source of calcium (usually calcium chloride) to achieve ECM mineralization.39,54 No deposition of mineral was detected by VK staining in ZFB1 cell cultures exposed to a mineralogenic cocktail lacking calcium, even after extended periods of treatment (results not shown). This suggests that the quantity of calcium initially present in the culture medium (0.1396 g/L or 1.8 mM) is not sufficient to trigger the formation of calcium phosphate crystals during osteoblastic differentiation of zebrafish cells. We propose that calcium is a general requirement for ECM mineralization in fish mineralogenic cell lines and that this is probably the consequence of the presence of high levels of calcium in fresh and sea water (averaged value are about 4 and 400 mg/L, respectively) and the need for a higher plasma calcium concentration in fish as a stimulus for initiating ECM mineralization. 39
Confluent cultures of ZFB1 cells were also treated for 3 weeks with a chondrogenic cocktail containing transforming growth factor-β1, a key factor in the regulation of cellular differentiation in cartilage formation. 55 Upon treatment, ZFB1 cells condensed into three-dimensional aggregates and produced highly sulfated proteoglycans, evidenced through Alcian Blue staining (Fig. 2J) and further confirmed through spectrophotometry analysis (Fig. 2L). No Alcian Blue staining was observed in cultures left untreated (Fig. 2K). These results demonstrate the ability of ZFB1 in differentiating towards a chondroblastic phenotype and producing a cartilaginous matrix. Similar results have been reported by Ogawa et al. 56 in a study where adipose-derived stem cells (cells with a mesenchymal origin, similar to chondrocytes and osteoblasts) treated with transforming growth factor-β1 resulted in three-dimensional aggregates positively stained with Alcian Blue.
Based on the above results we propose that ZFB1 cells have the capacity to differentiate towards osteoblast and chondroblast lineages upon appropriate stimulation. This was further confirmed by immunocytochemistry using antibodies specific for cartilage-associated proteins including Cspgs (chondroitin sulfate proteoglycans) and Col2a1 (collagen, type II, alpha 1) (Fig. 3A, C respectively). These proteins were located mostly at sites of cell condensation and deposit of mineralization-related proteins including Spp1 (secreted phosphoprotein 1/osteopontin) and Col10a1 (collagen, type X, alpha 1) (Fig. 3E, G respectively). No signal was detected in cells at week 0 (i.e., prior to treatment, data not shown). Positive staining for all markers was observed after 1 week (data not shown) and 3 weeks (Fig. 3) of treatment. Treated and untreated cultures, as well as negative controls, are shown in Supplementary Figure S1. To confirm the suitability of mouse antibodies to detect zebrafish proteins, whole-mount immunohistochemistry using zebrafish larvae (for Cspgs and Col2a1) and western blotting comparing mouse and zebrafish vertebra extracts (for Col10a1 and Spp1) were performed (Supplementary Fig. S2), thus validating the use of these antibodies in our study.
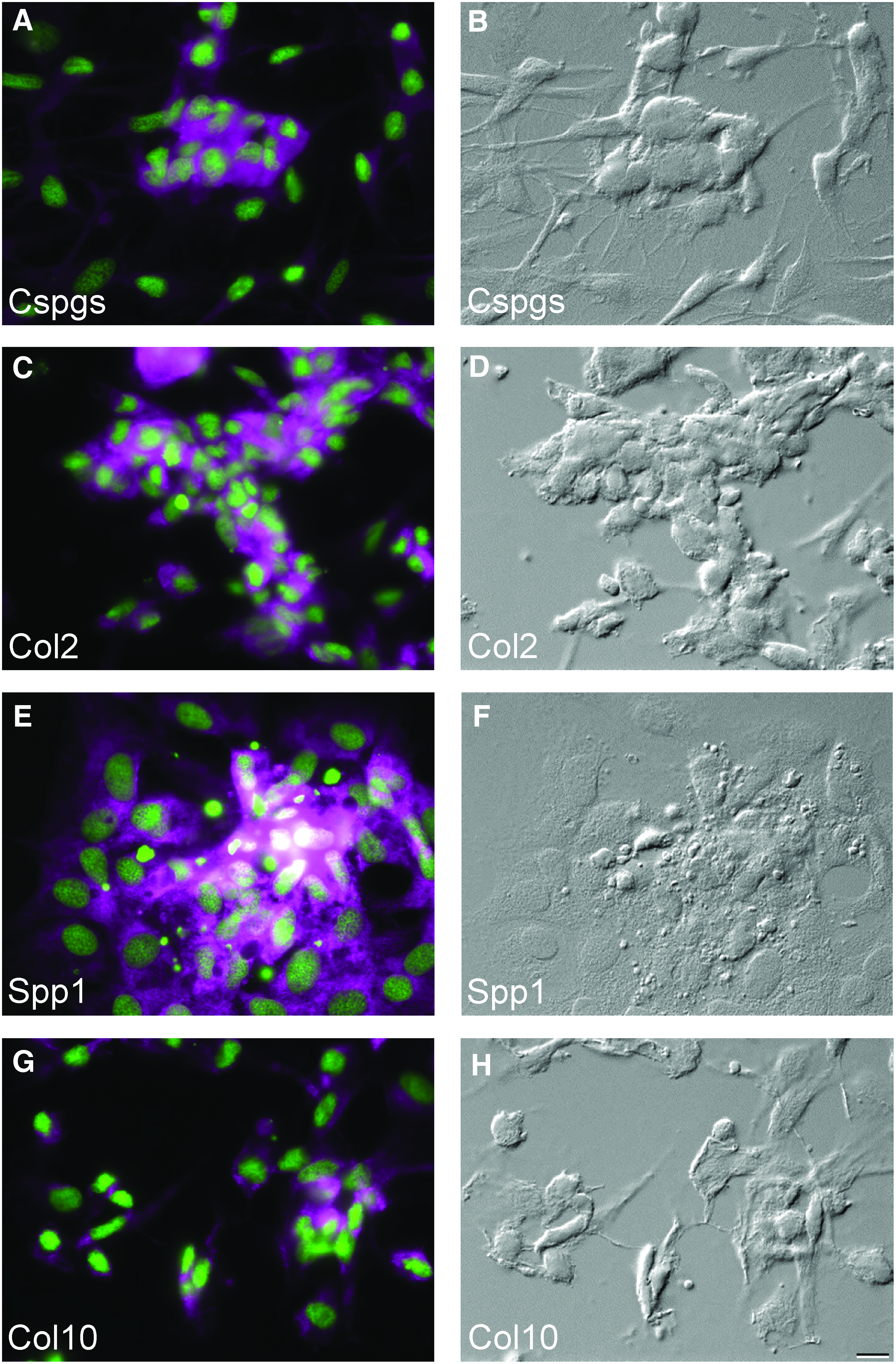
Osteochondroblastic differentiation of ZFB1 cells and detection of lineage-specific markers by immunocytochemistry.
Gene expression during in vitro mineralization of ZFB1 cells
