
Abstract
Caudal duplication syndrome is a rare and interesting entity with a reported prevalence of <1/100,000 births. Caudal duplication syndrome encompasses a diverse spectrum of anomalies primarily involving partial or complete duplication of organs comprising the gastrointestinal, genitourinary and distal neural tube systems. The term ‘caudal duplication syndrome’ was coined by Dominguez et al. in 1993, in a case series of six patients presenting with findings pertaining to duplication anomalies of genitourinary system, hindgut, lumbosacral spine and cord. We here report a unique case of caudal duplication presenting in late adolescence and briefly review the available literature on this rare abnormality.
Case report
A 17-year-old virgin female presented to us with complaints of irregular cycles, hypomenorrhoea and dysmenorrhea with known double urethral, vaginal and anal openings since birth, all of which were functional.
Two months prior, she had developed gradual abdominal pain and distension followed by vomiting.
Clinical examination revealed a healthy well-built female with no apparent spinal deformity or skeletal abnormality. Genital examination revealed normal labia with bifid clitoris and two separate urethral openings behind it located adjacent to each other. Two vaginal openings separated by a small palpable midline cystic structure could also be discerned just behind the urethral openings. Two separate anal openings were also noted in the perineum posterior to the vaginal openings. An illustrative example of the same has been provided (Figure 1).
