
Abstract
Rustrela virus (Rubivirus strelense) is a single-stranded, non-segmented RNA virus that causes brain diseases in animals, especially encephalitis and meningoencephalitis. Its rapidly mutating RNA genome complicates control, making a vaccine imperative. Immunoinformatics approaches were employed to make the polyepitope vaccine (PEV) for RuV control. Epitopes for cytotoxic T lymphocytes (CTL), helper T lymphocytes (HTL), and B cells were made from most immunogenic parts i.e., E1, E2, and capsid structural proteins of RuV. Epitopes that were antigenic, not allergic, IFN simulators, and not toxic were selected. This led to the selection of 6 CTL, 6 HTL, and 13 B cell epitopes, which were then used to construct the PEV with appropriate linkers and CTB as an adjuvant for immunological modulation. The physiochemical analyses demonstrated that the PEV was safe, stable, hydrophilic, and soluble. The secondary structure prediction has indicated mostly coiled and alpha helix in the PEV. The 3D scans revealed a solid and stable structure. The interactions and stability between the vaccine, TLR3, TLR4, and TLR7 were revealed by molecular docking. The vaccine candidate’s minimal deformability and high stability were verified by molecular dynamics modeling. The developed PEV vaccine demonstrated a strong immunological response in in silico immune simulation. RuV-PEV is predicted to be efficiently produced in E. coli following in silico codon optimization and vector cloning. It elicited strong antigen-specific humoral and cellular responses, indicating promise as a Rustrela virus vaccine. Its precise effectiveness, safety, and immunogenicity profile may be confirmed by further experimental validations.
Introduction
Rustrela virus (Rubivirus strelense), discovered in 2020, is a relative of rubella virus (RuV) that has been detected in cases of encephalitis and meningoencephalitis in diverse mammals. It was initially discovered in brain samples from a few animals in a German zoo, including a donkey (Equus asinus), a capybara (Hydrochloric hydrochaeris), and a red-necked wallaby (Macropus rufogriseus) with symptoms such as ataxia, apathy, and rhinorrhea (Mankertz et al., 2022). The Rustrela virus (RusV) was subsequently identified in the brain tissues of 8 of 16 yellow-necked field mice (Apodemus flavicollis) located on zoo grounds and within 10 km of the zoo (Pfaff et al., 2022). This mouse is considered to be a possible repository host for the virus (Grimwood et al., 2021).
RusV has a single-stranded, non-segmented RNA (+) genome that is approximately 9.6 kb nucleotides long and encodes two polyproteins (Matiasek et al., 2023): (i) the nonstructural polyproteins (nsPP) at the 5′ end of the genome, which cleaves into the RNA-directed RNA polymerase p90 (828 aa) and the protease p150 (1,093 amino acid residues [aa]); and (ii) the structural polyproteins (sPP), which cleaves into the envelope glycoproteins E2 (324 aa) and E1 (487 aa) and the capsid (C) protein (332 aa). There is an intergenic region of 290 nt between the open-reading frames for nsPP and sPP (Pfaff et al., 2022).
The RusV virus causes staggering illness, and the most prevalent clinical manifestation is hind leg ataxia with generalized muscular tone, resulting in a stumbling gait (de le Roi et al., 2023; Weiss et al., 2023). Furthermore, a range of prognostic neurological symptoms, such as the inability to retract the claws, hyperesthesia, and, on occasion, tremors and convulsions, may develop (Weiss et al., 2023). Behavioral vicissitudes include increased vocalization, despair, becoming more affectionate, and, in rare cases, hostility (Matiasek et al., 2023).
The primary challenges that doctors faced in managing viral infection were the lack of a vaccine and disease-prevention medications (Oker-Blom et al., 1984). Regardless of the substantial mortality rate, it was always believed that this agent causes no epidemic danger (Voss et al., 2022a). Presently, extensive research is being conducted to create virus-control techniques such as small interfering RNA, low-molecular compounds, and antibodies, medications based on monoclonal antibodies, and, of course, vaccinations (Bazhan et al., 2019).
To expedite the vaccine design process, researchers have recently focused on building multi-epitope vaccines using in silico immunoinformatics approaches without the requirement to cultivate pathogens (Yang et al., 2021). Multiple viral protein fragments with overlapping epitopes are used to create multi-epitope vaccinations (Mahdevar et al., 2022), which can be effective against viral infections and provide more effectiveness for clinical studies (Zhang, 2018).
The purpose of this study is to use reverse vaccinology to create an effective multi-epitope vaccination against Rustrela virus. Capsid, E1, and E2 epitopes were predicted and connected together using suitable linkers. Consequently, the vaccine’s physicochemical characteristics, antigenicity, and allergenicity were assessed. Several analyses were also carried out, including the prediction of secondary and 3D models of the vaccine construct, molecular docking, MD simulation, immunological simulation, and in silico cloning of the vaccine construct, which may now be examined in clinical trials.
Materials and Methods
Polyprotein sequence retrieval
The Rustrela virus sequence from the donkey isolate in Germany was selected on the basis of its high sequence homology to all other rustrela virus sequences available in NCBI at the time. The protein sequence of the virus was isolated from the NCBI (GenBank: QKO01649.2). The open reading frame of 9.6 kb Rustrela RNA encodes multiple proteins, including three structural proteins (Capsid, E1, and E2). Due to lack of 3D structure availability, the structure was predicted using Alphafold for designing the epitopes accordingly. Alphafold employs a deep learning based approach to predict protein structure by integrating deep leaning-based approach and multiple sequence alignment. Each of the four structural proteins also underwent B cell and T cell potential epitope prediction (Kotecha et al., 2017).
B cell epitope prediction
Both linear and discontinuous B cell epitopes were analyzed. To predict the linear B cell epitopes, SVM with Tri-peptide similarity and Propensity scores (SVMTriP) (http://sysbio.unl.edu/SVMTriP/prediction.phpp) web server was used, and the epitope length was determined to be 16aa. The server predicted the antigenic epitopes by utilizing the most up-to-date input sequence from the Immune Epitope Database (IEDB) dataset. Combining SVMTriP improves prediction accuracy (Sahu et al., 2022; Yao et al., 2012). SVMTriP uses a support vector machine (SVM)-based classification algorithm. The default parameters of score 0.5 and 6Å distance were utilized in the IEDB Analysis Resource application ElliPro (http://tools.iedb.org/ellipro/), which extrapolates protein 3D structures using protein shape as ellipsoid and calculating a protusion Index (PI) for each residue to predict discontinuous B cell epitopes. Scores are used to rank the output results. Only the highest-scoring epitopes were chosen (Triwijaya et al., 2022; Ponomarenko et al., 2008).
T cell epitope prediction
T cell epitopes are peptides from antigens presented to the TCR by APC-expressed MHC molecules. The IEDB’s analytical resource provides some of the best prediction methods for hundreds of alleles, including those for predicting T cell epitopes in accounts for vaccine development procedure. T cell peptides were predicted by TepiTool (http://tools.iedb.org/tepitool/). Tepitool predicts T cell epitopes using consensus-based algorithm that integrates multiple peptide-MHC binding prediction methods (e.g., stabilized matrix method [SMM], ANN, and combinatorial scoring) to estimate binding affinity across diverse HLA alleles. The FASTA format of the query sequence was provided as input, followed by uploading the 27 most commonly occurring HLA Alleles files. The length of 9mers and IC50 score of <250 were chosen. For MHC I and MHC II epitopes prediction, SMM and SMM align (also known as NetMHCII-1.1) were used, respectively (Nielsen et al., 2020).
Analysis of anticipated epitopes for antigenicity, allergenicity, and immunogenicity
The primary function of epitopes is to be selectively recognized by antibodies or T cell receptors to elicit immune responses. (Zhang and Tao, 2015). To determine the antigenic property of the selected epitope, VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used with a threshold filter of 0.5. Vaxigen is an alignment-independent algorithm that enables the classification of proteins.
To predict the allergenicity of epitopes, AllerTOP v. 2.0 was used (https://www.ddg-pharmfac.net/AllerTOP/index.html) (Dimitrov and Bangov, 2014). For IFN production analysis, IFNepitope (http://crdd.osdd.net/raghava/ifnepitope/predict.php) was used. The server predicts the potential IFN inducive epitope by using a library of MHC class II binders already submitted on the server (Mathew et al., 2022).
Population coverage analysis
Population coverage for the selected CTL and HTL epitopes was assessed using the IEDB Population Coverage tool (https://tools.iedb.org/population/). Finalized epitope sequences (6CTL, 6HTL) were submitted to population coverage tool. We focused on global populations prone to rustrela virus spillover risk using the combined MHC I and II mode.
Development of a poly-epitope vaccine sequence
Computational vaccinology relies on the construction of vaccine constructs, which entails randomly inserting epitopes and joining them with EAAAK, AAY, CPGPG, and KK linkers. A 6× His tag was incorporated at the C-terminus of the final vaccine construct to facilitate future protein expression and purification. The sequence consisting of adjuvant, linkers, and epitopes making a whole new protein was named poly-epitope vaccine (PEV) (Rana and Akhter, 2016; Sanches et al., 2021). Analysis was carried out to test the newly developed construct(s) for antigenicity, allergenicity, toxicity, solubility, and stability. The constructed PEV was analyzed for allergenicity and antigenicity (Nosrati et al., 2019).
Analysis of physiochemical properties
The physical and chemical characteristics of the PEV were studied with the help of the ProtParam tool (https://web.expasy.org/protparam/). Calculations for the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) score were all performed using the raw sequence of the poly epitope vaccination to predict the stability and efficiency of the PEV (Kaushik et al., 2022).
Vaccine efficiency via immune modeling
The activity and the immune response stimulation by the PEV were predicted by using the C-immsim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/?page=1). The simulation was performed by application of machine learning techniques for the prediction of immunological interaction and the use of a position-specific scoring matrix for the prediction of an immunogenic epitope. For PEV, a total of 300 steps were performed (1 step ∼8 h of real-life). Whereas the injection doses were provided with the duration of 1, 84, and 168 steps. The remaining parameters were selected as the default to perform the simulation. (Omoniyi et al., 2022).
Structure prediction and refinement of PEV
The secondary structure of the PEV was predicted using the Self-Optimized Prediction Method with Alignment (SOPMA) server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). The server predicted the protein secondary structure by taking in the query as the amino acid sequence and performing a series of neutral networking giving the prediction of alpha helix, beta sheets, and coils in the query sequence.
For the tertiary structure prediction, Alphafold was used (https://www.deepmind.com/research/highlighted-research/alphafold). After the tertiary structure prediction, GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) was used to refine the predicted structure of the PEV.
In addition, the refined structure was validated using the SAVES server’s ERRAT program. (https://saves.mbi.ucla.edu/). The ERRAT gives the score according to the structure quality. Furthermore, the PROCHECK, available on the same server, is used to predict the φ and ψ dihedral score in the energetically allowed and disallowed region by drawing the Ramachandran plot, which adds to the validation of the protein refined structure.
Disulfide bond engineering of PEV
To enhance proper folding and strengthen the structural stability of designed PEV, disulfide bond engineering was performed. For this purpose, Disulfide by Design 2.0 (DbD2) server (http://cptweb.cpt.wayne.edu/DbD2/) was used. This tool enables the identification of residue pairs that are not geometrically suitable for disulfide bond formation but are also likely to enhance the protein’s thermal stability.
Using the tool, disulfide engineering was performed with default parameters of Cα-Cβ-Sγ angle and χ3 value. Potential disulfide bonds were selected using strict criteria, requiring an energy level of less than 2.0 kcal/mol and χ3 angles within the range of −87° to +97° (±30°).
