
Abstract
The skin mucus of teleost fish harbors a complex microbial community that is continually interacting with the aquatic environment. Despite zebrafish, Danio rerio, serving as a model organism in a myriad of research fields, very little is known about the composition and role of the skin mucus microbiome. The purpose of this study was to determine a simple sampling method for the skin mucus microbiome, identify prominent bacterial members, and compare its composition to the microbial community of the surrounding environment. Next-generation sequencing of the V3–V4 region of the 16S rRNA gene was performed on skin mucus and filtered tank water samples. Results show that prominent bacterial members of the skin mucus in zebrafish include Actinobacteria (Mycobacteriaceae) and Gammaproteobacteria (Aeromonadaceae), followed by Alphaproteobacteria and Betaproteobacteria. The tank water contained much higher bacterial diversity and was clearly different from the skin mucus microbiome, despite continuous interaction. This study identifies a straightforward sampling method for the zebrafish skin mucus microbiome, enabling hypothesis generation on the role of ectosymbionts on host and microbiome health.
Introduction
Life in an aquatic environment involves continual interactions between the surface of the aquatic species and the extensive array of microbes that exist in the surrounding water. The skin is the outermost layer of the body and provides a unique innate defense in animals. It serves as a physical barrier against pathogens that could be harmful to the body and its physiological functions. 1 Aquatic species not only contain an outer skin later, but also a mucosal surface harboring bacteria that are thought to exist in mutualism with the fish host, known as a skin mucus microbiome.2,3 To understand the relationship between aquatic host species, their symbionts, and their environment, initial work must identify the bacterial community members to generate hypotheses on the functional roles of specific microbial players.
The skin microbiome contributes to the first line of immune defense and overall health of their host species. In humans, it has been hypothesized that the commensal microbial communities on the skin are shaped by the host immune defense network and modify host immune factors. 2 Similar to this function in humans, past research has shown that the surface microbes of aquatic species are crucial for protection against pathogens and may shape host immune responses. 3 Increased susceptibility to infection, environmental stressors, growth rates, and mortality are correlated with changes in the teleost skin bacterial composition (reviewed in Gomez and Primm4). A recent review outlines the potential role for zebrafish in understanding the purpose and importance of the teleost skin mucus microbiome, 4 yet to our knowledge, only one small study has been performed. 5
The purpose of our study was to determine a sampling method that could be used to compare the skin mucus microbiome to the surrounding water. Next-generation sequencing of a region of the 16S rRNA gene was performed to identify the bacterial community of each location. First, the effectiveness of two isolation techniques for capturing the zebrafish skin mucus bacterial community was tested. Next, bacteria present in the tank water were identified and compared with those skin mucus results.
Based on studies from other teleost fish species, we hypothesize that there will be a distinct bacterial species composition in the skin mucus microbiota compared with the environment.6–9 Research with other fish has shown that the skin mucus microbiome is host species specific, yet the Proteobacteria phylum is commonly present, as seen with other animal microbiomes. 4 Our work provides initial insights to the bacterial community of laboratory-reared zebrafish skin mucus, thus enabling the development of hypotheses on ecological roles and importance of individual symbiont species on host health.
Materials and Methods
Zebrafish
Healthy adult zebrafish, Danio rerio, were maintained in a recirculating system with a 14-h light–10-h dark cycle. Partial water changes occurred twice daily through mechanical, ultraviolet (UV), and activated charcoal filters. Salt and sodium bicarbonate were maintained by a dosing system and conductivity is 600–800 μS/cm. Fish were fed a combination of brine shrimp and standard dry food at least 6 days a week. Six fish (sex undetermined) were randomly netted and euthanized with an overdose of buffered 2% MS-222 until no longer responsive to tap stimuli (euthanasia procedure approved by the Goucher College IACUC Committee) before skin mucus sampling.
Mucus sample collection and processing
Euthanized fish were rinsed with sterile water to remove transient microbes and then separated into sterile Petri dishes for skin mucus collection. Previous research suggested that gentle rubbing with tissue paper removed epidermal mucus, whereas swabbing with a sterile cotton swab removed the epidermal mucus layer and uppermost epithelial cells.3,10 Our study compares these sampling methods.
In this study, three fish were sampled using a sterile cotton swab. Sterile forceps held the fish in place while a sterile cotton-tipped swab (Puritan, Guilford, ME) was gently rolled on both sides of the fish, operculum to peduncle and back again. The cotton swab tip was then directly placed into a ZymoBIOMICS BashingBead lysis tube for processing. The other three fish were sampled by a filter paper collection method. Each fish was wrapped with a sterile 0.22 μm polyethersulfone (PES) bottle-top filter that was removed with sterile blades and forceps (CELLTREAT, Pepperell, MA). Both sides of the fish were gently rubbed simultaneously with the filter paper. The filter paper was then cut in half and placed into separate ZymoBIOMICS BashingBead lysis tubes for processing.
