
Abstract
In this study, we assessed if the superimposition of incident sexually transmitted infections (STIs) on HIV phylogenetic analyses could reveal possible sexual behaviour misclassifications in our HIV-infected population. HIV-1 sequences collected between 1997 and 2014 from 1169 individuals attending a HIV clinic in Antwerp, Belgium were analysed to infer a partial HIV transmission network. Individual demographic, clinical and laboratory data collected during routine HIV follow-up were used to compare clustered and non-clustered individuals using logistic regression analyses. In total, 438 (37.5%) individuals were identified in 136 clusters, including 76 transmission pairs and 60 clusters consisting of three or more individuals. Individuals in a cluster were more likely to have a history of syphilis, Chlamydia and/or gonorrhoea (P < 0.05); however, when analyses were stratified by HIV transmission risk groups (heterosexual and men who have sex with men [MSM]), this association only remained significant for heterosexuals with syphilis (P = 0.001). Under closer scrutiny, this association was driven by six heterosexual men who were located in six almost exclusively MSM clusters. A parsimonious conclusion is that these six individuals were potentially misclassified as heterosexual. Improving the accuracy of sexual behaviour reporting could improve care.
Keywords
Introduction
Phylogenetic analysis of HIV sequences passively accumulated through routine drug resistance testing (DRT) can provide useful insights into local HIV transmission network characteristics when coupled with demographic, clinical and sexually transmitted infection (STI) data. These approaches have proven useful in highlighting HIV transmission networks at various geographical levels,1–3 thereby allowing the identification of hotspots within (sub)populations such as men who have sex with men (MSM). 4 A recent Belgian study by Chalmet et al. 1 demonstrated a correlation between syphilis and clustering. Recently, studies employing phylogenetic analyses of HIV clinical cohorts have highlighted probable occurrences of sexual behaviour misclassification among males,5,6 although it is still uncertain if these inferences about transmission linkages and sexual behaviours should be used to guide clinical follow-up or services at the individual level. 7
With the advent of increasingly user-friendly and robust bioinformatic tools, 8 it is becoming easier for clinicians and epidemiologists to apply these approaches to various cohorts and it is expected that phylogenetic analyses will be increasingly employed to inform and improve public health efforts. 9
HIV-infected individuals play a disproportionately large role in the current STI epidemics in Europe and elsewhere.10–12 HIV-infected MSM, in particular, contribute up to 49.8% of those diagnosed with certain STIs such as syphilis. 13 In 2015, an estimated 18,758 individuals in Belgium were HIV-positive (1.7 per 1000 people); of the individuals with a known HIV transmission route in 2016, MSM represented 52% of new HIV infections. 14 A resurgence of several STI epidemics including syphilis, hepatitis C and lymphogranuloma venereum has been observed in our HIV/STI clinics during the last two decades (15–17; V Maes, personal communication, 30/09/2016). For example, during a syphilis epidemic observed at our clinic between 1992 and 2012, over half of infections occurred in patients with a previous diagnosis of syphilis and almost all were HIV-infected MSM. 18 From 2000 to 2015, only 14/895 (1.6%) of syphilis episodes reported at our clinic occurred in females.
In this study, we aimed to ascertain if mapping STI diagnoses onto our local HIV phylogenetic tree could help us to better understand the sexual networks underpinning local STI transmission by investigating the possible role of sexual behaviour misclassification.
Methods
Study participants
A retrospective review of clinical data from HIV-1 infected individuals attending the Institute of Tropical Medicine’s (ITM) HIV clinic who had at least one plasma sample submitted for HIV sequencing for DRT from 31 December 1997 to 17 February 2014 was performed. The ITM has a medium-sized HIV-outpatient clinic with 3371 HIV-infected individuals in regular care during the study period. From 2008 onwards, DRT in the form of reverse transcriptase (RT) and protease (PR) gene sequencing was incorporated into the HIV diagnostic workup; prior to 2008 HIV sequencing was performed in the case of treatment failure.
