
Abstract
Background
Deep learning excels at processing raw data because it automatically extracts and classifies high-level features. Despite biology's low popularity in data analysis, incorporating computer technology can improve biological research.
Objective
To create a deep learning model that can identify nucleosomes from nucleotide sequences and to show that simpler models outperform more complicated ones in solving biological challenges.
Methods
A classifier was created utilising deep learning and machine learning approaches. The final model consists of two convolutional layers, one max pooling layer, two fully connected layers, and a dropout regularisation layer. This structure was chosen on the basis of the ‘less is frequently more’ approach, which emphasises simple design without large hidden layers.
Results
Experimental results show that deep learning methods, specifically deep neural networks, outperform typical machine learning algorithms for recognising nucleosomes. The simplified network architecture proved suitable without the requirement for numerous hidden neurons, resulting in effective network performance.
Conclusion
This study demonstrates that machine learning and other computational techniques may streamline and expedite the resolution of biological issues. The model helps identify nucleosomes and can be used in future research or labs. This study discusses the challenges of understanding and addressing simple biological problems with sophisticated computer technology and offers practical solutions for academic and economic sectors.
Introduction
Machine learning (ML) approaches, which use algorithms to categorize data, discover patterns, and provide explanations for events, have proven effective in solving scientific challenges. 1 Although heterogeneous biological data is complicated, modern deep learning algorithms are capable of extracting features and making predictions based on the data, including classification. 2 The DNA is tightly condensed inside the chromatin fiber, which consists of nucleosomes. 3 The nucleosomes maintain very stable locations over the whole genome. The study has examined the involvement of remodelers and transcription factors, which are two elements that determine this placement. The exact function of this role is unclear, however it is probable that the arrangement of this sequence plays a part in defining the location of the nucleosome. The issue at hand involves the task of finding AI methods that can accurately distinguish between sequences where a nucleosome is present and sequences where it is not. 4 If the model achieved sufficient accuracy in classifying sequences, it might enable researchers to elucidate the role of the DNA sequence in nucleosomal placement.5–7 The primary goal of the research is to develop a machine learning model that can accurately categorize a DNA sequence as either a nucleosome or not, with significant performance. This would enable the automation of determining if a DNA sequence would be able to locate a nucleosome. At this juncture, it is important to note that our objective is to extract information from the supplied data. We also want to do appropriate preprocessing of the data and develop a model that might potentially offer a solution to the original issue. 