Illustration depicting spectrum of anomalies in our patient comprising of two separate hemi UBs located side by side (starred), separate Mullerian ductal system (marked by thick lines) and duplication of descending and sigmoid colon (diamond marked) and anal canals (marked by thin lines). Duplicated sigmoid colons pass to separate ipsilateral recti and anal orifices.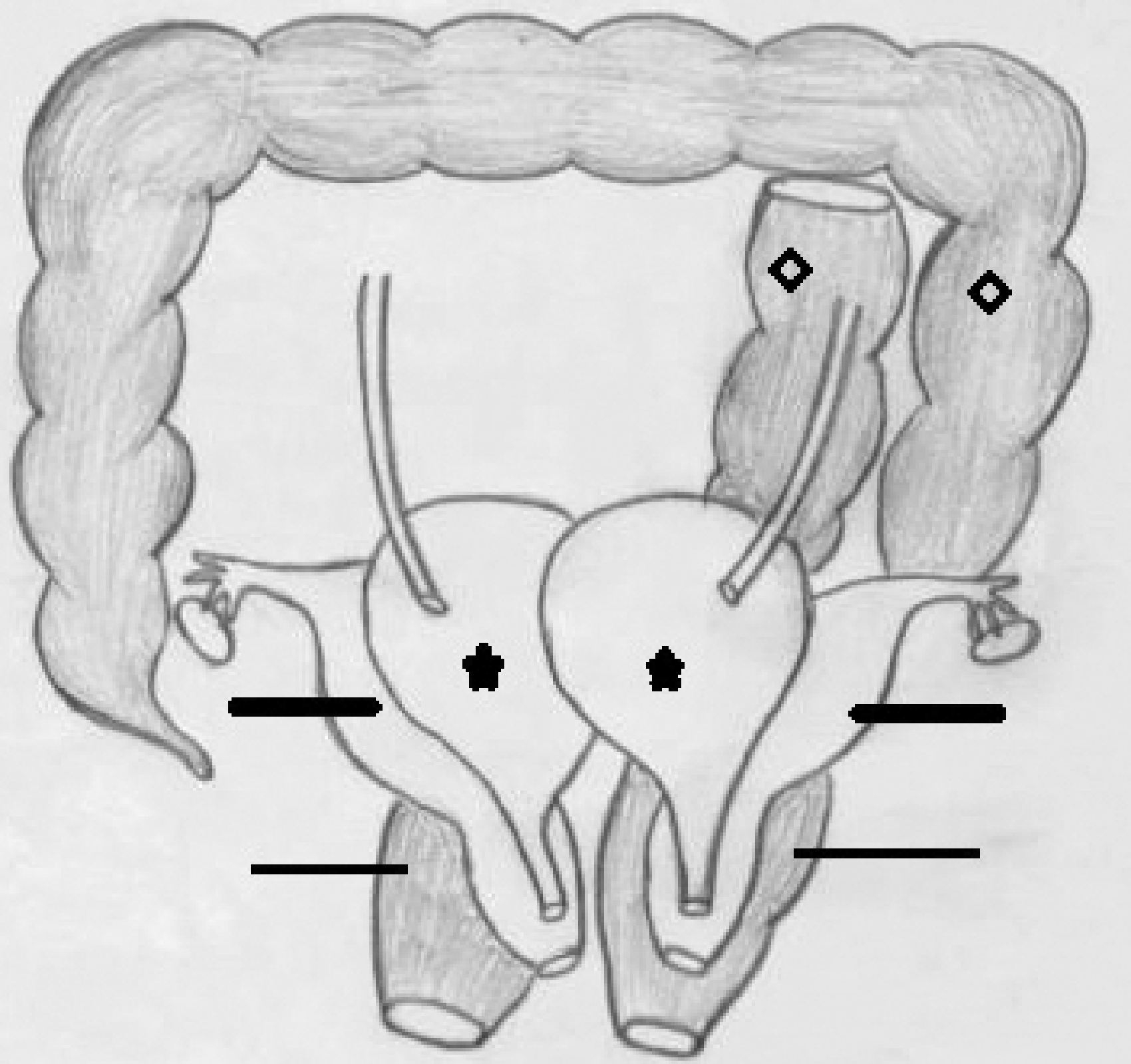
Clinical signs coupled with radiological and ultrasound findings were suggestive of subacute intestinal obstruction at level of sigmoid colon. Our patient underwent an exploratory laparotomy with operative findings confirming a duplication of the sigmoid colon with reversed bowel rotation. A temporary end transverse colostomy was established.
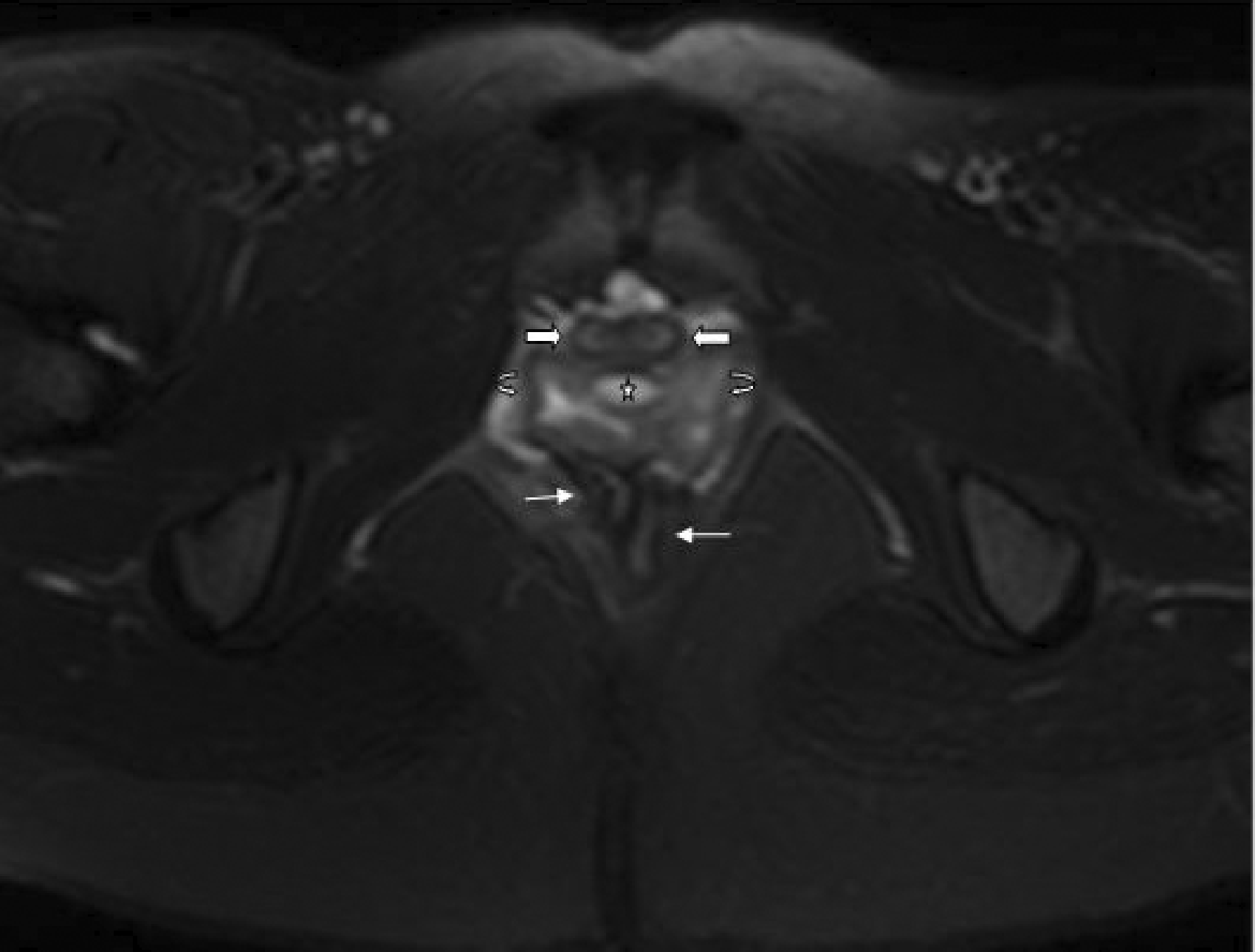
Subsequently, an abdominal MRI scan delineated completely duplicated urinary bladders (UBs) separated by a well-defined fibrous septum; complete duplicated and widely divergent uteri were seen (Figure 2). Individual urethrae, cervical and vaginal canals were seen on either side with a small well-defined midline cyst separating them (Figure 3). The sigmoid colon, rectum and anal canal were also found to be duplicated with two separate anal orifices. Both kidneys and lumbosacral spinal cord were normal.