Demographic and clinical data were extracted from our clinical database and pseudonymized. Self-reported HIV transmission risk was recorded by the HIV care physician at the time of diagnosis. Individuals were grouped into categories: ‘heterosexual’, ‘MSM’, ‘homo/bisexual (females only)’ and ‘other’, which included intravenous drug user (IVDU), mother-to-child transmission, blood transfusion and occupational risk transmissions. The sexual behaviour characteristics of the IVDUs were not specified in the clinical database, thus were regarded as unknown. Sexual preference classification was extrapolated from the transmission risk categories, with the exception of the ‘other’ classification for which no sexual behaviour group was assigned. MSM were defined as males ever reporting sexual contact with someone of the same sex. All episodes of gonorrhoea, Chlamydia and syphilis that were diagnosed at the ITM laboratory from August 1993 to June 2014 were extracted from the laboratory database for each individual.
STI laboratory testing
STI testing is typically performed at the ITM every 3–12 months for patients in routine HIV follow-up care depending on their risk profile. 19 During follow-up visits, MSM are tested for syphilis, Chlamydia and gonorrhoea, even if they are asymptomatic. Treponema pallidum ssp. pallidum infection was dual tested with a non-treponemal Macro-Vue rapid plasma reagin (RPR) card test (Becton, Dickinson and Company, United States [US]) and treponemal antibody detection was performed via the Cellognost Syphilis H Combipack T. pallidum haemagglutination assay (Dade Behring, Germany) up until November 2002, thereafter testing was performed with the SERODIA T. pallidum particle agglutination (TPPA) assay (Fujirebio, Inc., Japan). Syphilis episodes were defined as previously described 18 ; briefly, initial syphilis was defined as an RPR titre ≥1/8 and a positive TPPA test on serum. Repeat episodes of syphilis, including syphilis reinfections and reactivations, were defined as an episode in a person who had a ≥ 4-fold increase in RPR titre compared to a previous RPR titre.
Neisseria gonorrhoeae (NG) and Chlamydia trachomatis (CT) presence in first void urine and/or (anal/pharyngeal/vaginal) swabs were assessed with a nucleic acid amplification test from 1999 onwards for CT and 2010 onwards for NG following manufacturer’s instructions. Before 2010 NG presence was tested by culture.
HIV RNA extraction, amplification and sequencing
Viral RNA was extracted from plasma using the QIAamp Viral RNA Mini Kit (QIAGEN, Germany) following the manufacturer’s instructions. The resistance genotyping was performed with the Trugene HIV-1 genotyping kit (Siemens Healthcare Diagnostics, US). The PCR primers covered a 1.3 Kb fragment of the pol gene. Full length PR and partial RT sequences were sequenced as previously described. 20 Sequencing products were analysed using the OpenGene DNA Sequencing System (Visible Genetics, Inc., Canada) following manufacturer’s instructions. Sequences were stored using the Integrated Database Network System software package (SmartGene, Switzerland).
Sequence analyses
HIV subtyping was performed using REGA version 3.021 based on the RT gene since the short PR sequence was found to have insufficient signal for subtyping. Sequence alignments were performed for both the PR and RT genes separately. Sequences were placed in-frame using Mesquite version 3.1022 and then aligned using the codon model of MUSCLE version 3.723 implemented in MEGA7.24 Trailing sequence ends were trimmed using MEGA7 and then the two gene alignments were concatenated using Mesquite. A maximum likelihood phylogenetic tree of all samples was created using a GTR+Γ model of evolution with each gene as a separate partition and 1000 bootstrap replicates as implemented RAxML version 8.2.9. 25 Patients for whom their multiple samples did not cluster on the phylogenetic tree (patristic branch length distance >0.1) were identified using a custom python script implementing DendroPy. 26 Such large distances are likely the result of either multiple HIV-1 infections or a sample switch error. The above alignment and tree building steps were redone using only the first sample for all individuals. Trees were visualized using FigTree version 1.4.2. Cluster Picker 27 version 1.2.3 was used to identify clusters of all individuals. Clusters were defined as those with >90% bootstrap support for the clade and <4.5% genetic difference between all patient samples. These settings have been found to be optimal for similar data previously. 27 Mixed transmission clusters were defined as clusters consisting of two or more individuals, whereby at least one individual was a heterosexual male who clustered with at least one MSM individual and regardless of the presence of female(s) in the cluster. Potentially misclassified HIV sexual transmission cases were defined as heterosexual males with a history of syphilis who were present in a mixed transmission cluster. We chose a history of syphilis as a predictive factor for possible misclassification of sexual behaviour because syphilis almost exclusively occurs in MSM in our cohort. 18
Statistical analyses
Continuous variables are expressed in median and interquartile range (IQR). Clustered and non-clustered individuals were compared using Chi square (χ2) test or Fisher’s exact test for categorical variables. Bivariate logistic regression analyses using maximum likelihood ratio for the whole cohort (Stage 1 analysis) were performed. Penalized Multinomial Logistic Regression28,29 analyses were performed using the STATA module ‘firthlogit’ 30 when stratifying STI (syphilis/Chlamydia/gonorrhoea) diagnosis by HIV transmission risk groups (heterosexual versus MSM) (Stage 2 analysis). Multivariate analyses were not performed due to the small number of individuals in multiple categories. A P-value of ≤0.05 was regarded as significant. All analyses were performed in STATA version 13 (StataCorp LP, College Station, TX, US).