Tank water sampling
The bacterial community in the tank water was sampled in a collection tank at the base of zebrafish system, which collects the water from all the zebrafish tanks and serves as a biofilter before additional filtration. One liter of water was passed through an 0.22 μm PES bottle-top filter (CELLTREAT). The filter paper was then removed from the apparatus with sterile blades and forceps, cut in half, placed into separate ZymoBIOMICS BashingBead lysis tubes and stored at −20°C until processed. Three replicate water collections were performed.
DNA isolation, sequencing, and analysis
DNA was isolated from all samples using the same ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA). The manufacturer's protocol was followed with the inclusion of Appendix B for cheese- and protein-rich biofluids (addition of Proteinase K). DNA was then assessed for purity using a Thermo Scientific NanoDrop One Microvolume UV-Vis spectrophotometer. All DNA samples had a 260/280 wavelength value between 1.8 and 2 and were sent to GENEWIZ, Inc. (South Plainfield, NJ) for 16S EZ (V3–V4 region) sequencing and analysis (GENEWIZ Supplied Methods in Supplementary File S1).
Briefly, Illumina MiSeq was used to sequence the V3–V4 region of the 16S rRNA gene using the forward primer GTGYCAGCMGCCGCGGTAA and reverse primer CTTGTGCGGKCCCCCGYCAATTC. Sequencing read lengths ranged from ∼250 to 300 bp with a sequencing depth ranging from 114,636 to 453,690 reads with a median of 163,330 reads per sample.
Paired-end reads were assembled, and chimeric sequences removed before analysis. Trimmed, quality-checked paired-end reads were ∼450 bp (Supplementary Fig. S1). QIIME and R were used for microbiome analysis. Sequences with 97% identity were clustered and classified as operational taxonomic units (OTUs), identified, and categorized using the Silva 119 database and Ribosomal Database Program classifier. Sequencing statistics are listed in Table 1. All fish and water samples were collected and processed between September 14 and October 17, 2018. Raw data files can be found at NCBI BioProject ID PRJNA759847.
Sequencing Statistics
OTU, operational taxonomic unit; PE, paired-end.
To compare bacterial diversity within each sample, samples were rarefied to account for differences in sequencing depth before alpha and beta diversity analyses. Several alpha diversity indices were calculated to determine diversity within individual samples (Table 2). Abundance-based coverage estimator and Chao1 indices estimate bacterial richness (i.e., number of different observed OTUs). Shannon-Weaver and Simpson diversity indices measure bacterial diversity by incorporating both richness and evenness (i.e., relative abundance of each OTU type within a sample). It is important to note that the Shannon-Weaver index is biased toward richness, and it increases as richness and evenness increase. The Simpson index weighs evenness more and ranges from 0 to 1; higher numbers correspond with less diversity. See Kim et al. 11 for a more detailed explanation of these parameters.
Alpha Diversity Indices of the Zebrafish Skin Mucus Microbiome and Surrounding Water Calculated from Operational Taxonomic Units Generated from 16S rDNA V3–V4 Region Sequences
Microbiome beta diversity between tank water and skin samples were compared several ways. Principal coordinate analysis (PCoA) plots created a two-dimensional visualization of microbiome community composition differences between sample types.
Unweighted pair group method with arithmetic mean (UPGMA) clustered samples with taxonomically similar microbial communities into a dendrogram. UniFrac distance matrices use phylogenetic information to cluster samples qualitatively, based on presence/absence of OTUs (unweighted), and quantitatively, based on relative abundance of OTUs (weighted). Microbiome community variation between all fish skin mucus samples and tank water samples were statistically evaluated on weighted and unweighted UniFrac distance matrices using PERMANOVA. These tests were conducted in R, using micropower wrapper for adonis.12–15 PERMANOVA tested the null hypothesis that there was no significant difference between all fish and water samples. A second PERMANOVA tested the fish mucus collection method samples. PERMANOVA was run with all nine samples, as well as run with only eight samples where outlier sample AJ04 was removed.
Results
Tank water contains greater bacterial diversity than the skin mucus
To compare the bacterial communities present in the tank water and zebrafish skin, DNA was isolated from filtered tank water and fish skin mucus. The mucus was collected by gently rubbing filter paper or swabbing the sides of euthanized fish, which were previously rinsed with sterile water to remove transient microbes. Bacterial community composition was determined by sequencing a region of the 16S rRNA gene and taxonomic classifications were assigned to OTUs using the Silva 119 reference database (Supplementary File S1).
Sequence coverage was similar across all samples. A Goods Coverage16,17 at or close to 1 indicated that the sequencing was sufficient to reliably describe the bacterial community in the skin mucus using both collection methods, as well as the tank water (Table 2). AJ01, a skin mucus swab replicate had approximately double the number of reads than the other samples.