MRI image of pelvis (axial T2-weighted fat saturated) revealing duplication of UB with a T2 hypointense septum (starred) separating the two entities. Two well-formed uterine cavities with complete zonal differentiation (arrows) can be seen posterior to the duplicated UBs. MRI pelvis image (axial T2-weighted fat saturated) at level of perineum showing duplication of urethrae (block arrows); duplicated vaginal canals (curved arrows) with interposed midline bright (T2 hyperintense) cyst (starred) and duplicated anal canals (straight arrows).
Discussion
CDS is the outcome of a complex pathophysiology with most cases being diagnosed asneonates or young children1 owing to obvious morphological or functional abnormalities.
The cloaca is the common embryological source of origin of both the genitourinary and gastrointestinal tracts which is the reason behind the frequent association of anomalies involving these systems.2,3 Classical CDS describes complete or incomplete duplication of the structures derived from the primitive gut, which forms at approximately the fourth week, with concurrent spinal column defects owing to defective development of the neural tube.4
Since there were no spinal column defects in our patient, it can be inferred that the initial anomaly occurred only at the level of the primary intestine.
Less than 100 cases of CDS have been published worldwide with most cases being reported at birth. A vast majority of these cases undergo treatment in infancy or early childhood. To date, only four cases have been reported in adults, including a patient who had a cloacal malformation repaired during infancy.5–7
In fact, except for a case of CDS reported in 2013,8 no previous presentation in adolescence has been reported. Additionally, complete duplication of the bladder, urethra, vagina and uterus with partial/complete duplication of the hindgut is an even rarer manifestation confined only to a few case reports.9,10
The genitourinary anomalies in CDS vary from isolated partial duplication of the UB to complete duplication of the lower urinary tract. The associated genital tract anomaly may range from bicornuate uterus to complete duplication of the uterus and vagina.11 Hindgut anomalies frequently involve the anorectal regions with manifestations including anal stenosis, ectopic anus and others.
The first reported case in India5 was of an asymptomatic adult female with duplication of colon, rectum, anus, UB, urethra, uterus, cervix, vagina and external genitalia.
Brief summary of cases illustrating CDS spectrum.

Conventional treatment of CDS consists of staged correction of duplication anomalies with emphasis on reasonable anatomic, functional and cosmetic reconstruction.11 Septal resection in UB is done to convert it into a single chamber. The duplicated colons are usually not fused and have separate blood supply, so either mucosal stripping or resection of duplicated colon and rectum may be undertaken.10 Untreated duplication of the hindgut may be a prelude to intestinal obstruction or even neoplastic transformation.
Otherwise, reconstructive surgery is essentially aesthetic in nature as normal menstruation, coitus and pregnancy may be expected in most female patients.6
However, late presentation may adversely affect prognosis as there is a predisposition to gastrointestinal and genitourinary tract obstruction. Our patient had menstrual complaints which make reconstructive surgery a necessity rather than a luxury. Although it has been demonstrated that patients are capable of successful vaginal delivery after surgical repair of cloacal malformation,7 our patient opted to delay genitourinary and gastrointestinal reconstructive surgery till after completing her impending high school examinations.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.