Results
General description of study cohort
In total, 1173 HIV-infected individuals who submitted at least one plasma sample for HIV sequencing out of 3371 patients in follow-up over the study period were initially included in the study. Of the 183 individuals that submitted two samples for DRT, four were excluded from the study because the two samples did not cluster on the phylogram. A total of 1169 individuals were retained in the study. General cohort characteristics are presented in Table 1.
Characteristics of study cohort of HIV-infected individuals (n = 1169).

ART: antiretroviral therapy; MSM: men who have sex with men.
‘Other’ transmission risk category includes: n = 8 intravenous drug users, n = 3 mother-to-child transmission, n = 1 occupational and n = 2 blood transfusion HIV transmissions.
Calculated from date of initial HIV diagnosis to end of study inclusion date.
High incidence of STIs among HIV-infected MSM
Syphilis was the most common STI, with a total of 298 episodes reported in 198 individuals. Most (133/198; 67.2%) only had one episode and 65/198 (33.9%) individuals had two or more episodes. Almost all syphilis episodes were reported in MSM (290/298; 97.3%). Chlamydia was the second most common STI with a total of 173 episodes in 131 individuals, mostly occurring in MSM (116/131; 88.5%). With regards to gonorrhoea, 173 episodes were reported in 104 individuals – most were MSM (98/104; 94.2%). When considering all study individuals, 324/1169 (29.3%) had at least one STI episode diagnosed during their HIV follow-up and this increased to 42.8% (299/699) when only considering the MSM population.
HIV transmission clustering analysis reveals a high number of mixed transmission clusters
The HIV phylogram constructed placed a total of 438/1169 (37.5%) study participants into 136 transmission clusters, including 60 larger clusters defined as three or more individuals and 76 transmission pairs (also defined as clusters) (Table 2; Figure 1). Larger clusters contained a median of four individuals (IQR 3–6) with a maximum of 17 individuals (n = 1 cluster; MSM only). Details of the clusters defined based on the HIV transmission risk category of the individuals are reported in Table 2, including 24 clusters (containing 94 individuals) that were mixed transmission clusters.