Contrastingly, the filter sample, AJ04, had the lowest number of analyzed reads. To eliminate any bias due to read numbers in downstream diversity analysis, sample read number was rarefied (Supplementary File S1). A total of 395 unique OTUs were found in this study (Supplementary File S2). The rarefaction curve, which compares the relative bacterial richness (i.e., number of different OTUs observed) at different sequencing depths 11 leveled off, providing evidence that the sequencing sufficiently detected the majority of OTUs in each sample (Fig. 1).
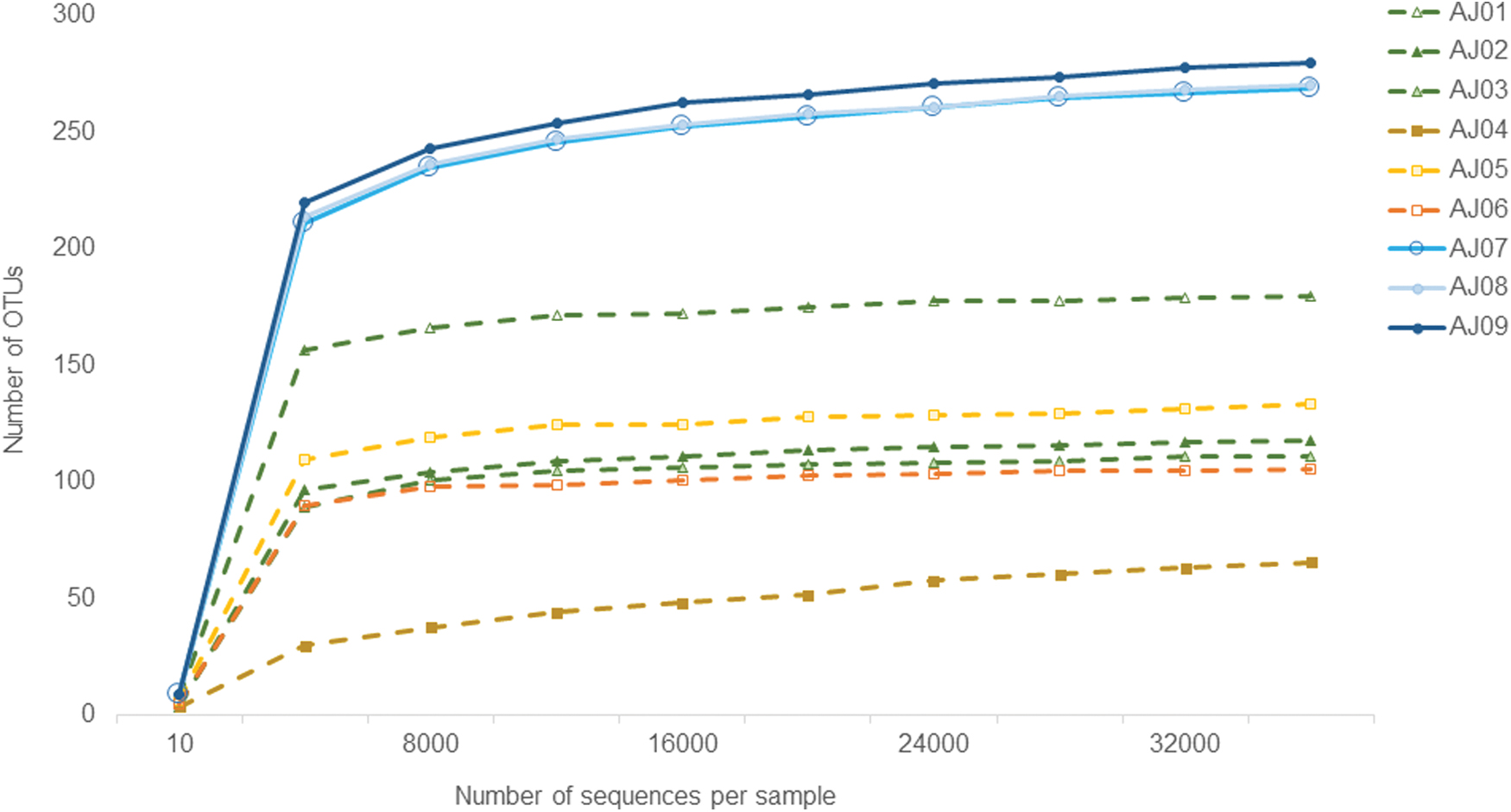
Rarefaction curves representing the relative number of observed OTUs in each sample within this study. Samples include skin mucus swabs (AJ01–3), skin mucus filters (AJ04–6) and filtered tank water (AJ07–9). OTUs, operational taxonomic units. Color images are available online.