8 Another objective is to get a comprehensive comprehension of a biological issue. Acquire knowledge of the biological foundations, articulate it, and address material that is beyond the realm of computer science. The last goal is to acquire the skills to develop deep learning models, a subject that has not been covered in the course but is very intriguing owing to its potential applications.
Machine Learning (ML) and Deep Learning (DL) are data-driven approaches used to execute tasks such as clustering, classification, or regression.9,10 By integrating these two branches of artificial intelligence with the techniques of bioinformatics, it becomes possible to forecast certain outcomes without the need of conducting several laboratory tests and their subsequent implementation. The compaction function performed by chromatin is significant. Additionally, it functions as a regulatory mechanism for controlling access to the DNA sequence by other macromolecules, thereby acting as a “traffic light.” This regulation governs crucial activities like transcription, replication, recombination, and repair. Therefore, the regulation of chromatin involves the alteration of histones, the activity of chromatin remodeling enzymes, and the presence of structural proteins. The location of the nucleosomes is intricately linked to these events. There are established patterns of nucleosome presence or absence that either enhance or impede the action of certain agents on DNA. 11 In recent years, there has been a surge in experiments investigating the positioning of nucleosomes. 12 This is mostly owing to the development of advanced methods that enable the creation of nucleosome maps, which accurately pinpoint the locations of nucleosomes throughout the genome. These maps allow us to see the inclination of nucleosomes towards certain sections of the genome, as well as the presence of proteins or chromatin remodelers. There is variation in the occupancy of nucleosomes in areas close to the transcription start site among different animals. It has been observed that this phenomenon may be attributed to the quantity of activated genes. In multicellular organisms, only the required genes are activated while the unused genes remain dormant. In contrast, unicellular organisms have a lower level of complexity and a smaller variety of cells. As a result, the majority of their genes are always active and hence need easier accessibility via exposed portions of nucleosomes. 13
Sequences of nucleosomes derived from organisms in their natural environment have been compared to nucleosomes and DNA sequences that were previously isolated and studied in a controlled laboratory setting. The arrangement of nucleosomes in the genome is comparable between in vivo and in vitro conditions. The only notable observation is an increased presence of nucleosomes in vivo.14,15 The correlation between both models varies throughout the genome, with an increase seen in intergenic regions after the end of a transcribed gene, and a decrease observed at promoters or coding regions. The findings indicate that in living organisms, the placement of the nucleosome is influenced by several factors other than the DNA chain itself, including transcription factors, chromatin remodelers, and an active transcription process. 