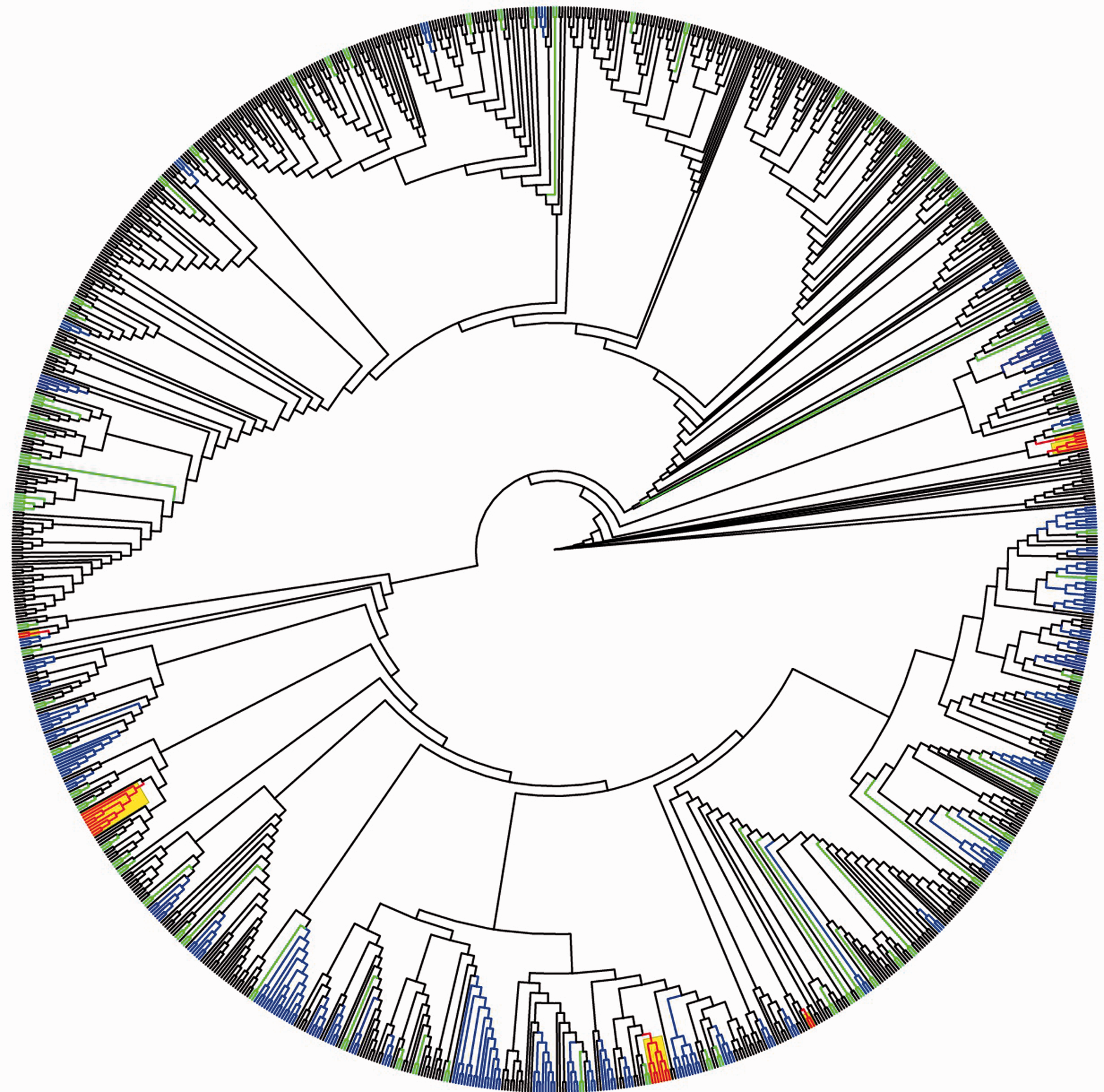
HIV transmission cladogram of 1169 individuals in this study. Clusters (n = 136) are depicted in colour: blue represents clusters containing ≥3 individuals, green represents cluster pairs and red/yellow depicts mixed transmission clusters with possible misclassified HIV sexual transmission among heterosexual males
HIV transmission clusters identified by Cluster Picker and STI history of individuals in each cluster category.