Molecular docking
The protein–protein docking was performed between the refined PEV and TLR3 and TLR7 receptors of the Homo sapiens (humans). The structures of both TLR3 and TLR7 of humans were retrieved from the Alphafold database (A0A3S5ZPK8 and A0A3Q1MK24, respectively). The LZerD (https://lzerd.kiharalab.org) was used to dock the PEV with receptors. The structure with the lowest score was selected, and Molecular simulation was performed. Protein–protein interactions of the docked proteins were done using PDBsum (Christoffer et al., 2021). To identify stable interatomic contacts and binding hotspots at PEV-TLR interfaces, docked complexes (TLR3-PEV, TLR4-PEV, TLR7-PEV) were analyzed using COCOMAPS 2.0 webserver (https://aocdweb.com/BioTools/cocomaps2/). This tool is available online for atomic-level interface characterization in protein–protein complexes (Chawla et al., 2025).
Structural dynamic analysis
When it came to ensuring the steadiness of the docked-complex protein inside the cell, molecular simulations were used. To check the deformation probability of the TLR3-PEV and TLR7-PEV docked complex, an NMA simulation was performed. iMODS (https://imods.iqfr.csic.es/) opted to perform the simulation with default parameters by simply providing the PDB-ID or the atomic coordinates in PDB format to investigate the collective movements of proteins and nucleic acids using NMA in internal coordinates (torsional space). A method known as “normal mode analysis” has been used to characterize the flexible states that are available to a protein around an equilibrium point. It is based on the physics of tiny oscillations. In Addition, NMA creates the crucial eigenvalue parameter, which is an indication of stability (López-Blanco et al., 2014).
Codon optimization and in silico vector construction
After completing and validating the PEV structure, the reverse translation and codon optimization were done using OPTIMIZER http://genomes.urv.es/OPTIMIZER/). It uses a codon usage bias based algorithm that replaces synonymous codons according to host-specific frequency tables. The amino acid sequence was inserted as the query, and the codon optimization was performed according to the codon usage database, using prokaryote as the expression system. The codons were then optimized as the standard genetic code and one amino acid, one codon method. The restriction sites were added at the flanking region of the optimized sequence by using Snapgene software. BamHI and XhoI restriction sites were added at the 5′ and 3′ ends, respectively, to facilitate the restriction insertion of the desired fragment in the expression vector (pET-28 a).
Results
B cell prediction
There were over 40 continuous B cell epitopes predicted using ABC pred, and 19 discontinuous B cell epitopes predicted using ElliPro tool. After screening the epitopes for antigenicity, allergenicity, and toxicity using VaxiJen v. 2.0, AllerTOP v. 2.0, and Toxinpred, respectively, total of 13 continuous and discontinuous B cell epitopes were finalized as given in Table 1.
The Sequences of Potential B Cell Epitopes along with Their Position and Antigenicity
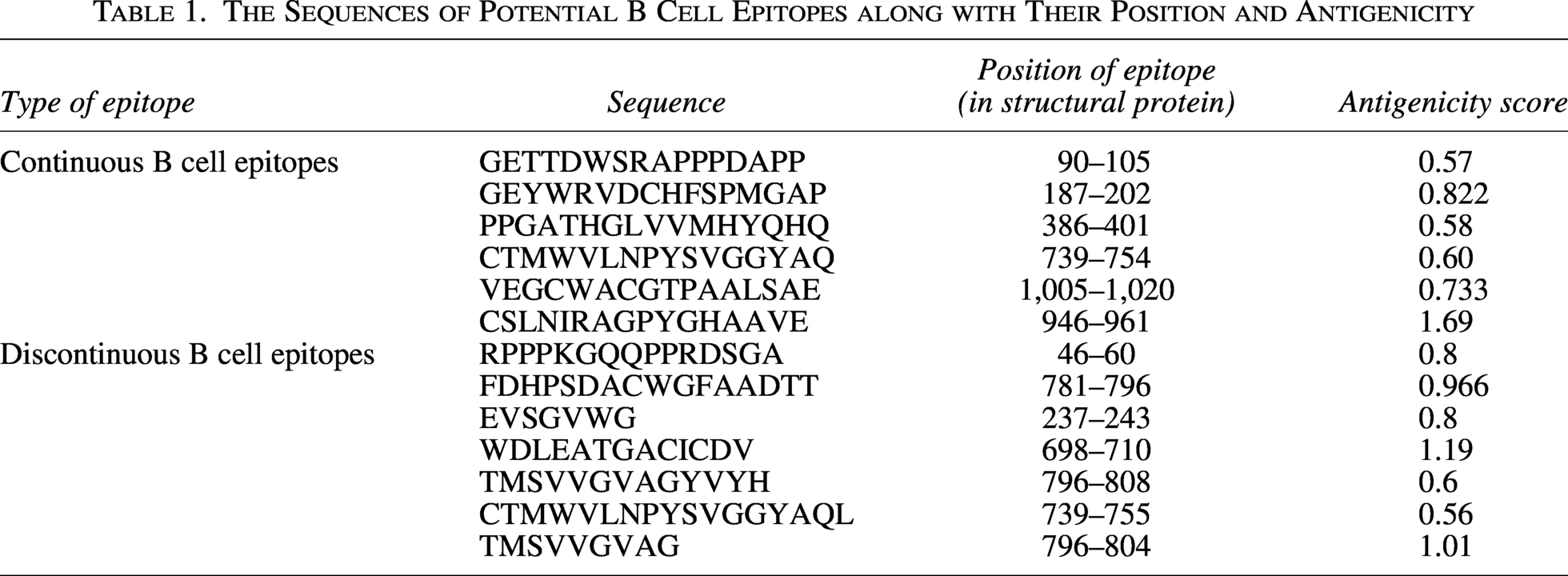
T cell prediction
CD8+and CD4+epitopes are predicted using IEDB database, and initially around 150 MHC I and 170 MHC II epitopes were obtained. After screening for allergenicity, antigenicity, toxicity, and IFN-inducing capabilities of all the epitopes, 6 CD8+ and 6 CD4+ epitopes were shortlisted and included in designing the PEV (Table 2).
Potential T Cell Epitopes with Their Sequences, Antigenicity Scores, Position, Alleles, and IFN Induction Ability
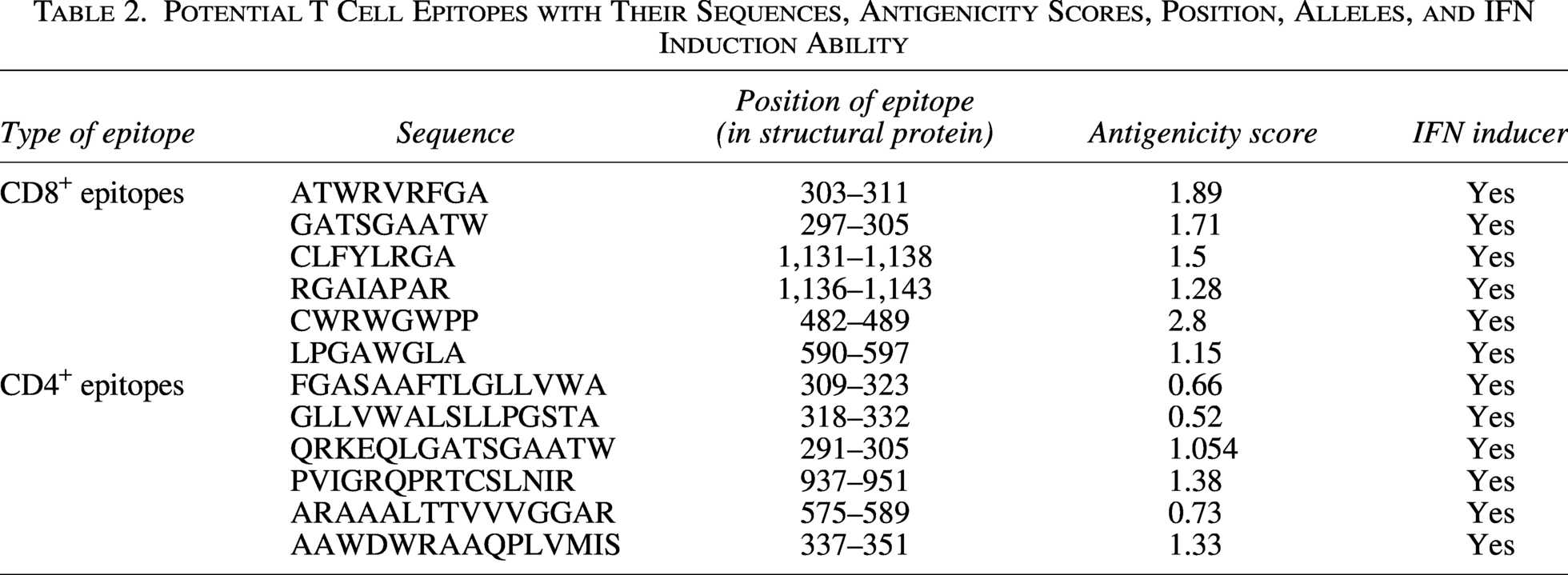
Population coverage of the T cell epitopes
Both CD8+ and CD4+ alleles having IC50 value < 100 were chosen for the population coverage analysis by IEDB. This analysis revealed that population coverage was 86.71% worldwide, which suggests that epitopes selected for this vaccine cover most of the population of the world and can be effectively utilized. It also suggests that this vaccine has broad immunogenicity potential (Fig. 1).