16 In order to examine the impact of the DNA sequence on nucleosome placement, we can only consider the in vitro sequences that are solely defined by the DNA and are not affected by other factors or biological processes. The prediction of areas abundant in nucleosomes and regions devoid of nucleosomes was made possible via the use of a statistical model. Therefore, it was discovered that the raw DNA sequence has the ability to promote the binding of transcription factors and biological activities like transcription. 17 The exorbitant expense associated with the representation of nucleosome maps has prompted the scientific community to explore computational remedies for identifying nucleosome sequences. As shown, the DNA sequence is a crucial factor in determining location.
A significant quantity of data has been gathered and has undergone preprocessing techniques, since a deep learning model cannot accept textual input directly but instead requires numerical values. Ultimately, AI models optimize a mathematical function that necessitates the manipulation of numerical values. It is one of the assignments with the lengthiest development time. 18
Various methods have undergone testing using distinct hyperparameter search spaces for each algorithm. 19 The ML algorithms and DL models follow their respective hyperparameters for the optimal scenario. The measure might be deceptive since it estimates the proportion of positive outcomes, and there may be an unequal distribution of data across categories. This could lead to an improvement in the measure if more data is added to the category that is well-classified. However, in this study, the confusion matrices of the models are compared with their respective counterparts to prevent any ambiguity in the measure. The algorithms were trained using a balanced measure of the target data, ensuring that each target class contributed about 50–60% of the data. This approach was used to prevent the models from making unbalanced predictions.
Method, materials and tools
The DNA sequences dataset is derived from Gene regulation. 20 DNA sequences are neither stochastic and chaotic, but rather consist of character sequences composed of four deoxyribonucleic acid bases (A, C, G, T). The analysis of these sequences might be likened to deciphering a language, where the arrangement of words and letters is vital for comprehending the significance. Machine learning algorithms are unable to directly interpret DNA sequences. Figure 1 presents the block diagram for predicting DNA Nucleosomal Sequences using deep learning. Therefore, it is essential to convert the sequences into numerical values in order to create an input matrix. 21 Preprocessing the data is crucial for accurately training the machine learning models. Figure 1
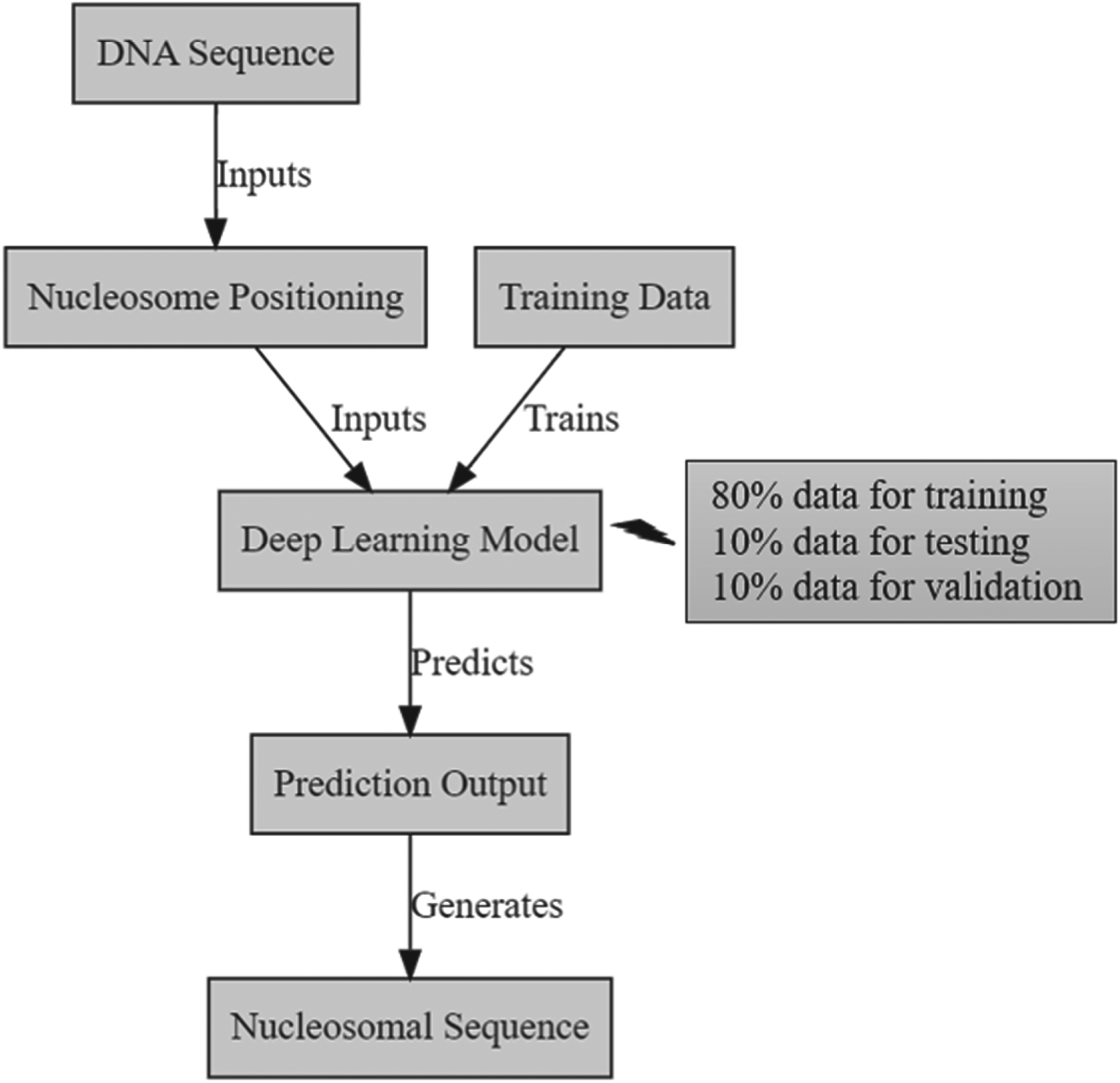
Block diagram for predicting DNA nucleosomal sequences using deep learning.
The techniques used in this study are explained below.
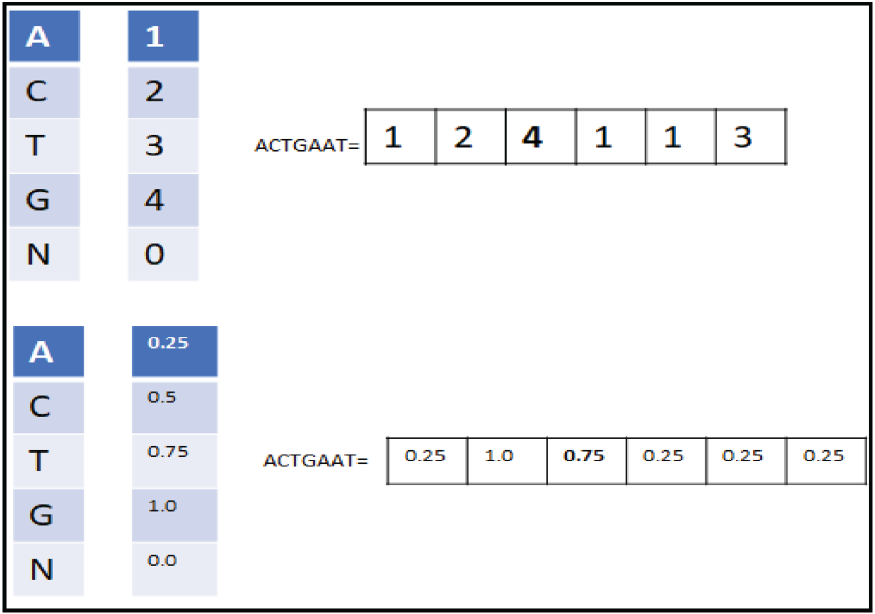
Ordinal coding.