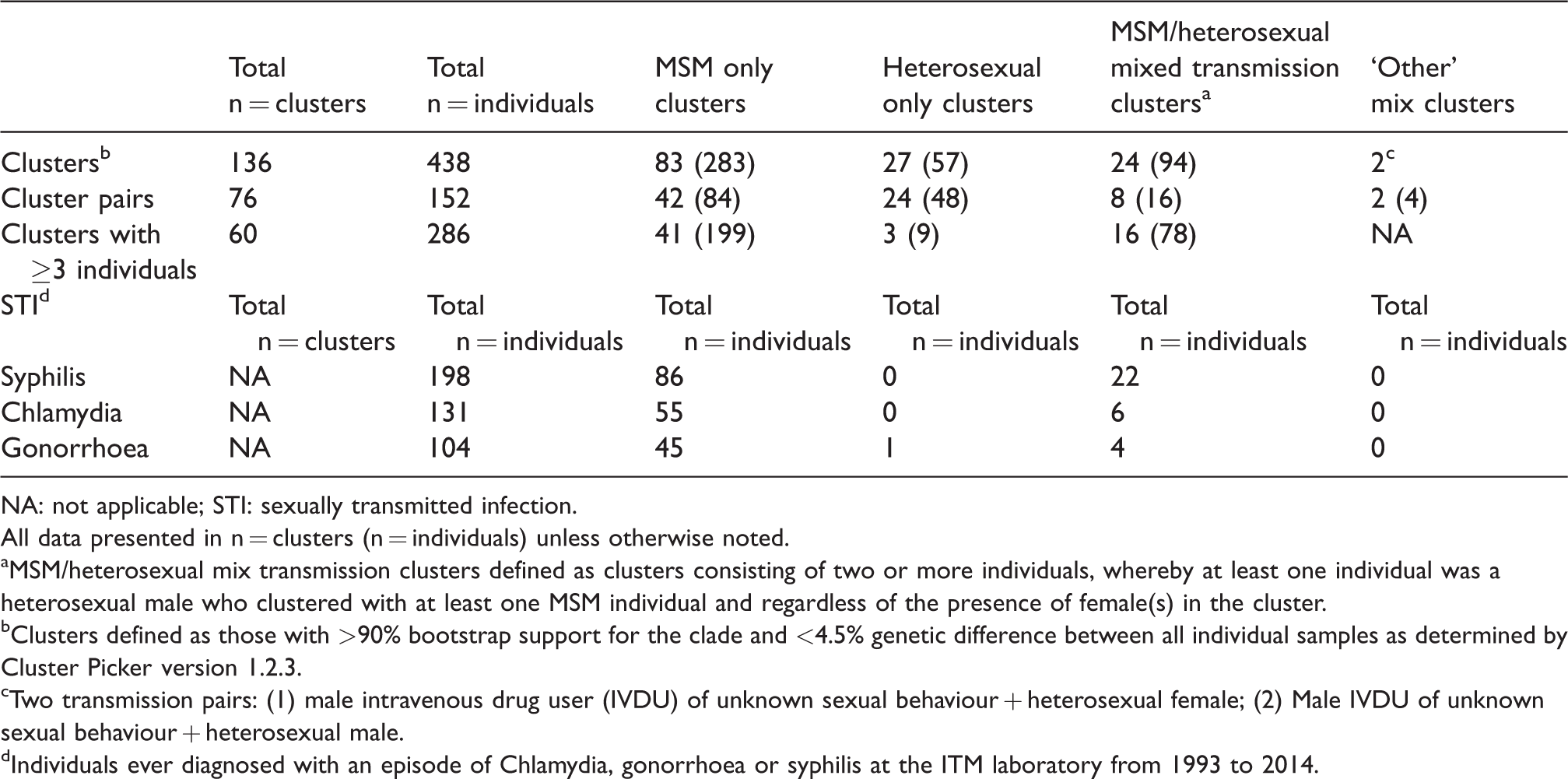
NA: not applicable; STI: sexually transmitted infection.
All data presented in n = clusters (n = individuals) unless otherwise noted.
MSM/heterosexual mix transmission clusters defined as clusters consisting of two or more individuals, whereby at least one individual was a heterosexual male who clustered with at least one MSM individual and regardless of the presence of female(s) in the cluster.
Clusters defined as those with >90% bootstrap support for the clade and <4.5% genetic difference between all individual samples as determined by Cluster Picker version 1.2.3.
Two transmission pairs: (1) male intravenous drug user (IVDU) of unknown sexual behaviour + heterosexual female; (2) Male IVDU of unknown sexual behaviour + heterosexual male.
Individuals ever diagnosed with an episode of Chlamydia, gonorrhoea or syphilis at the ITM laboratory from 1993 to 2014.
Table 3 outlines various bivariate associations with HIV transmission clustering. There was a strong positive association between male gender (odds ratio [OR] 4.96 [CI 3.4–7.3]; P < 0.001), HIV B-subtype (OR 4.7 [CI 3.6–6.1]; P < 0.001), HIV sequencing performed at diagnosis (OR 2.3 [CI 1.7–3.0]; P < 0.001) and MSM transmission (OR 3.9 [CI 3.0–5.2]; P < 0.001) with transmission clustering.