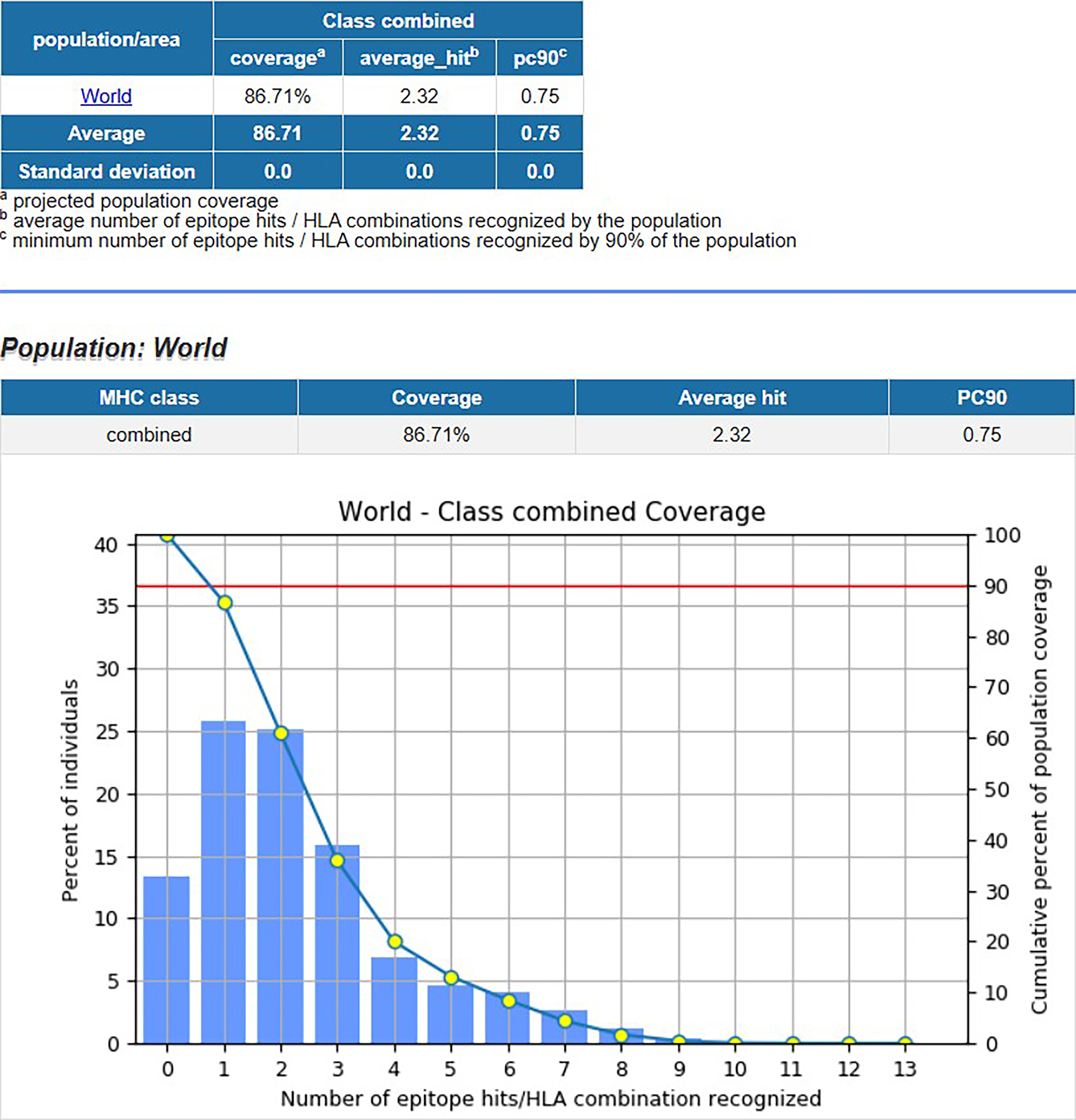
Population coverage analysis graph.
Constructing polyepitope protein
After shortlisting all the B and T cell epitopes, each epitope is linked together using specific linkers. At the N-terminal of the protein, CTB adjuvant is added by using EAAK linker to increase the immunogenic response of the protein. Followed by adjuvant, CTL epitopes are added using AAY linkers and HTL ones using GPGPG linker. The B cell epitopes are linked using KKK linkers, followed by EAAAK linker, linking the 6x His-tags at the C-terminal of the protein. The combination of these linkers ensures structural stability, appropriate MHC presentation, efficient proteasomal processing, and enhanced epitope accessibility, optimizing overall immunogenic potential of the designed vaccine. After constructing PEV, antigenicity, allergenicity, and toxicity analyses show that the vaccine is antigenic, with antigenic score of 0.7, non-allergen, and nontoxic (Fig. 2).
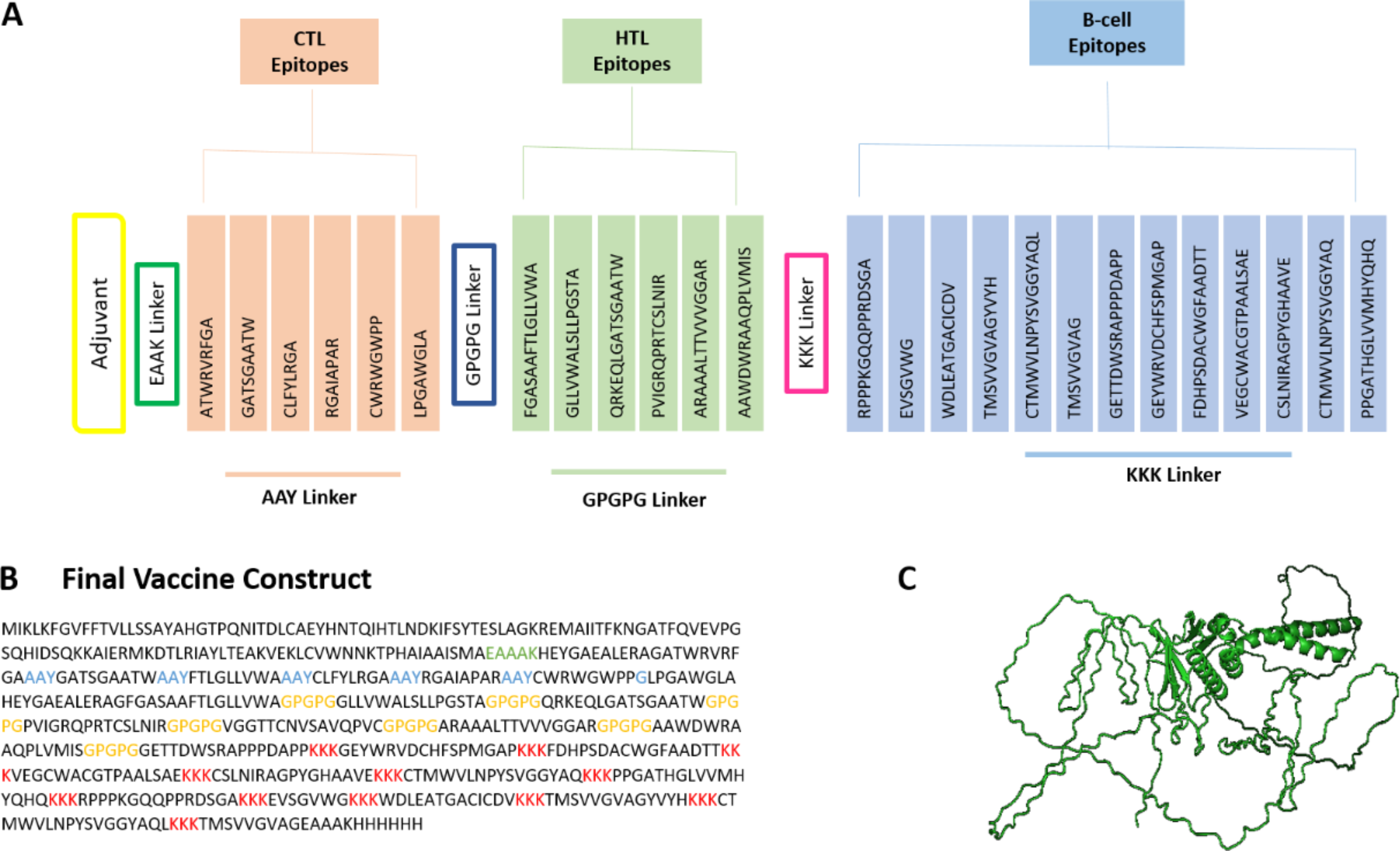
Physiochemical properties of the vaccine
The physiochemical analysis of the protein was performed using Expasy ProtParam to validate the efficacy of the vaccine. The protein is stable with the instability index of 28.9, half-life of 30 h in mammals and aliphatic index of 68.07. The molecular weight of the vaccine is 6.3 kDa and with theoretical PI of 9.66. Major physiochemical parameters are given in Table 3.
Physiochemical Characteristics of Polyepitope Vaccine
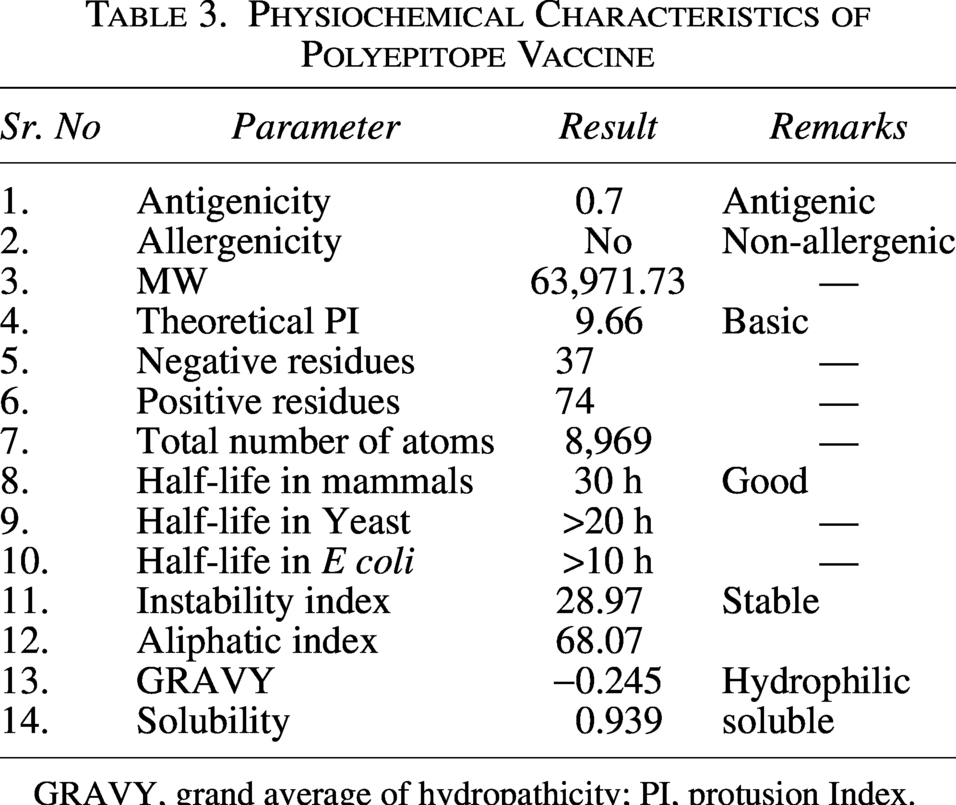
GRAVY, grand average of hydropathicity; PI, protusion Index.
Immunogenic response
The C-immsim server was used to run an immunological simulation of PEV. According to the simulation results, the PEV triggered a more significant secondary and tertiary response than primary response. In addition, after the tertiary response, the vaccine induced a robust level of IgM+ IgG (Fig. 3). After the third vaccination dosage, the total number of B cells and the number of active B cells both increased significantly. Dendritic cell function has revealed that the MHC I and II epitope is highly represented and internalized. Shortly after PEV administration, cytotoxic CD8 T cell numbers dropped as well.