- The presence of the dinucleotide was confirmed. - The exact location of the dinucleotide in the chain was verified.

One-hot encoding (OHE).
This study follows a data distribution where 80% of the data is used for training, 10% for testing, and the remaining 10% for validation. Upon examining the two graphs, it is evident that there is a phenomenon known as ‘overfitting’ when using the Adam optimizer. However, this issue is much mitigated when employing the SGD optimizer. Starting now, the SGD optimizer was considered when ‘overfitting’ was detected in certain results. Figure 4 displays the accuracy graphs as a function of the number of epochs trained using the Adam and SGD optimizers. The gradient (partial derivative) is not computed for every input data and characteristic, but rather selects a data point randomly. This approach significantly reduces the processing burden.

Graphs of accuracy versus the number of epochs trained. (a) Using Adam optimizer (b) Using the SGD optimizer.
Figure 4(a) shows that the accuracy graph of the Adam optimizer often increases quickly before leveling off or steadily increasing. While training, Adam, an adaptive learning rate optimizer, modifies the learning rate for every parameter. In comparison to gradient descent, the convergence and performance are often faster and better. Lower batch sizes improve generalization and increase noise, whereas larger batch sizes expedite convergence and smooth gradients.
As shown in Figure 4(b), SGD accuracy graphs often exhibit fluctuations, with spikes followed by plateaus or dips. Using the gradient of the loss function for a single training sample, SGD, a simpler optimizer, modifies parameters. This could lead to less refined updates and less rapid convergence compared to Adam. SGD has to be careful when choosing a learning rate so it doesn't hit local minima. While mini-batches may boost generalization, they impede convergence and introduce noise.
Figure 5(a) demonstrates little overfitting, with just a slight deviation seen from epoch 22 onwards. Prior to that point, the model exhibits satisfactory progression. However, in Figure 5(b), we see ‘overfitting’ as the accuracy of the validation set is somewhat lower than that of the training set. Nevertheless, the performance is not significantly poor. It is important to note that both of these models, when trained using the ‘Adam’ optimizer, had much worse outcomes, resulting in overfitting in both instances. It is worth mentioning that the second instance was executed with 100 epochs, whereas the first case was executed with 25 epochs. The trial-error-based models were evaluated at various time points to see the evolution of the metrics. In the instance of One-Hot Encoding (OHE), the progression was consistently improving without encountering the issue of ‘overfitting’. Therefore, the process was prolonged until a threshold was reached where further enhancements were no longer possible. In order to evaluate the model's classification performance, the confusion matrix (Figure 6) was used in both scenarios to assess its accuracy and the real negative rate. Confusion matrix evaluates the performance of the classification model by calculating metrics like True positive, true negative, False positive and false negative which has been classified as No-nucleosome or Nucleosome in this research.

Performance in terms of loss and accuracy vs. epochs (a) with OHE preprocessing (b) with dinucleotide preprocessing.

Confusion matrices (a) with OHE preprocessing (b) with dinucleotide preprocessing.