Expression of several bone-related genes was evaluated by qPCR in ZFB1 cell cultures undergoing osteoblast differentiation (1 week) and ECM mineralization (3 weeks) and compared with control cells (Fig. 4). Alp is thought to play a role in the early mineralization process through the break-down of pyrophosphate, a known inhibitor of tissue mineralization, 57 thus inducing production of phosphate groups for crystal formation. 58 While alp expression was down-regulated in control cells at both time points, it was up-regulated in cells undergoing osteoblastic differentiation (approximately 2-fold increase over control) and down-regulated in cells undergoing ECM mineralization to levels found in control cells. In agreement with previous reports, 59 Alp seems to play a more important role during ZFB1 cell differentiation and matrix production than during ECM mineralization. Runt-related transcription factor 2 (Runx2) and osterix (Osx) are two transcription factors involved in osteoblast differentiation and bone formation in mammals60,61and in fish. 62 Expression of both genes was stable in control cells and strongly up-regulated in cells undergoing differentiation (2.7 and 5-fold increase at 1 and 3 weeks, respectively) and ECM mineralization (13.3 and 66.5-fold increase, at 1 and 3 weeks respectively) upon exposure to the mineralogenic cocktail. Osteopontin (Spp1) and osteonectin (Sparc) are two non-collagenous proteins present in osteoblast ECM.63,64 Spp1 is a highly phosphorylated sialoprotein and a prominent component of bone and teeth mineralized ECM, 65 while Sparc is a matricellular glycoprotein with calcium binding domains which has been associated with ECM mineralization in osteoblasts. 66 These two proteins play a role in the regulation of mineralization, 63 attachment of osteoblasts and osteoclasts to the bone matrix, and/or attraction of cells to the bone matrix. 67 Expression of both genes was stable in control cells and up-regulated in cells undergoing differentiation (1.8 and 1.4-fold increase at 1 and 3 weeks, respectively) and ECM mineralization (9.3 and 3.7-fold increase, at 1 and 3 weeks respectively) upon exposure to the mineralogenic cocktail. While an increase in spp1 expression in differentiating ZFB1 cells is in agreement with previous reports in mammalian68,69 and fish 70 cell systems, up-regulation of sparc expression in zebrafish cells is contradictory to previous expression data in gilthead seabream bone-derived VSa16 cells. 71 qPCR data are however consistent with immunocytochemistry data showing a stimulated production of Spp1 and Sparc in the ECM of ZFB1 cells undergoing mineralization. Type X collagen (Col10a1) is a structural protein of the ECM essential for mineral nodule deposition. 72 While col10a1 expression is used as a marker for chondrocyte hypertrophy in higher vertebrates,73,74 Avaron et al. 75 and Renn and Winkler 76 recently reported the expression of col10a1 in early osteoblasts, during intramembranous and perichondral ossification of fish bone and therefore suggested that it could be a marker of pre-osteoblasts in teleost fish. Expression of col10a1 was stable in control ZFB1 cells and strongly up-regulated during differentiation (16.2-fold increase after 1 week of treatment) and in cells undergoing ECM mineralization (14,700-fold increase after 3 weeks of treatment, respectively). Thus, expression data related to col10a1 pointed towards an osteoblastic differentiation of ZFB1 chondro/osteoblast progenitor cells upon exposure to mineralogenic cocktail. Type II collagen, a homotrimer of the α1(II) chain (Col2a1), is a major protein of cartilage ECM and is synthesized primarily by proliferating chondrocytes but not by hypertrophic chondrocytes. 