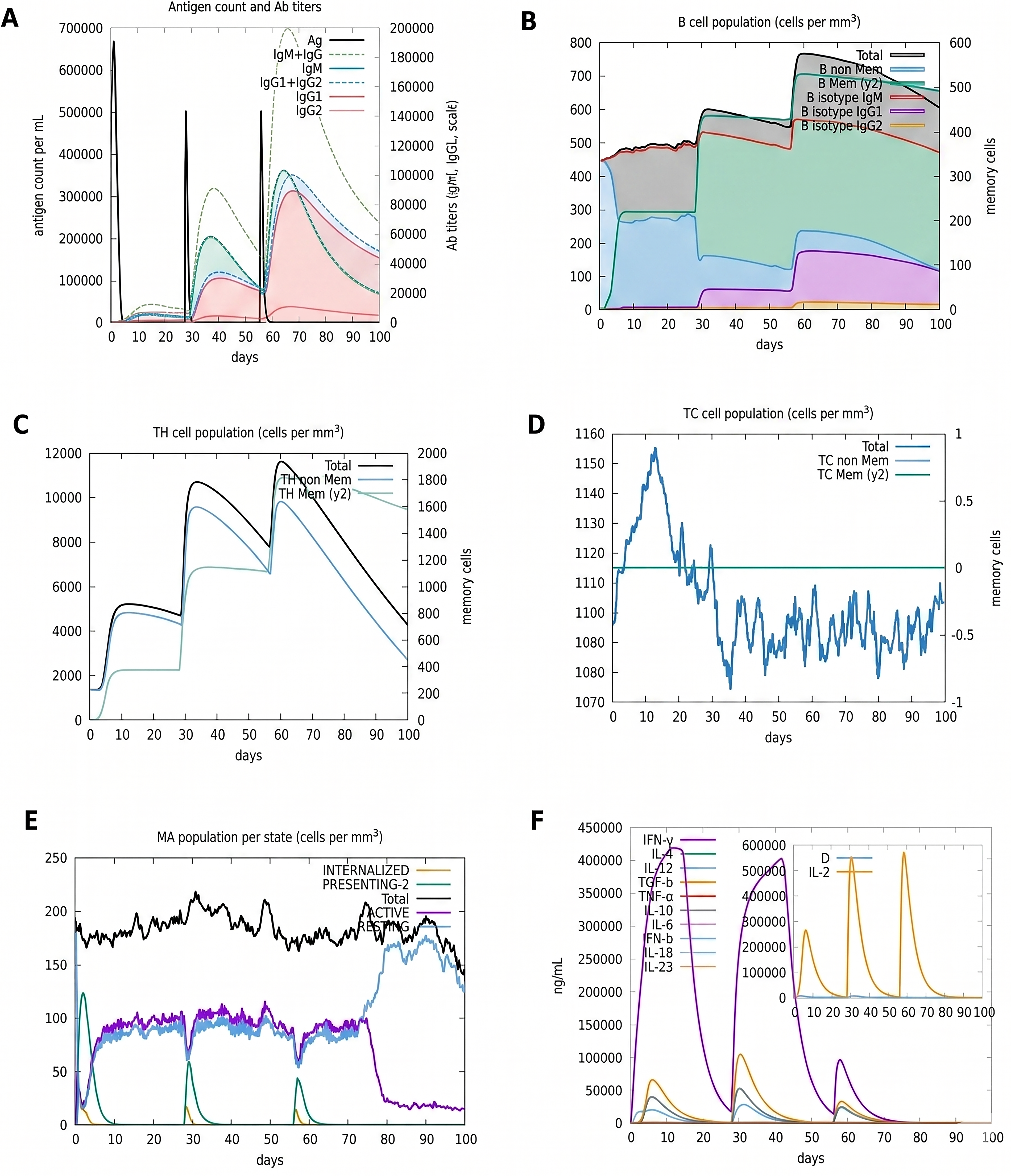
Three doses of the vaccine were simulated using the c-ImmSim server, and the findings were promising.
Structure prediction
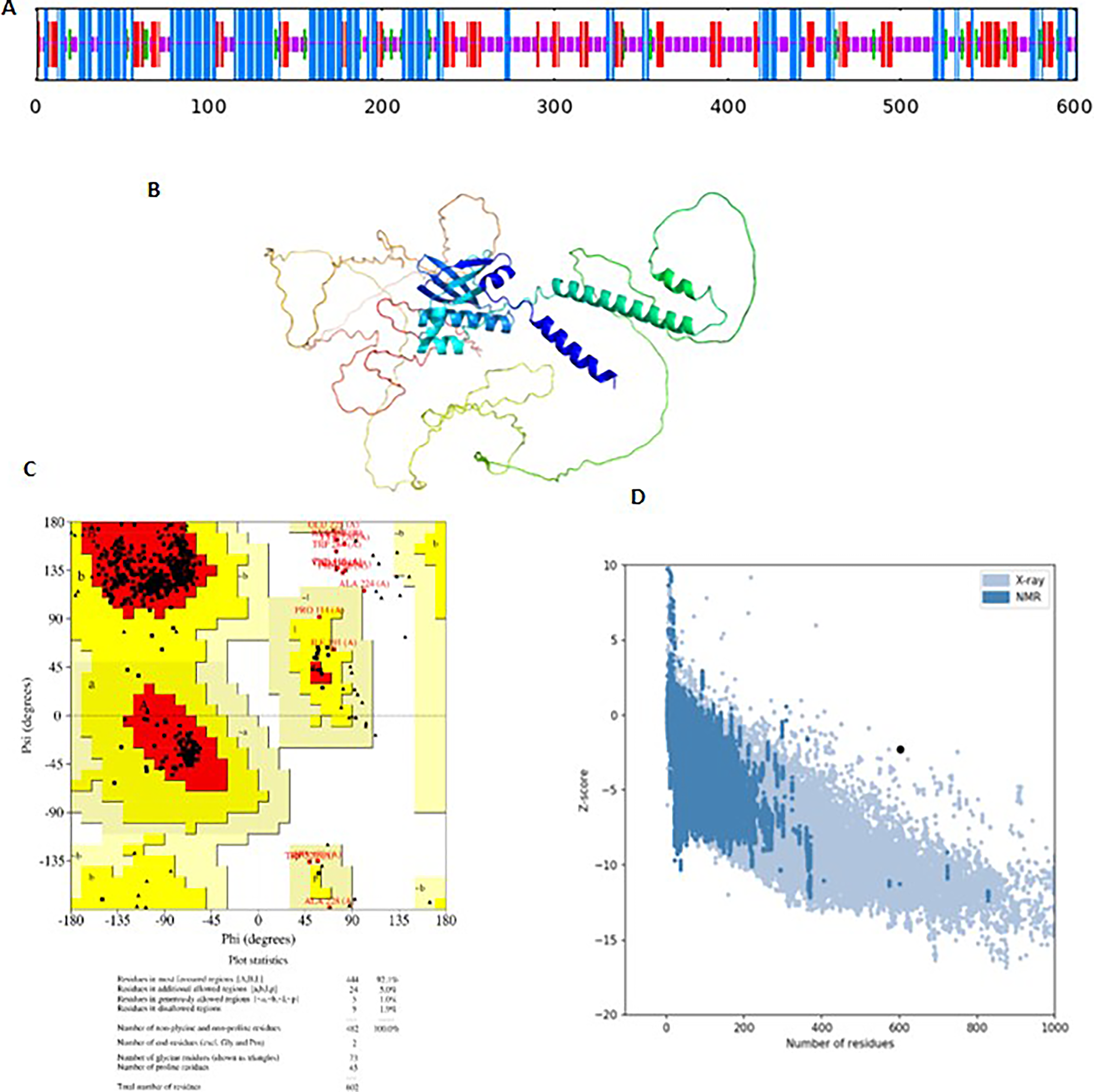
The secondary structure of the protein was predicted using Sopma, indicating that the protein contains 32.8% alpha helix, 20% extended strand, and 40% random coils (Fig. 4A). While tertiary structure was predicted using AlphaFold2 and refined for side chain modification using GalaxyRefine (Fig. 4B). The model 4 was shortlisted with the Rama favored score of 95.2, RMSD 0.76, and GDT-HA 0.832 (Fig. 4C). The refined structure was then validated using the ERRAT and have ERRAT score of 94.3 (Fig. 4C). The Ramchandran plot of the protein was designed using ProCheck, indicating that the most residues were in favored region (92.1%) and very few in the additionally allowed (5%) and disallowed regions (1.2%). Furthermore, the Z-score predicted by using Prosaweb server has −2.39 value indicating that the precited structure has close resemblance with that of the X-ray–generated structure.