To compare overall similarities among samples, beta diversity analyses were performed and statistically tested. A PCoA represents the microbiome diversity in each microbiome community with a single point. Samples with most similar microbiome communities cluster together on the PCoA plot. The PC1 axis explains over 65% of the variation in microbiome community diversity between fish and tank water samples (Fig. 2). While there was slightly more variation between fish mucus samples, this is expected since samples were taken from six different fish (biological replicates). There was no separation between mucus collection methods, suggesting that both are reliable methods. Instead, swab samples (AJ01, AJ03) and filter mucus sample (AJ05) clustered together on the PC2 axis.
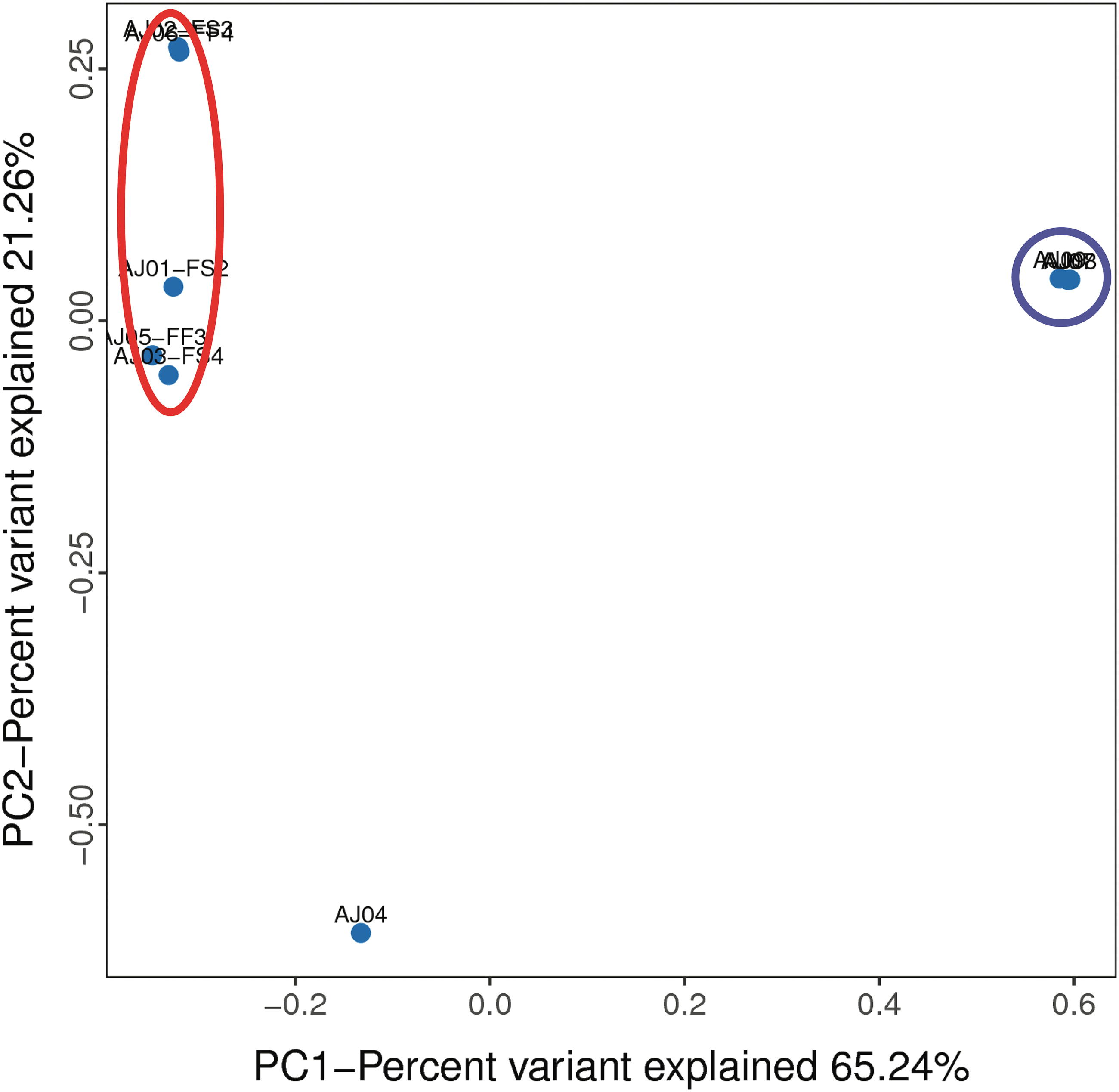
PCoA plot of PC1 versus PC2 cluster samples with the most similar microbial communities together. With the exception of AJ04, all fish samples are encircled in red. All three water samples cluster together (blue circle). PCoA, principal coordinate analysis. Color images are available online.
UPGMA clustering and UniFrac (weighted, using abundance of OTU present and unweighted, using presence or absence of OTUs) distance matrices produced similar results (Supplementary Figs. S2–S4).