77 Upon exposure to mineralogenic cocktail, col2a1 expression was down-regulated (about 5-fold decrease) in differentiating (not significantly different from control cells) and mineralizing (20.9-fold decrease after 3 weeks of treatment) cultures. Expression data are consistent with immunocytochemistry data, where Col2a1 was detected in few cells at the sites of condensation during the ECM mineralization (Fig. 3E) and further confirm that ZFB1 cells do not differentiate towards a chondroblast phenotype upon exposure to mineralogenic cocktail. Sox9 is a transcription factor with a high-mobility-group (HMG-box) DNA-binding domain exhibiting a high degree of homology with that of the mammalian testis-determining factor, SRY. It was reported that during chondrogenesis, sox9 is expressed in all chondroprogenitors and all differentiated chondrocytes.78–80 In ZFB1 cells, expression of sox9a remained low and was not regulated during cell differentiation and ECM mineralization thus further excluding chondrogenic differentiation, while qPCR analysis of osteoblast marker gene expression revealed the up-regulation of osteoblast-specific transcription factors (runx2 and osx), non-collagenous ECM proteins (spp1 and sparc), and fibrilar collagen (col10a1) demonstrating the osteoblastic potential of ZFB1.

Real-time PCR analysis of bone-related genes in ZFB1 cells treated for 3 weeks with osteoblastic cocktail. alp, alkaline phosphatase; sox9a, SRY (sex determining region Y)–box9; runx2, runt-related transcription factor 2; osx, osterix; spp1, osteopontin; sparc, osteonectin; col10a1, type X collagen; col2a1, type II collagen. C: control cultures; M: mineralizing cultures. Values are presented as mean±standard deviation calculated from three biological replicates (the value of each biological replicate is the mean of at least three technical replicates). Asterisks indicate values statistically significant from T0 and cardinals indicate values statistically significant from mineralization and control (one-way ANOVA p<0.05).
Conclusions
This is the first report of the successful development of a bone-derived stable cell culture from zebrafish. Our results clearly show the presence within the ZFB1 cell population of cells able to differentiate into two different bone lineages, that is, chondroblast and osteoblast, with the capability of ECM mineralization. ZFB1 cells represent the first zebrafish-derived cell model with such characteristics. As detailed above, the ZFB1 cells are cultured and manipulated using regular reagents (medium and serum) and methods (cryopreservation and transfection), and thus its use can be easily implemented in virtually any laboratory equipped with cell culture facilities. The availability of such in vitro cell systems represents a promising model for investigating bone lineage cell differentiation in fish and will contribute to promote the use of zebrafish as a model organism to study specific pathways involved in human skeletal-related diseases.
Footnotes
Acknowledgments
This work was funded by the FISHCELL project (PTDC/MAR/105313/2008) financed by the Portuguese Science and Technology Foundation (FCT) and received the support of the Association of European Marine Biological Laboratories through the ASSEMBLE project (FP7/227799). This work was cofunded by the European Regional Development Fund (ERDF) through the COMPETE Program and by the National Fund through FCT under the PEst-C/MAR/LA0015/2011 project. P.V. is supported by a postdoctoral grant from FCT (SFRH/BPD/39189/2007). We thank Joana Rosa and Cátia L. Marques for their assistance in western blot analysis. The monoclonal antibodies used in this study were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA. Flow cytometry and fluorescence imaging were performed in the facilities of the Department of Biomedical Sciences and Medicine, University of Algarve.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.