Molecular docking
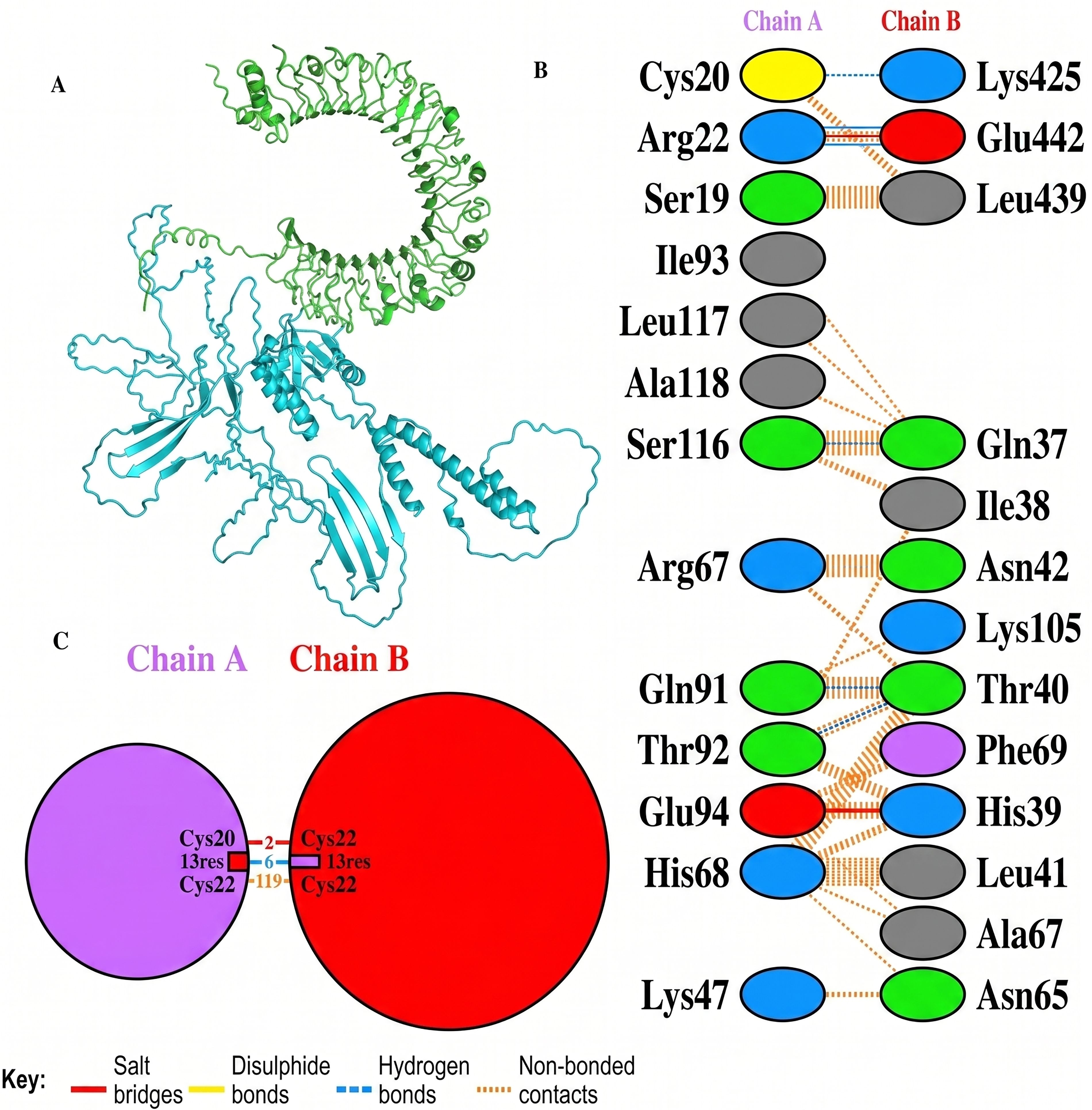
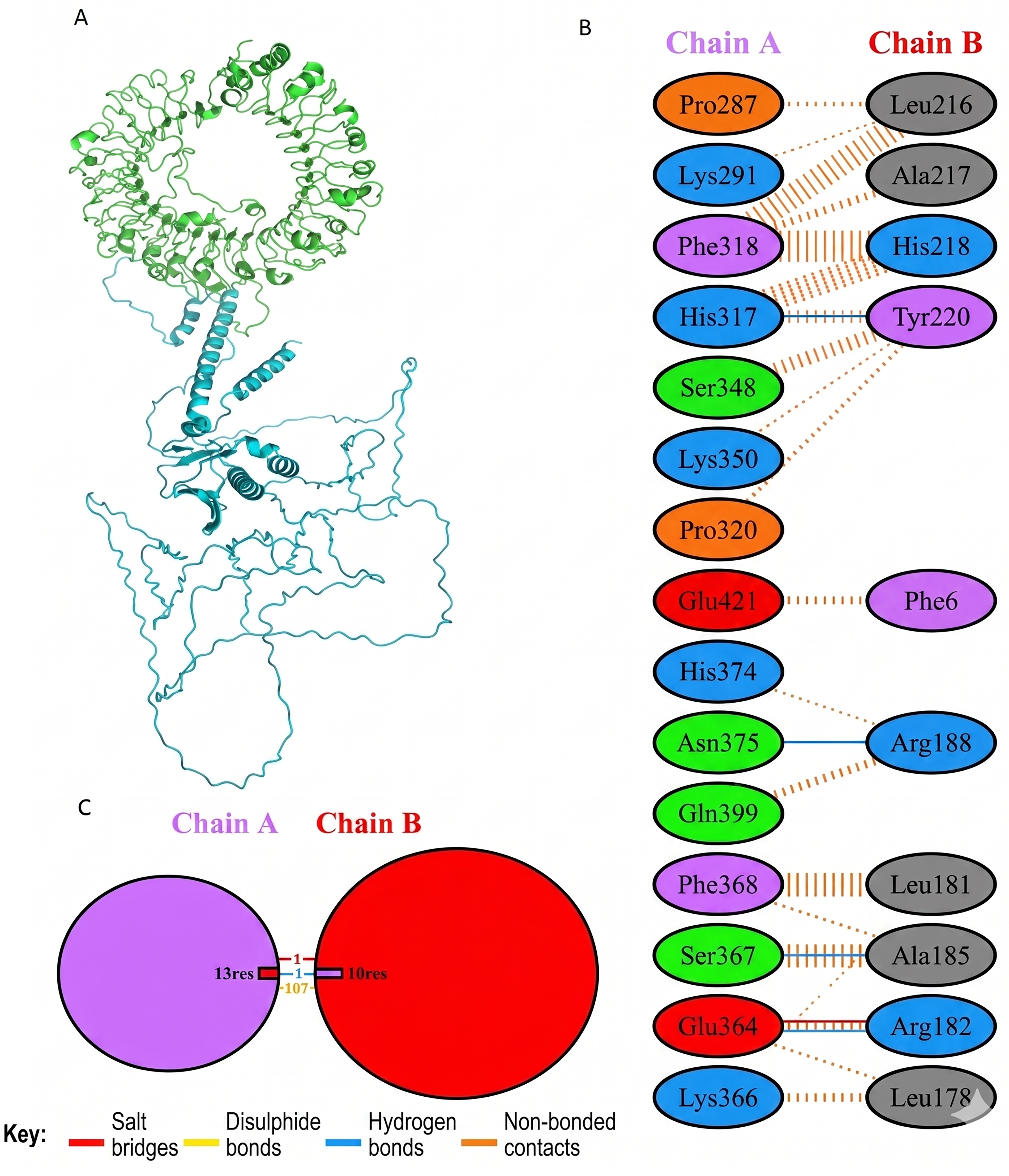
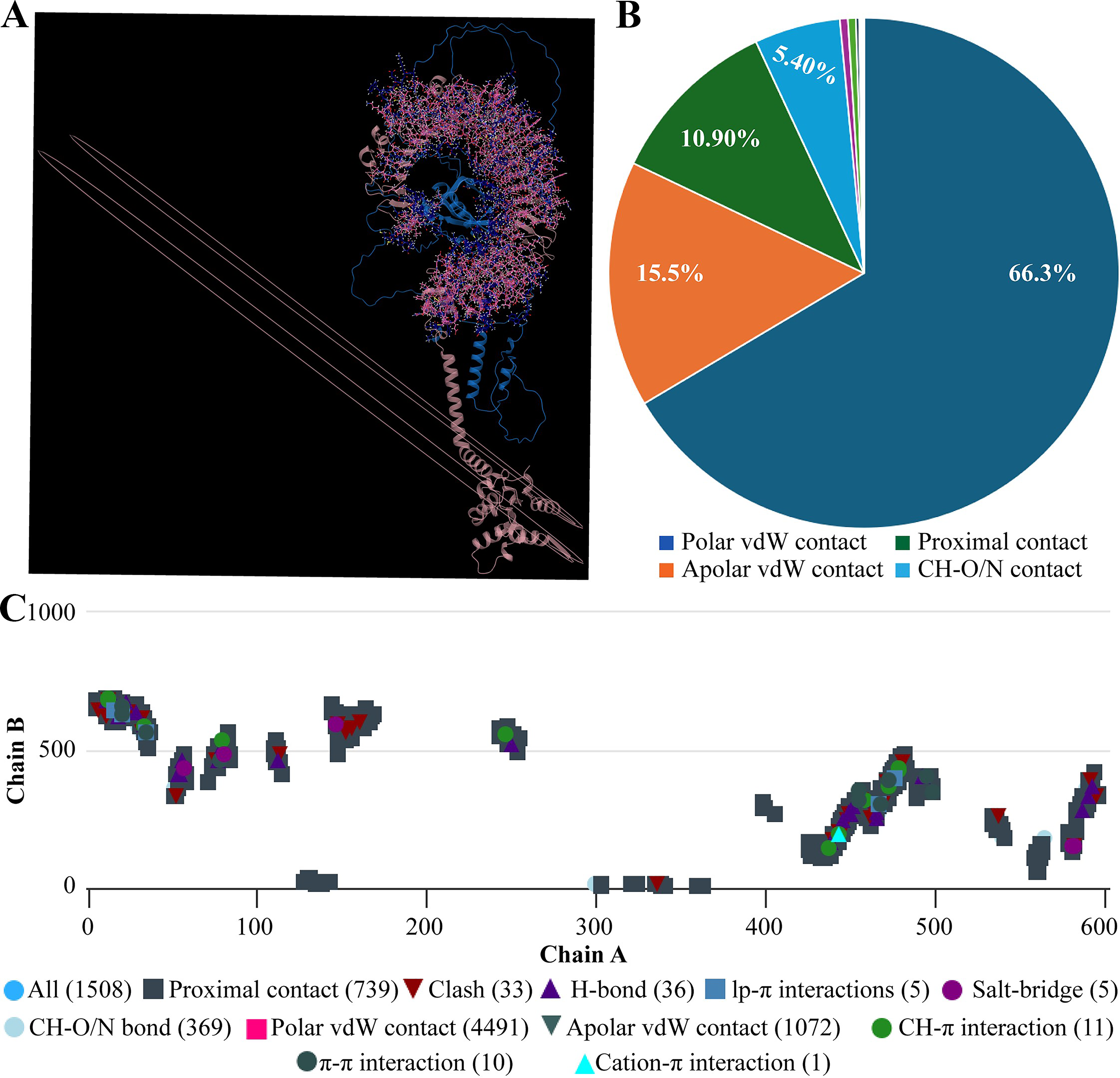
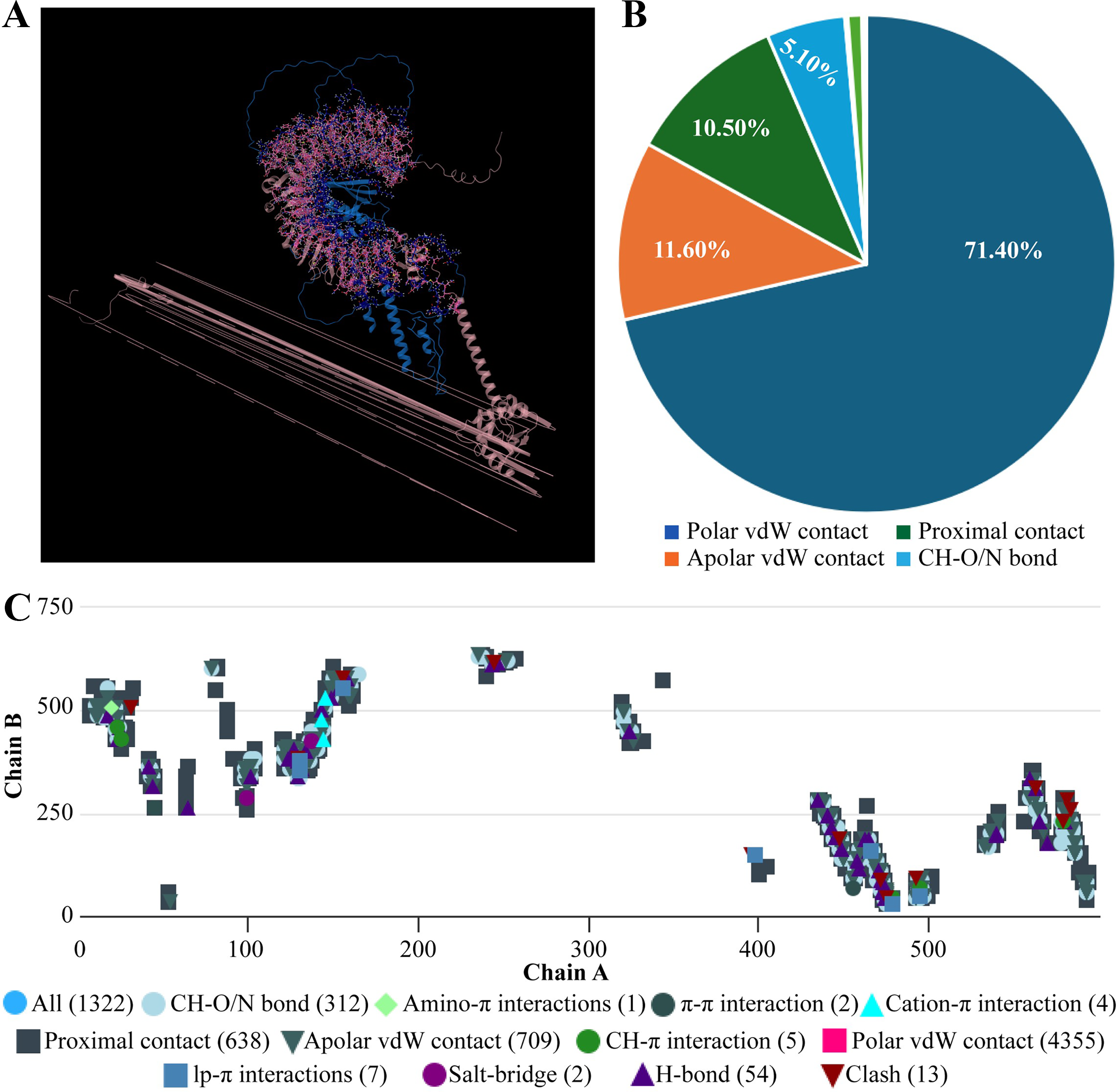
Molecular docking of designed multiepitope vaccine was performed with the three TLR receptors, TLR3, TLR4, and TLR7, using Hawkdock, and then PDBsum was used to determine the interaction between ligand and the receptor. The TLR3-PEV docking shown 2 hydrogens bind interaction, 2 salt bridges interactions, and 71 nonbonded interactions (Fig. R5). Whereas TLR4-PEV shown 6 hydrogen bond interaction, 2 salt bridges, and 119 nonbonded interactions (Fig. 6). While TLR7-PEV docked structures have shown 4 hydrogen bond interaction, 1 salt bridge, and 107 nonbonded interactions (Fig. 7). COCOMAPS 2.0 analysis confirmed docking stable binding of multi-epitope vaccine construct with TLR3, TLR4, and TLR7 receptors. Quantitative analysis demonstrated that polar van der waals interactions were predominant in all three complexes, accounting for 66.3% in TLR3 complex, 71.40% in TLR4 complex, and 67.90% in TLR7 complex (Figs. 8–10).