Fish skin mucus bacterial community samples were not significantly different (PERMANOVA, unweighted UniFrac p = 0.2 and weighted UniFrac p = 0.8) when collected using swab or filter paper methods and were grouped for downstream analyses. Comparison of the fish skin mucus and tank water bacterial communities were significantly different (PERMANOVA, unweighted UniFrac p = 0.006 and weighted UniFrac p = 0.036). Sample AJ04 appeared to be an outlier, thus all analyses were repeated with this sample removed, producing similar results (Supplementary File S4). PERMANOVA statistical tests of weighted and unweighted UniFrac matrices provide additional support that the fish skin mucus 16S rRNA sequences were distinct from the tank water. While our sample size is low, the clustering of the data suggest that the fish mucus microbiomes are different from the water.
Bacterial community in tank water is distinct from skin mucus
To examine the bacterial community composition more closely, the sequenced OTUs were identified and then categorized at each taxonomic rank. All alpha diversity indices show that the tank water has significantly higher OTU richness and diversity than the zebrafish skin mucus samples (Table 2). The microbial composition of the tank water and fish skin mucus were distinct at every taxonomic level (Figs. 3 and 4 and Supplementary Figs. S5–S8, taxonomic identification as determined by Silva 119 database in Supplementary File S3). Bacteria were the main Kingdom identified, but two Archaeal OTUs, identified as Cenarchaeaceae and Nitrososphaeraceae, were consistently harbored at low levels within all tank water samples (Supplementary File S3). No archaeal OTUs were identified within the skin mucus samples.
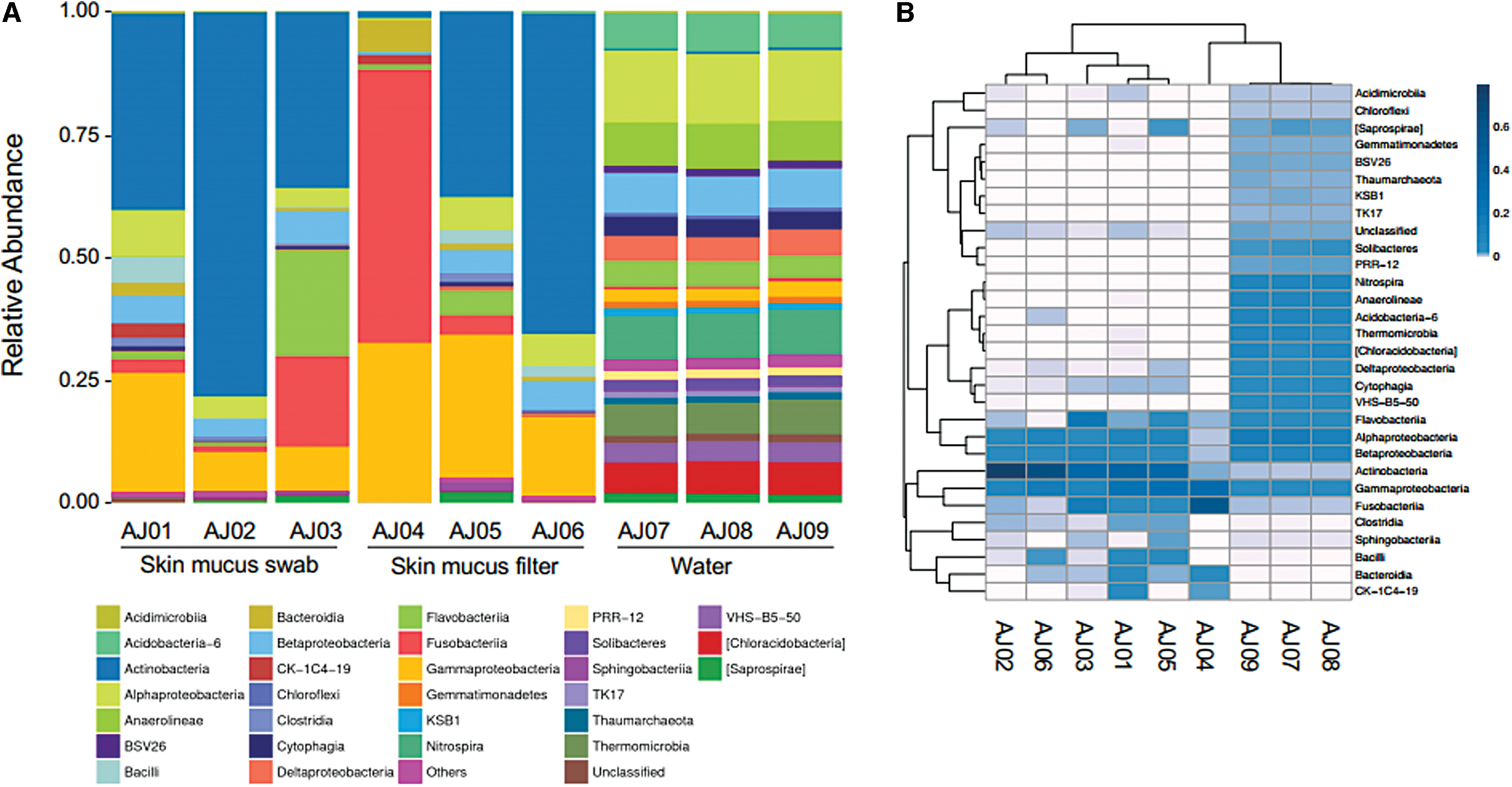
Microbiome relative abundance identified by Class.