Characteristics of HIV-infected individuals who were identified in HIV transmission clusters compared to non-clustering individuals.
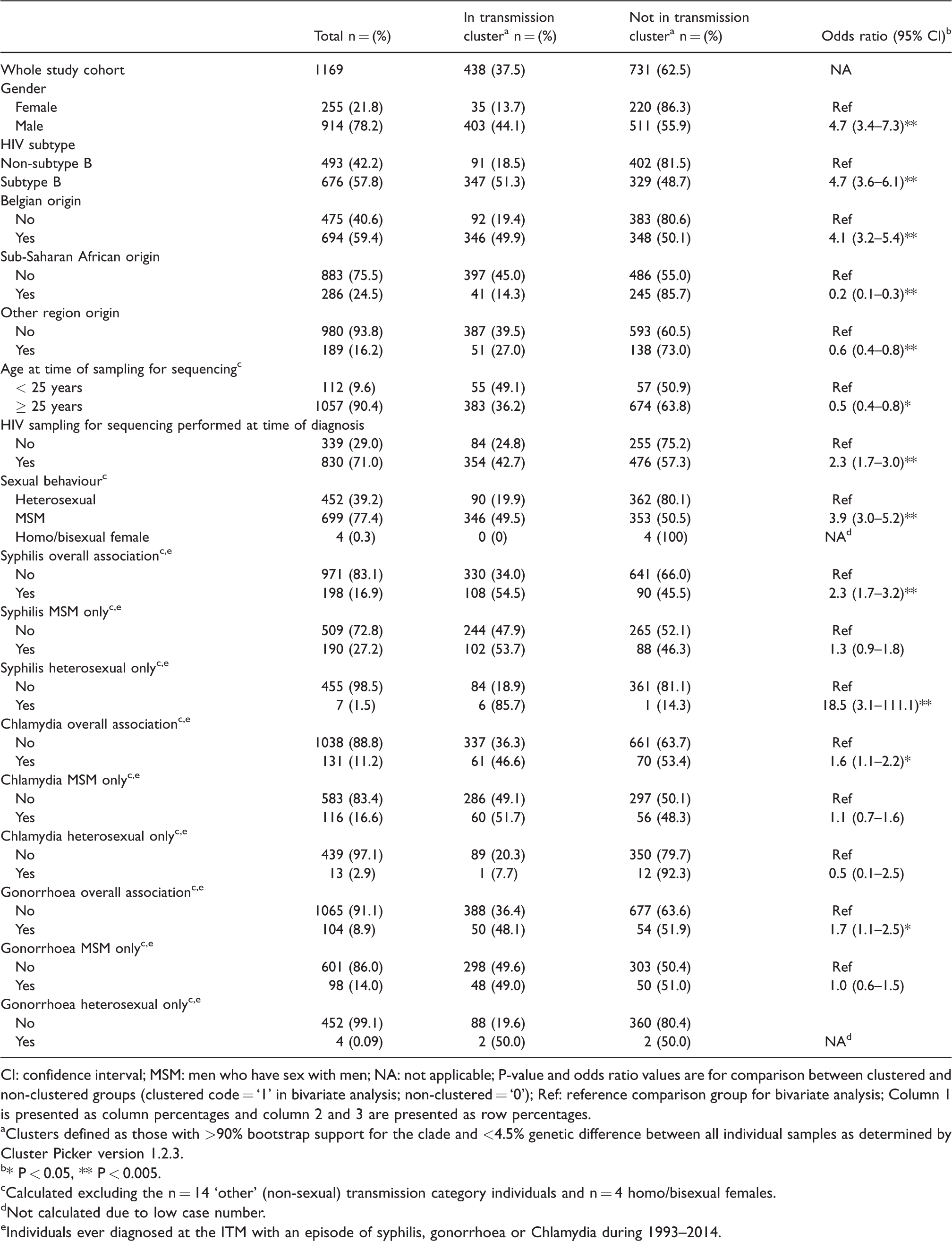
CI: confidence interval; MSM: men who have sex with men; NA: not applicable; P-value and odds ratio values are for comparison between clustered and non-clustered groups (clustered code = ‘1’ in bivariate analysis; non-clustered = ‘0’); Ref: reference comparison group for bivariate analysis; Column 1 is presented as column percentages and column 2 and 3 are presented as row percentages.