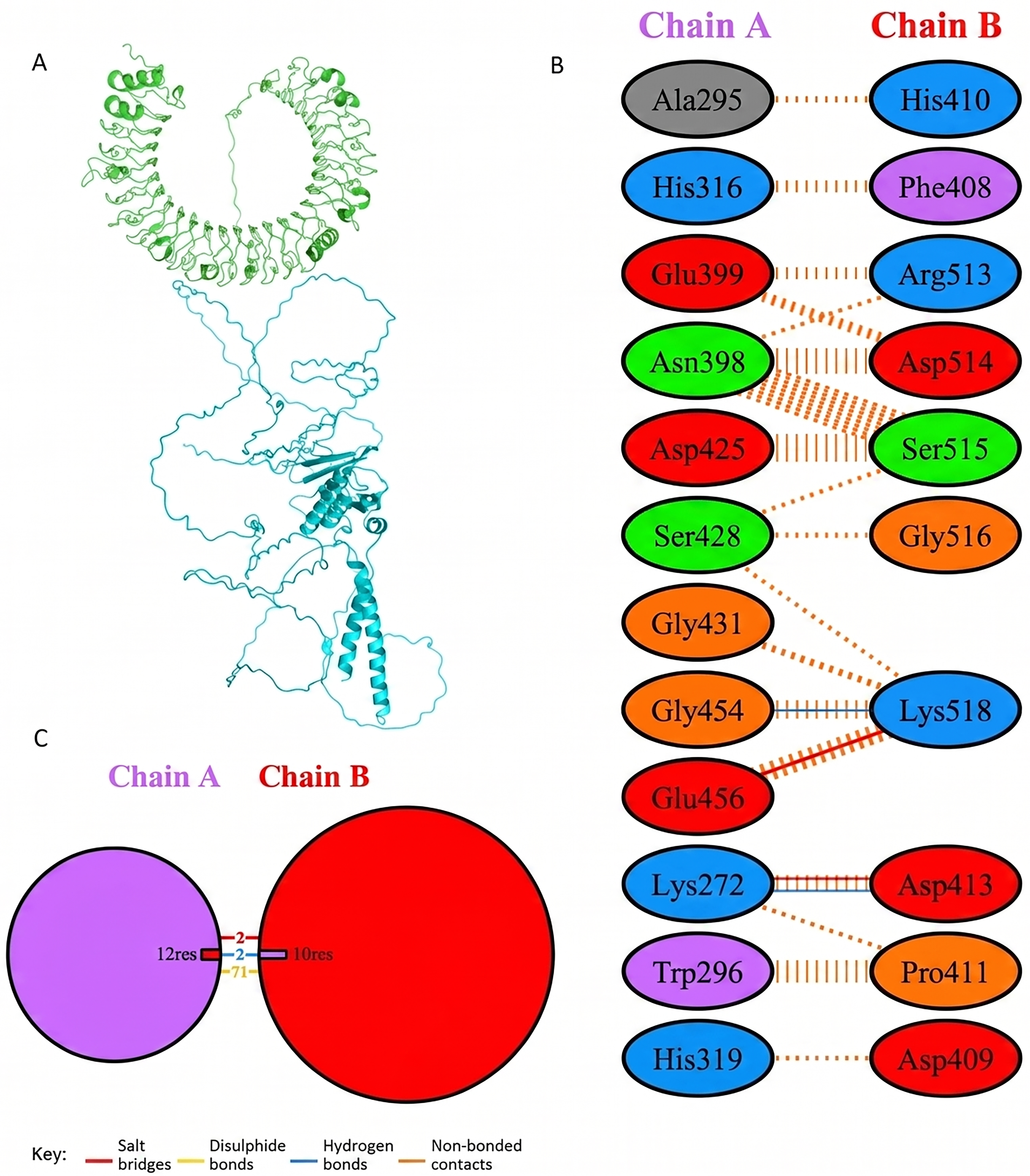
PEV construct docking with human TLR3.
PEV construct docking with human TLR4.
PEV construct docking with human TLR7.
Interaction analysis of the vaccine construct with TLR3 receptors.
Interaction analysis of the vaccine construct with TLR4 receptors.
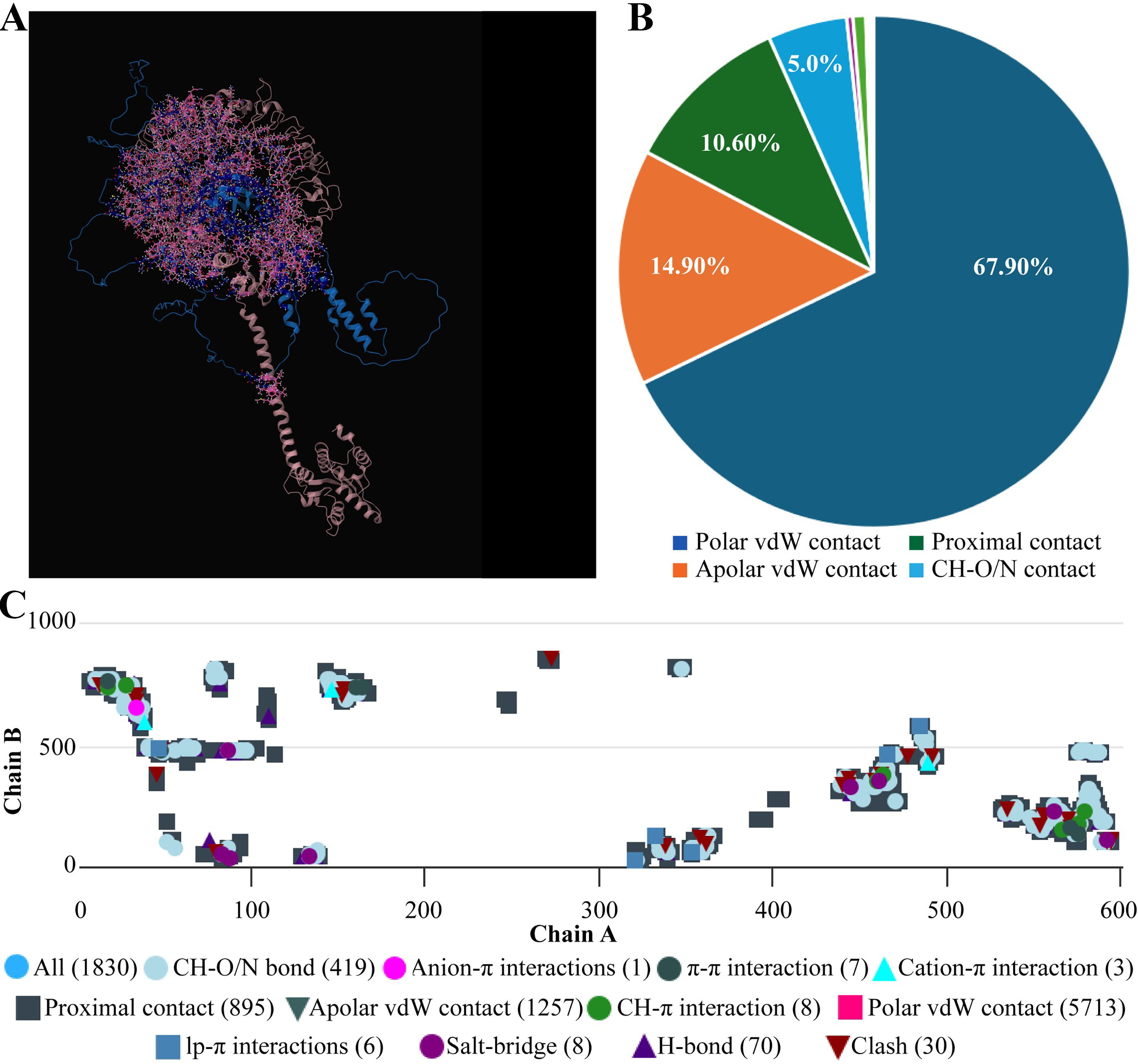
Interaction analysis of the vaccine construct with TLR7 receptors.
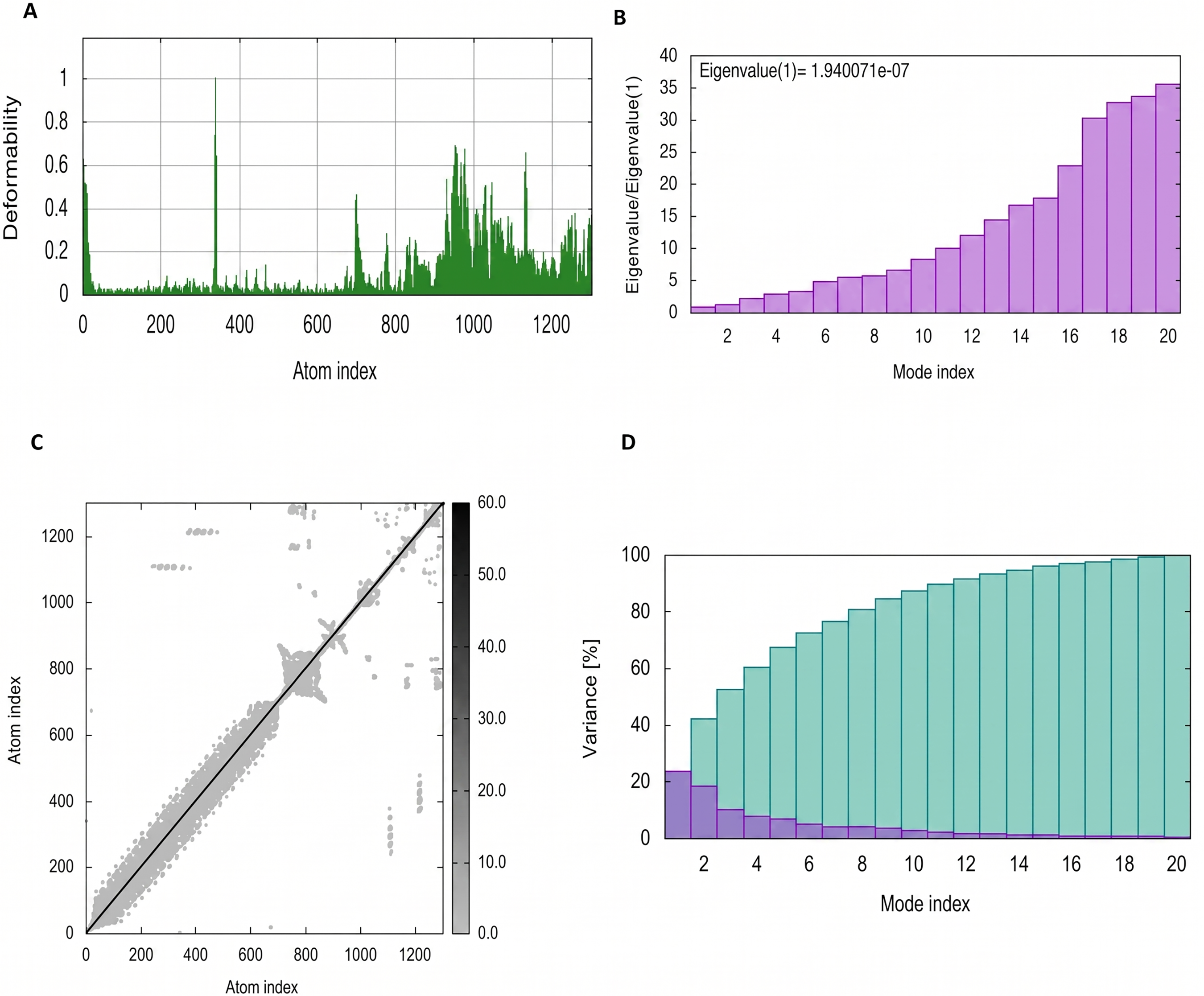
NMA Simulation of PEV-TLR3 complex.