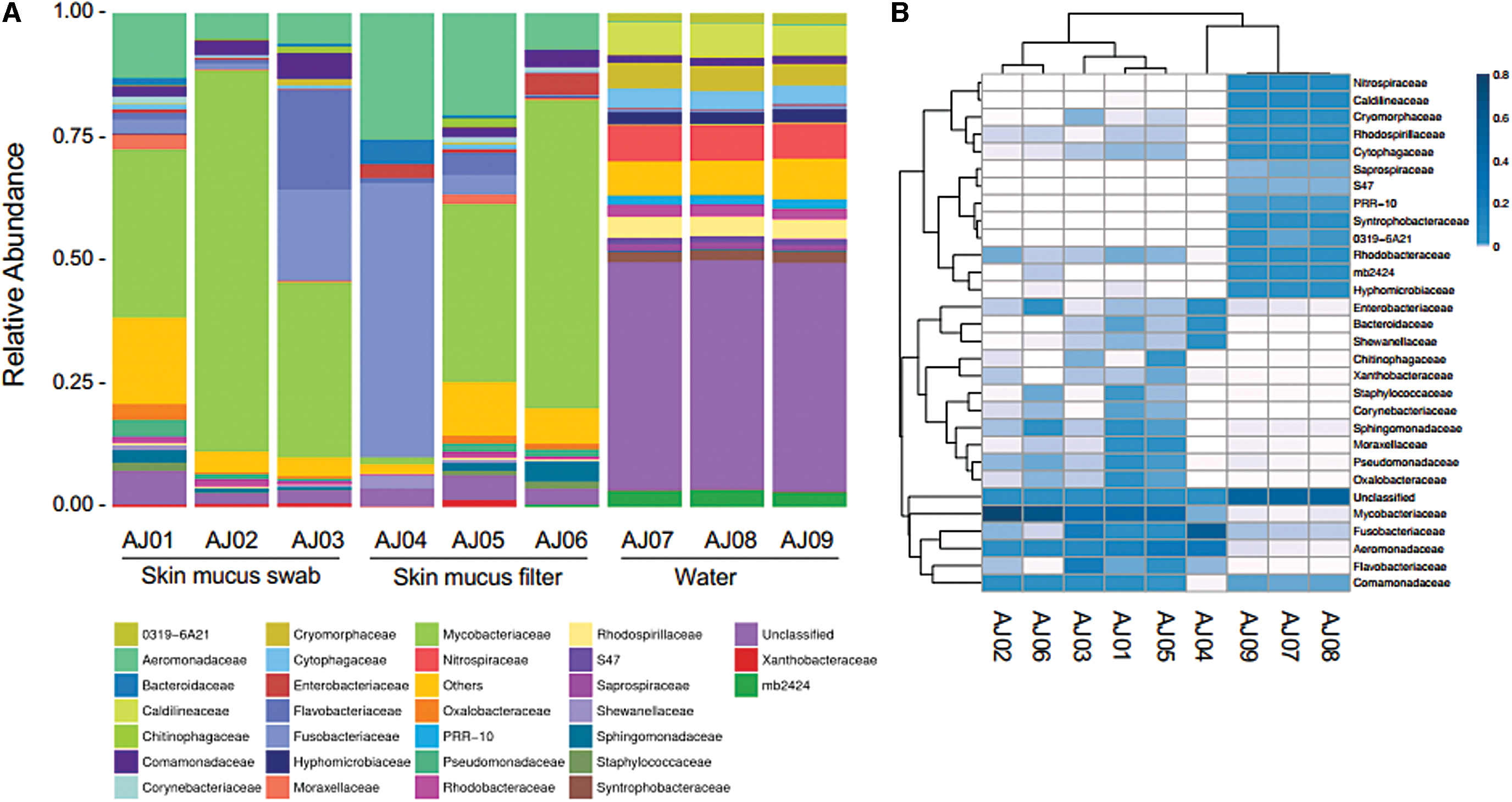
Microbiome relative abundance identified by Family.