The initial confusion matrix corresponds to One-Hot Encoding (OHE) with an accuracy of 67.18% and a loss of 0.59. Despite its less than optimal accuracy, it is capable of performing a comparable categorization for both categories. The preprocessing of dinucleotides yields a modest improvement, achieving an accuracy of 77.22% with a corresponding loss of 0.48. Nevertheless, this outperforms the machine learning model to some extent; more experimentation using convolutional neural networks (CNN). Figure 7 depicts a box plot that shows the relationship between sensitivity and specificity in predicting DNA sequencing using OHE. Figure 8 depicts a box plot that shows the relationship between sensitivity and specificity in predicting DNA sequencing using Di-nucleotides.

Predicting DNA sequencing using OHE (a) box plot for OHE based sequencing (b) sensitivity vs. specificity.

Predicting DNA sequencing using Di-nucleotides (a) box plot for Di-nucleotides based sequencing (b) sensitivity vs. specificity.
Initially, preliminary experiments were conducted to determine the optimal hyperparameters using a trial and error approach. Once the hyperparameters were finalized, the model was trained using preprocessed data consisting of dinucleotides and one-hot encoding (OHE). The model's accuracy improved as the epochs progressed, therefore we tested it with up to 30 epochs. The precision is around 80%. Both situations exhibit fluctuating validation results characterized by peaks and troughs, indicating non-linear training progress. The accuracy reached is 82.85% for dinucleotides and 81.7% for OHE. While there are similarities between the two, dinucleotides are ultimately superior.
The model's performance experiences a significant decline, characterized by a decrease in accuracy and a rise in the loss function. The variation in results across different preprocessing methods is minimal, with an average accuracy of around 75% and no instances beyond 80%. Upon examining the confusion matrices, one may see a comparable proportion of true positives and true negatives, indicating a fair categorization of the categories.
After deciding that the best model was the Deep Learning model, a small website was made as a presentation in which a model test can be carried out by inserting a file with a ‘CSV’ extension with the 150 nucleotide sequences to be predicted. Deep learning models for nucleosome identification already exist in the literature. An example is CORENup, 23 which processes a DNA sequence as input with an associated numerical representation and combines a convolutional network with a recursive one. The results presented in this process are based on the AUC metric, so in order to compare with our result, the AUC of the 1D convolutional network model was calculated. The results of the present work with an AUC of 0.908 is some point below the performance shown in CORENup of 0.933. 24
Its predecessor is probably LeNup, 24 a deep learning model that mixes Inception-type networks to identify nucleosome sequences. This model was trained with genomes from Homo sapiens, Caenorhabditis elegans, Drosophila melanogaster, and Saccharomyces cerevisiae. The performance produced with LeNup was accuracy of 0.89 for H. sapiens, 0.92 for C.elegans and 0.88 for D.melanogaster. These data are higher than the one obtained in our case, 0.82; even so, it cannot be directly comparable because we are talking first about different genomes, ways of dealing with different input data, and networks with a different architecture distribution.
Determining the precise location of nucleosomes on the DNA strand is crucial for unraveling the mechanisms behind gene regulation. There is no one factor that determines the optimal positioning of a nucleosome in the genome. The most notable components are chromatin remodelers, transcription factors, and the DNA strand. 24 In this instance, the study has concentrated on the impact of the latter. An endeavor has been undertaken to use machine learning techniques to ascertain the impact of the unprocessed DNA sequence on placement. The use of DL and ML approaches has facilitated the development of a solution that effectively categorizes DNA sequences into two distinct groups: nucleosomes and non-nucleosomes.
Once the decision was made to use the Deep Learning model, a concise website was created to serve as a presentation. This website allows users to do model testing by uploading a ‘CSV’ file containing 150 nucleotide sequences that need to be predicted. There are currently existing deep learning models in the literature that can be used for nucleosome identification. The approach employs a combination of Inception-type networks to accurately detect nucleosome sequences. 25 The training of this model included the use of genomes from Homo sapiens, Caenorhabditis elegans, Drosophila melanogaster, and Saccharomyces cerevisiae. The collected data in our instance, 0.82, is greater than the data being compared. However, direct comparison is not possible due to differences in genomes, handling of input data, and variations in network architecture distribution.