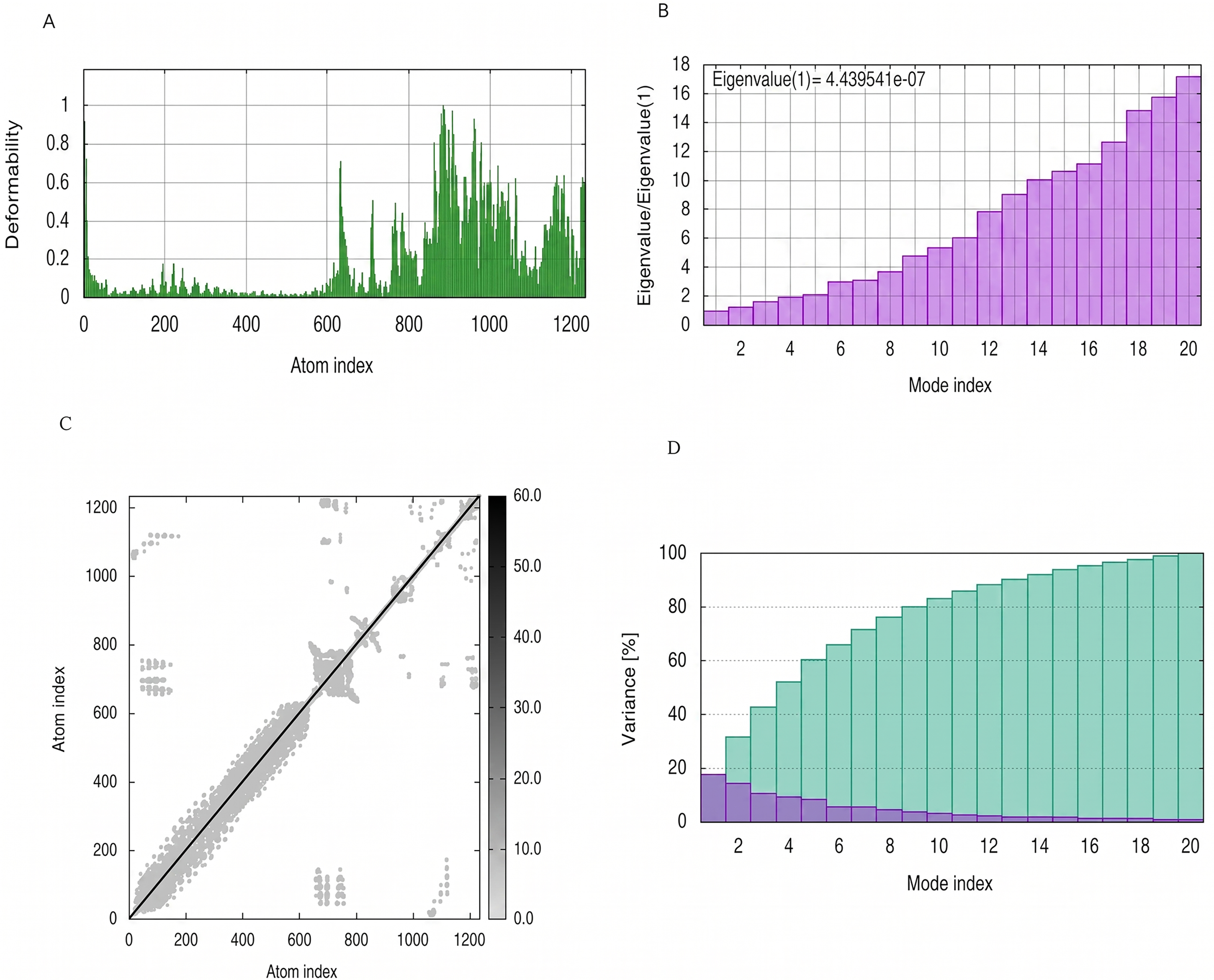
NMA Simulation of PEV-TLR4 complex.
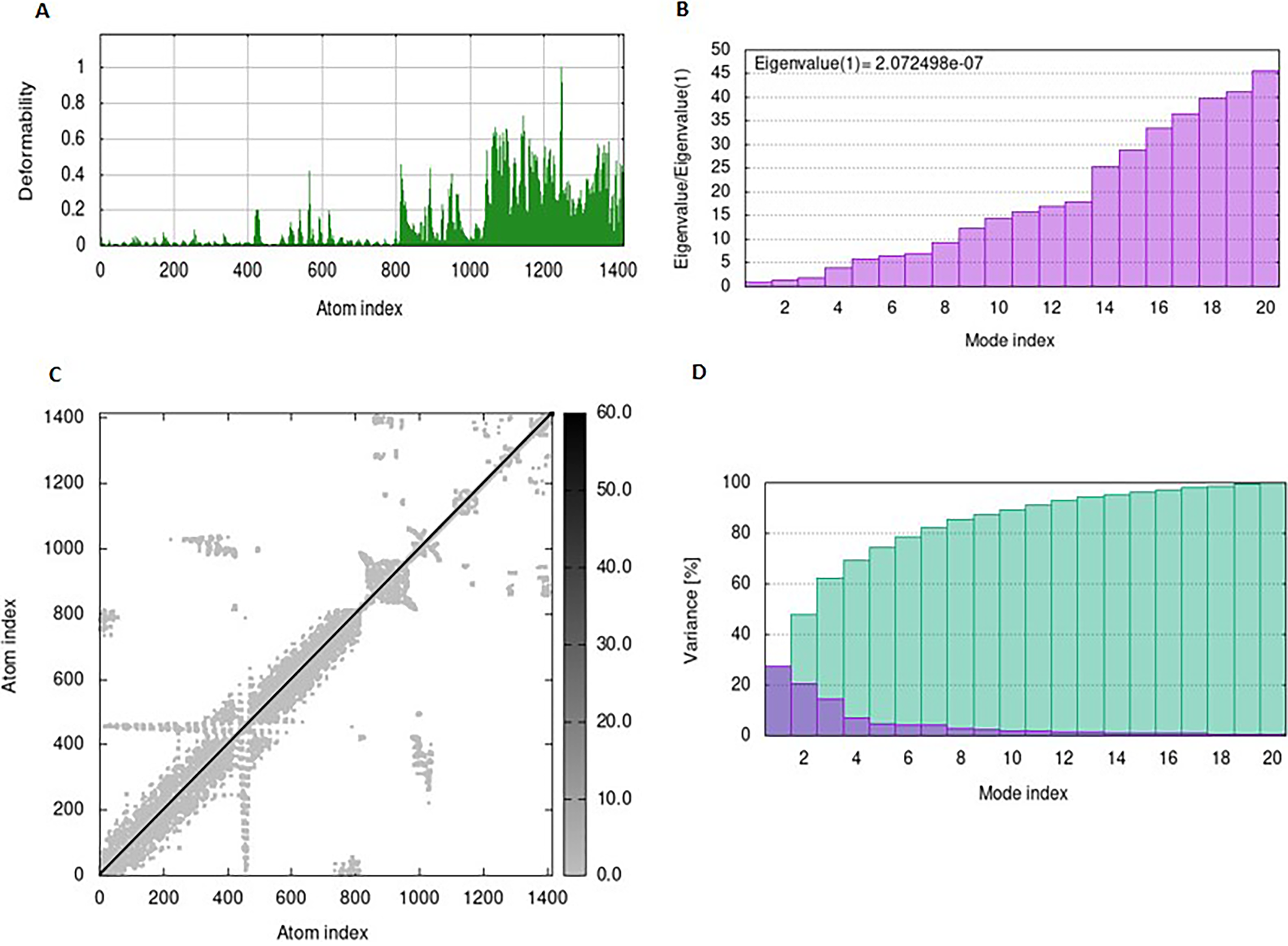
NMA Simulation of PEV-TL73 complex.
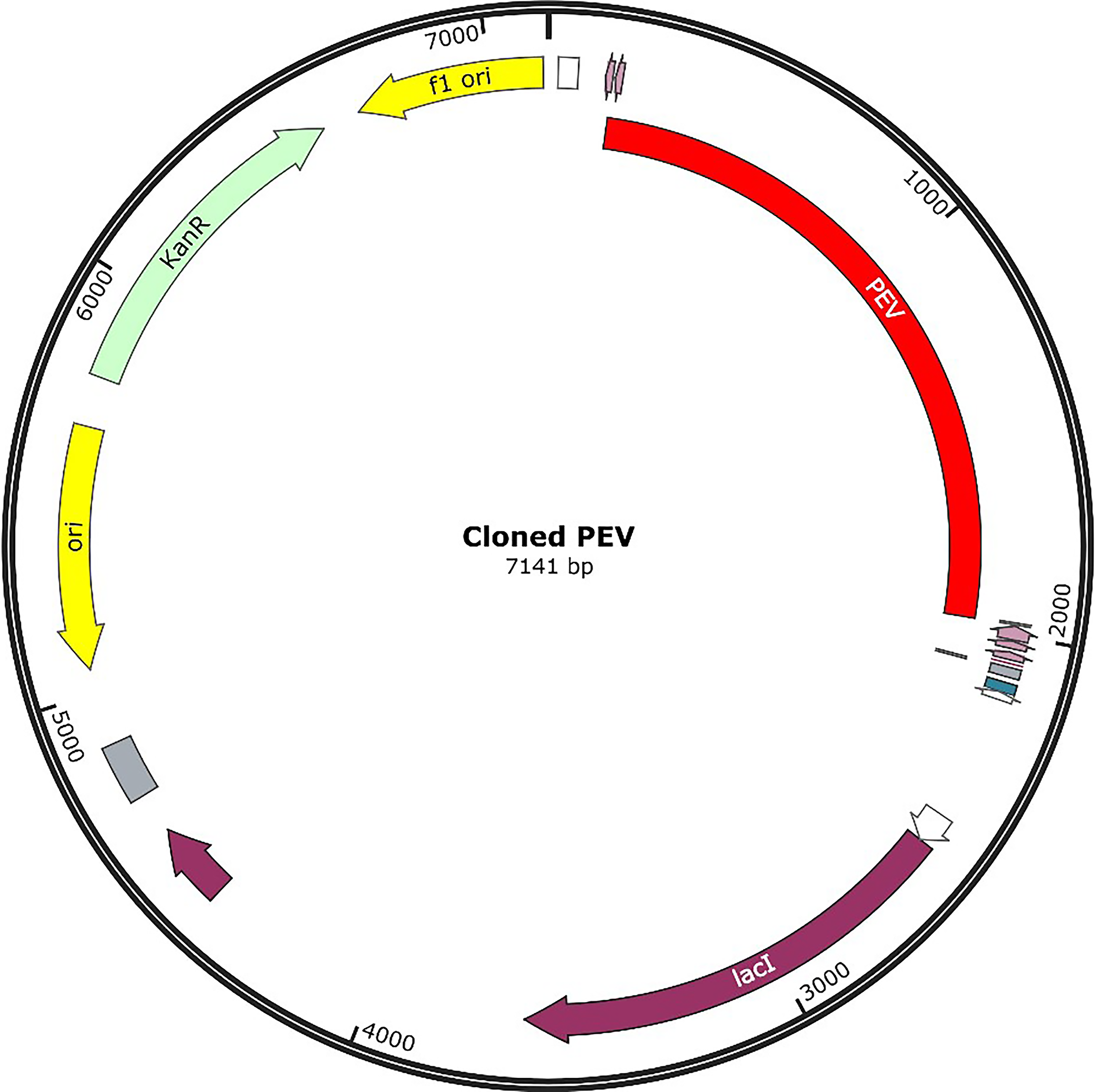
In silico vector construction of Rustrela virus vaccine-PEV. PEV, polyepitope vaccine.