Similar bacterial communities were identified in all fish skin mucus samples, regardless of isolation method, except for AJ04 (Figs. 2–4). When examining OTUs by Class, the most abundant bacteria present were Actinobacteria and Gammaproteobacteria, followed by Alphaproteobacteria and Betaproteobacteria (Fig. 3A). Distribution of the top 30 OTUs by Class reinforces the clear distinction between the composition of the tank water and skin mucus bacterial communities (Fig. 3B). Actinobacteria was almost exclusively found in the skin mucus, while Gammaproteobacteria was present in both locations. Examination of OTUs by Family identification further supports that the water and skin mucus are dissimilar communities (Fig. 4).
Within this taxonomic rank, about half of the OTUs from the water were unclassified. The most abundant OTUs in the majority of the skin mucus samples were Mycobacteriaceae and Aeromonadaceae. The most prominent OTU was identified at the genus level of Mycobacterium (no species level identification) between 33% and 77% relative abundance in most fish samples, 1% relative abundance in AJ04, and less than 0.1% relative abundance in all water samples. The second most abundant OTU was identified to family rank as Aeromonadaceae and ranged from 5% to 25% relative abundance in the skin mucus and less than 0.3% relative abundance in the tank water.
The fish sample, AJ04, was unique from the other samples. Over half of the sample reads (55.3%) were identified at the species level from a single OTU, which aligned with the Fusobacteria Cetobacterium somerae. 18 Notably, while this OTU was detected in all samples, it was ∼10 × more prevalent in sample AJ04 than the other skin mucus samples and ∼100 × more prevalent than in the water samples (Supplementary File S3). Additional OTUs present in AJ04 were identified as Gammaproteobacteria (Plesiomonas shigelloide, Shewanella, Vibrio, and Aeromonadaceae [unclassified genus]) and Bacteroidetes (classified in the family Bacteroidaceae).
There were many bacterial phyla that were exclusively in the tank water environment, including Acidobacteria, Chlorobi, Chloroflexi, and Nitrospirae. Within Bacteroidetes, the families Cryomorphaceae and Cytophagaceae were consistently observed in only tank water. Within the phylum Proteobacteria, the Deltaproteobacteria were specific to the water, while Gammaproteobacteria were mostly present in low abundance in all samples. Alpha- and Beta-Proteobacteria were detected in all samples, yet there was a clear distinction at lower taxonomic ranks, as most OTUs in these groups were rarely detected in the tank water and skin mucus simultaneously. In summary, the skin mucus bacterial community appears to be distinct and not a subset of the tank water in which they were housed.
Discussion
First, this study shows that the two skin mucus isolation methods tested, gentle swabbing and filter paper rubbing, produce similar results. The lack of grouping by sampling method supports the conclusion that the two isolation methods both capture the complete microbiome and either could be used in future studies.
Second, this study supports the hypothesis that the zebrafish skin mucus microbiome encompasses a bacterial community that was different from the surrounding water and begins to address an issue presented by Gomez and Primm, 4 where water samples are typically sequenced at greater depth than the skin mucus samples. Here, sequencing depth was similar across all samples, the rarefaction curves leveled off, and Good's Coverage was comparable for all samples enabling reliable alpha and beta diversity indices to be calculated. Nonetheless, future work should include the incorporation of negative controls to identify any contaminating DNA throughout the isolation, amplification, and sequencing process.
The higher bacterial diversity and richness in the tank water is likely due to several factors. First, the filtering procedure of the water likely captured more bacteria than sampling the mucus from a single fish. Next, the water sample was taken from the area that collects the entire system water. This water contains the shedding from various zebrafish microbiomes, for the example from the gastrointestinal tract, the brine shrimp and dry food added almost daily and the biofilter. Due to the flow through nature of the individual tanks, this area was chosen as a reliable and repeatable location, as individual tanks are small and change based on experimental needs.
In future studies, housing the fish in an isolated tank and examining tank water is necessary to determine the microbiome of the aquatic environment without the presence of the biofilter. This research aligns with other studies showing limited or no correlation with the bacterial community of fish skin mucus and the surrounding water.4,7–9
The consistent community composition of the zebrafish skin suggests that the prominent bacterial members of the skin mucus in zebrafish include Actinobacteria (Mycobacteriaceae), Gammaproteobacteria (Aeromonadaceae), Alphaproteobacteria, and Betaproteobacteria. Research has shown that teleost skin microbiome composition is species specific, yet similarities in bacterial groups are present.4,9,19 Actinobacteria has been shown as a prominent member of rainbow trout microbiome (Oncorhynchus mykiss). 20 Nontuberculosis Mycobacterium is now considered ubiquitous in aquatic environments, known for its biofilm production and ability to survive phagocytosis, even living in endosymbiosis with amoeba hosts.21,22
The phylum Proteobacteria has been described as a dominant group in fish skin mucus in farmed Atlantic salmon (Salmo salar), 7 rainbow trout, 20 six fish species in a large Gulf of Mexico survey, 9 and all other studies included in a review by Gomez and Primm. 4 Notably, the classes gamma, alpha, and beta within Proteobacteria were most common, which aligns with the findings from this study. Aeromonas has been one of the most abundant bacterial groups to be cultured from freshwater fish external mucus. 23
To our knowledge, only one other study has been published on the zebrafish skin mucus microbiome. 5 This was a preliminary study examining the link between skin pigmentation and skin microbiome from pet shop zebrafish. In that study, the rarefaction curves and Good's Coverage estimation showed that the entire bacterial community was not sampled at the sequencing depth performed. Three main phyla detected were Proteobacteria, Fusobacteria, and Bacteroidetes and all corresponded with the normal zebrafish gut microbiome.