The accuracy measure is observed in the diagram(Figure 9), a measure that has been chosen as a contrast throughout all the models. It could be a misleading measure since it calculates the percentage of positive results, and there may be an imbalance in the number of data linked to the categories that make this measure improve if the input data of the category that classifies well is increased. However, in the present work, they are compared with their corresponding confusion matrices presented in each of the corresponding models to avoid confusion in the metric. All algorithms were trained with a balanced or rather balanced measure of the target data, that is, around 50–60% of data from each target class, to avoid unbalanced predictions of the models as presented in Figure 9.

Model comparison diagram used in the work.
Deep learning algorithms predict nucleosome locations more accurately than traditional methods. Advanced deep learning methods can find DNA sequence patterns and connections that simpler methods cannot. The location of nucleosomes affects chromatin architecture and gene expression. Accurate predictions may help researchers understand DNA packing and gene expression. Up to now, aberrant nucleosome organization has been linked to several illnesses. A study of nucleosome formation may reveal disease causes and therapy targets. Nucleosome arrangement may alter DNA accessibility to transcription factors and downstream proteins. Accurate gene expression disease predictions may help discover treatment targets. Knowing the cellular location of nucleosomes may help create drugs that affect chromatin structure and gene expression. Nucleosome placement may identify regulatory components like enhancers and promoters. A complete knowledge of nucleosome placement may help explain gene expression-affecting epigenetic modifications. This work introduces deep learning algorithmic or architectural methods for nucleosome spatial prediction. Therefore, this work greatly improves our knowledge of disease, gene regulation, and chromatin arrangement.
The primary outcome of this study is the creation of a robust deep learning model that effectively addresses the nucleosome identification challenge. The test findings substantiate the notion that often, a smaller quantity yields superior outcomes. Data analysis approaches have mostly been used in the realms of economics and business. Biology is not a prevalent domain for data analysis. However, a tool has been developed that might be beneficial for use in labs or future research endeavors. Computer science has shown its use in supporting research disciplines like biology. Tackling a minor biological issue and using computational methods to resolve it has proven to be a significant endeavor. In order to comprehend the factors that contribute to the superior performance of a model, it is necessary to engage in the construction of many models and conduct a thorough comparison among them. Engineering endeavors to identify a solution to the issue, but at times encounters challenges in determining the underlying cause of the ultimate outcome. Consequently, a major obstacle in developing new algorithms has been the challenge of determining the most suitable hyperparameters and providing a valid rationale for their selection. As a result, there has been a reluctance to explore the creation of very complex networks that are challenging to elucidate. Data science is a very valuable tool, yet its basic workings for mathematical model improvement may be quite confusing. This issue emerges when proposing a solution to a biological problem that demands a higher level of precision than computer science, given its following consequences. The efficacy of AI model training heavily relies on robust software capabilities, but the acquisition of suitable hardware posed its own set of difficulties. The model's training durations may be limited due to a lack of emphasis on optimizing the model within a certain timeframe, with all effort directed towards performance. The main finding of this study is that the integration of deep learning, a machine learning method, facilitates the discovery of answers to many issues, including those in the field of biology. This approach not only reduces the time required to find a solution but also offers practical tools that can be included into existing systems.
5.1 Limitations and future work
Both the amount and quality of training data have a significant influence on the accuracy of deep learning models. Accuracy of the model may be affected by data that is either insufficient or inconsistent. The training and deployment of deep learning models for large datasets may require a significant number of resources. Scalability and applicability may be hindered as a result of this. There is a possibility that complex models may overfit, which will lead to poor generalization on new data. Forecasting nucleosomal sequences is a challenging biological challenge, and deep learning methods may fail to include all relevant biological aspects.
In the future, we will investigate methods to enhance the training data to increase model generalization and mitigate overfitting. Future incorporation of domain-specific data, such as chromatin accessibility or histone alterations, may augment the model's predictive capability.
Footnotes
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at the Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2024-1584-05”
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received funding from Northern Border University, Arar, KSA; Project number: NBU-FFR-2024-1584-05.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data used to support the findings of this study are included in the article.