Molecular simulation
The mobility and stability of the proteins in the vaccine-receptor complex were analysed with NMA. Each residue's deformation determines the overall shape change of the PEV-TLR3, TLR4, and TLR7 complex (Figures 11,12 and 13A) . The rigidity of the molecule is described by the Eigenvalues associated with its normal modes; the smaller the Eigenvalue, the more easily the structure can be deformed. Figures 11,12, and 13B show that the Eigenvalue of the PEV-TLR3 complex is 1.94 x 10−7, that of TLR4 complex is 4.43 x 10−7 and that of the TLR7 complex is 2.07 x 10−7. It also spawned an elastic-network model that pinpointed the atomic pairs linked by springs. Each “dot” in the graph stood in for one “spring” between their respective atomic pairs, and the colour of the dots indicated the rigidity of the structure (Figures 11,12, and 13C): the darker the spot, the stiffer the structure. The amount of variance a mode carries (Figures 11,12, and 13D) is a measure of how much that mode contributes to the overall equilibrium motions. Since the PEV structure contains coils and loops, the overall iMODS results indicated little deformation at the terminal residues of both docked complexes. Insilco vector designing The amino acid sequence was codon optimized using the OPTIMIZER database. The optimized sequence resulted in 1806 base pairs nucleotides and 27.6 % GC content with a 1.00 CAI score. Following the addition of BamHI and XhoI restriction sites. The in silico restriction ligation was performed and the fragment was ligated in the targeted pET-28a vector (Figure 14).
In silco vector designing
The amino acid sequence was codon optimized using the OPTIMIZER database. The optimized sequence resulted in 1,806 base pair nucleotides and 27.6% GC content with a 1.00 CAI score. Following the addition of BamHI and XhoI restriction sites. The in silico restriction ligation was performed, and the fragment was ligated in the targeted pET-28a vector (Fig. 11).
Discussion
Vaccines are essential for protecting the host organism from a certain disease, which helps to prevent millions of deaths worldwide each year. The processes involved in developing vaccines are typically time-consuming, expensive, and labor-intensive (Ozger and Cihan, 2022; Khan et al., 2024). Computational biology has been heavily expanded upon, which assists many different kinds of study and reduces predicted time consumption. So, by utilizing different immunoinformatics parameters, a bioinformatics-based method, it is more convenient and fast to create vaccines. It involves identifying potential pathogen antigenic proteins, and using this knowledge, it further aids in the identification of immune-dominant epitopes that are in charge of producing humoral and cell-mediated immune responses against the pathogen inside the host organism (Naderian et al., 2025). Therefore, by identifying the B and T cell epitope regions on possible antigen proteins of a pathogen, multi-epitope-based subunit vaccines can be developed. Neisseria meningitidis was the first vaccine created using an immunoinformatics technique, and it was successfully developed later (Narula et al., 2018). Later, similar approaches were used to create vaccines for different diseases (Bhutta et al., 2025; Pajand et al., 2026; Emadi et al., 2026).
Multi-epitope vaccinations reduce undesired components that might either trigger abnormal immune responses or have negative effects in comparison with conventional vaccinations. Potential benefits of epitope-based vaccinations include better safety, cost effectiveness, the ability to rationally edit the epitopes for greater potency and breadth, and the capacity to concentrate immune responses on conserved epitopes (Shey et al., 2019). Researchers have long worked to reduce the expense, duration, and side effects associated with developing vaccines. Different methodologies, based on immunoinformatic methods, are currently readily available for designing and developing effective and competent new generation epitope-based vaccinations (Ali et al., 2017; Naderian et al., 2025).
Rustrela virus acts as a neuropathogenic and is detected in tissues of brain and is associated with mortal neurological diseases, including meningoencephalitis in marsupial and placental animals (Matiasek et al., 2023). Animals infected with this virus developed acute neurological symptoms, which include lethargy, stiff gait, tetraparesis, hind limb weakness, and ataxia. These symptoms make this disease often fatal. Within weeks to months, animals suffer from worsened paralysis and neurological decline. Some animals can recover partially, but in most cases they lose their motor abilities, making quality of life very poor and eventually, resulting in death (Fox et al., 2024; Aftab et al., 2024). The virus is considered endemic in these rodent populations, which likely serve as reservoirs maintaining viral circulation, although the exact transmission routes to spillover hosts remain unclear. Researchers in Austria performed retrospective studies and found that RusV is native to the region, and it was responsible for the staggering disease in cats from 1994 to 2016 (Camp et al., 2025). It is currently unclear how the virus reaches the spillover hosts, but researchers believe it is present in rodent populations and uses these rodents as reservoirs to keep the virus going. The identification of this RusV variant in a mountain lion in Colorado, USA, indicates that the virus can infect and spread in broader host range further than was previously understood (Fox et al., 2024).
Detection of Rustrela virus and related diseases is done in affected animals using methods like Real-Time PCR, Immunohistochemistry, and in situ hybridization, which detect viral RNA and antigen in brain tissues of afflicted cats and a diversity of wild and zoo animals. Detection using these techniques limits early intervention and outbreak control. In this context, development of a prophylactic vaccine is of substantial translational value. RusV has been found to be the cause of nonsuppurative meningoencephalitis in numerous species in Europe through the study of preserved tissue samples (de le Roi et al., 2023). Surveillance of this virus and associated diseases focuses on reservoir hosts to track evolution and viral circulation. Reservoir hosts include yellow-necked field mice (Apodemus flavicollis) and wood mice (Apodemus sylvaticus) (Nippert et al., 2023). This strategy provides support in comprehending viral variety, monitoring illness incidence, and guiding public and animal health interventions. Hence, with all the available information, we intended to construct vaccine against this virus using in -silico approach, as currently there is no commercially available vaccine for it (Voss et al., 2022b; Pfaff et al., 2022). This approach can ultimately contribute to the preclinical development pipelines and transition toward experimental validation in animal models. Validation of this approach can contribute to preventive strategies in veterinary medicine and reduce the burden of fatal neurological disease associated with Rustrela virus.
The final vaccine construct contains 602 amino acids whose antigenicity, allergenicity, and physicochemical properties were determined. It has molecular weight of 6.3 kDa, which makes it fall in acceptable range for vaccine. This vaccine is stable with a stability index of 28.9 whereas the low GRAVY value depicts its hydrophilic nature. It is thermostable due to its high aliphatic index. Moreover, its PI score is 9.66, which shows that it has basic nature (Dey et al., 2022).
With the help of Sopma, the protein’s secondary structure was predicted, showing that the protein includes 32.8% alpha helices, 20% extended strands, and 40% random coils (Sritharan and Elengoe, 2022). Following this, tertiary structure of protein was predicted using alphafold 2 and refined by GalaxyRefine (Pangabam et al., 2023). Results from the Ramachandran plot demonstrated that the overall model is plausible because more than 99% of the residues were found in the allowed region. In addition, high ERRAT score (94.3) confirmed the stereochemical accuracy, while −2.39 Z Score indicated that the modified vaccine’s overall structure is adequate. In addition, the values for GDT-HA and RMSD showed the fine quality of our developed vaccine construct (Sayed et al., 2020). Also, the vaccine was docked with the TLR-3, TLR-4, and TLR-7 receptors to see if it might trigger a significant immune response (Yılmaz Çolak, 2021).
We performed data-driven molecular dynamics (MD) simulations to assess the immune interaction and stability between the vaccine protein and TLR3, TLR4, and TLR7 receptors. To achieve conformational stability in the docked complex of the vaccine protein with TLR3, TLR4, and TLR7, we conducted energy minimization. This process reduced the potential energy of the entire system, rectifying any inappropriate structural geometry by adjusting individual protein atoms. Consequently, this enhanced the structural stability of the complex while maintaining proper stereochemistry. The derived eigenvalue served as an indicator of the complex’s rigidity in motion and the energy required for deformability. One of the initial steps in validating a potential vaccine is the immunoreactivity testing using serological evaluation. (Gori et al., 2013; Ahmad et al., 2025; Hameed et al., 2025). The dynamic integrity of the TLR-polyepitope complex was evaluated using Normal Mode Analysis (NMA) rather than classical MD. It is important to clarify that NMA solves for the collective vibrational motions inherent to the structure, whereas classical MD simulates atomic trajectories over time. Consequently, conventional metrics such as time-dependent RMSD plots and MM-PBSA binding energies, which require extensive sampling of an MD trajectory, cannot be directly generated from the iMODS server. However, NMA provides equivalent stability metrics: the eigenvalue indicates the complex’s mechanical stiffness, and the deformability profile serves as an analog to RMSF, highlighting regions of flexibility. Our results show low deformability at the TLR-vaccine interface, suggesting a stable interaction. While we acknowledge that full-scale explicit-solvent MD simulations remain the gold standard for atomic-level validation, the computational expense of such simulations for a large protein complex placed them beyond the immediate scope of this study. Nonetheless, the current NMA results offer a strong theoretical basis for the complex’s structural stability, warranting future long-timescale MD simulations to obtain precise energetic data.
To achieve greater extent of expression, codon optimization was performed. CAI score (1.00) and GC content (27.6%) were propitious for high expression level. To check for the expression, this constructed PEV (recombinant protein) needs to be engineered in a suitable expression vector (Rosano and Ceccarelli, 2014).
Limitations and future directions
Our immunoinformatics approach yielded a promising multiepitope vaccine (PEV) construct. However, it has certain limitations that must be acknowledged. This is a purely computational study lacking experimental validation of epitope binding, protein expression, and immune response. Moreover, epitope prediction with the help of IEDB/SVMTriP has risks of false positives and false negatives, particularly due to availability of limited training data for the emerging Rustrela virus. In addition to this, in vivo immunogenicity remains untested. Furthermore, 86.71% worldwide population coverage leaves 13% nonresponders due to rare HLA-alleles and overlooks nonhuman MHC diversity in reservoir hosts and spillover species.
In future, expression and purification of PEV protein in E. coli (pET-28a) system will be carried out. Animal studies would follow Apodemus flavicollis mice for immune response profiling and antibody titers. To reach veterinary trials, we will boost coverage >95% with additional promiscuous epitopes, validate against viral variants, and optimize adjuvants beyond CTB for stronger Th1 responses.
Footnotes
Author Disclosure Statement
The authors report there are no competing interests to declare.
Funding Information
No funding was obtained for this study.