Notably, their sampling methods used nonanesthetized fish held in nets, yet the authors do not indicate whether the fish were rinsed to remove transient microbes or if the nets where sterilized between fish. Due to the differences in sampling methods and sequencing depth between this study and Coetzer et al., 5 a comparison between the results of these studies is uninformative.
One skin mucus sample, AJ04, had a different bacterial community compared with the other five samples. In-depth examination of this sample revealed that over half of the sequences aligned with C. somerae of the phylum Fusobacteria, a species associated with the intestinal tract of teleost fish.18,24,25 This could indicate that either the fish was not rinsed well and transient gut microbes were present in fecal matter attached to the fish or that this fish may have had a compromised skin mucus microbiome, which was recolonized by free-floating gut microbes.
A lower Actinobacteria population size might also suggest dysbiosis or immunocompromised state, yet this hypothesis requires further investigation. The abundance of the Actinobacteria population could serve as a potentially useful indicator of skin mucus microbiome health in future studies, as this taxonomic Class, particularly the genus Mycobacterium, was almost exclusive to the skin mucus and devoid in the water (Supplementary File S3).
The Mycobacterium in this study could only be classified at the genus level, but species identification and comparison to known opportunistic pathogens of zebrafish would prove informative. As the fish used in this study were healthy (determined through observation of behavior and appearance), this might indicate a commensal or mutualistic role for this genus in the microbiome, yet additional studies comparing across laboratory colonies and wild-caught fish would help determine whether this genus is part of the normal skin microbiome.
Factors that contribute to zebrafish skin mucus community assembly may include transmission mode (vertical or horizontal), host immune factors, metabolic status, the presence of colonizer species, and mutualistic interactions within the microbiome.8,19,23,26 Host environment, such as pH, dissolved oxygen, salinity, and temperature, might also contribute to community composition.27–30 To examine how the host environment alters the zebrafish skin microbiome, broad sampling across laboratories with different husbandry practices and wild caught fish is needed. This survey would also inform which bacteria are consistent members of the skin mucus microbiome across colonies, which is important to discover links with the microbiome and host biology.
Changes in the teleost skin bacterial composition have been correlated with an increased susceptibility to infection and environmental stressors, decreased growth rates, and mortality.4,20,31,32 The functional roles of individual microbial taxa are currently unknown. The skin mucus microbiome in this study was determined through 16S rRNA sequencing and cannot lead to hypotheses regarding functional capabilities of specific OTUs due to the diverse nature of the microbial groups identified. Research from other fish species has begun to uncover its role in preventing infections through competitive exclusion, antimicrobial production, and host immune stimulation.3,20,33,34 The Actinobacteria and Gammaproteobacteria associated with the mucosal surface of amphibians and fish have been shown to produce antibacterial and antifungal substances.20,33,35–37
The antibacterial activity of antimicrobial peptides in mucus can modulate B cell functions thus contributing to the larger functions of the innate immune system. 3 To learn the specific roles individual bacterial symbionts play in host health, future studies will need to culture bacteria to uncover their biochemical functions and capabilities. Once cultured, these microbes can be utilized in host health and developmental studies to test their role in protection from infections, environmental stressors, and longevity.
Conclusion
The zebrafish skin mucus microbiome is unique from the surrounding water and consists of a complex community dominated by Actinobacteria and Gammaproteobacteria. Zebrafish provide a well-established model system to discover the functional roles and importance of specific bacterial members of the skin mucus microbiota. This study provides an initial overview to the diversity and richness of the microbial community within the skin mucus and sampling methods for use in future research.
Footnotes
Acknowledgments
The authors thank Dr. Jenny Lenkowski for providing zebrafish and tank water. They are grateful to Dr. Jennifer Kerr for her critical reading of the article. They appreciate Dr. David Hearn's assistance in R wrangling.
Authors' Contributions
A.K.S.J. conceived and coordinated the study. A.K.S.J. and W.W. designed and performed the experiments. A.M.E. interpreted data. All authors participated in data analysis, article preparation, and gave final approval for publication.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was made possible by Goucher College start-up and summer science research program funding. A.L. was supported by the Helen B. Funk Award at Goucher College.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.