Clusters defined as those with >90% bootstrap support for the clade and <4.5% genetic difference between all individual samples as determined by Cluster Picker version 1.2.3.
* P < 0.05, ** P < 0.005.
Calculated excluding the n = 14 ‘other’ (non-sexual) transmission category individuals and n = 4 homo/bisexual females.
Not calculated due to low case number.
Individuals ever diagnosed at the ITM with an episode of syphilis, gonorrhoea or Chlamydia during 1993–2014.
Those diagnosed with syphilis were more likely to be in a cluster (overall association; OR 2.3 [CI 1.7–3.2]; P < 0.001). No syphilis episodes were reported in heterosexual only clusters (Table 2). When the analysis was stratified by sexual transmission categories (‘MSM’ and ‘heterosexual’), clustering of syphilis only remained significant in the heterosexual group (Penalized Multinomial Logistic Regression OR 18.5 [CI 3.1–111.1]; P < 0.001). Individuals with a diagnosis of Chlamydia and gonorrhoea were also more likely to cluster; however, when stratified by transmission category, this relationship was no longer significant.
Six possible cases of HIV sexual transmission misclassification
Twenty-five heterosexual males were present in mixed transmission clusters, of which 6/25 had a diagnosis of syphilis and 5/6 clustered exclusively with other males. According to our definition, these males were categorized as possible HIV sexual transmission misclassifications. We estimate the misclassification rate in our cohort to be 0.7% of total males (6/914) and 3% of total heterosexual males (6/204). When only considering males in clusters, which are the only individuals assessed for misclassification, this rate increased to 1.5% (6/403) for all males and 11% for heterosexual males (6/55). Figure 2 is a graphical depiction of these six possible misclassified heterosexual males.

Six HIV transmission clusters containing possible misclassified HIV sexual transmission heterosexual males with a history of syphilis. MSM: men who have sex with men.
Discussion
This study exemplifies the utility of coupling phylogenetic analyses of HIV collected through DRT with clinical data to gain insights into local HIV transmission network characteristics and the degree of possible sexual behaviour misclassification of MSM. Simplified bioinformatic analyses revealed 136 transmission clusters encompassing 37.5% of the study cohort. STIs were highly prevalent among MSM, with almost half (42.5%) having a diagnosis of at least one STI episode. Individuals with a STI diagnosis were more likely to cluster in the close genetic proximity of one another. However, when analyses were stratified per risk transmission category, this relationship was attenuated and only remained significant for heterosexual males with a history of syphilis. This finding was unexpected since almost all syphilis infections in our cohort occur in MSM. 18 The phylogenetic analyses provided a possible explanation for this finding, the fact that five out of six of these men were in clusters that only contained MSM suggested that they might have been misclassified as non-MSM.
A recent HIV phylogenetic study in the United Kingdom estimated that 18.6% (249/1341) of all clustered heterosexual men were potential non-disclosed MSM. 6 This estimation is higher than the 11% suspected misclassifications in our cohort, a discrepancy that could be attributed to the larger national population study cohort that would likely cover sexual networks more extensively than our single-centre analysis. Further sources of misclassification could be that doctors in our clinic are not taking accurate sexual histories and clinical databases are incorrectly filled in and/or not regularly updated if new patient information is obtained.
There are a number of caveats to this analysis, for example the single institution setting and retrospective nature of the study. Only 34.6% of HIV-infected persons in our cohort were included in this study. Nonetheless, approximately a third of study participants still clustered with other individuals, indicating a moderate coverage of the local HIV transmission network. One must also take into account the high diversity of the cohort population. A quarter of the participants originated from sub-Saharan Africa; they were less likely to cluster (P < 0.001) with others, which could be indicative of transmission events outside the cohort or even the region. Moreover, the long study period spanning 17 years means that individuals diagnosed and sequenced early in the study period who then went on to transmit HIV years later within the same cohort may not be genetically linked due to the continued viral evolution that would slowly abolish phylogenetic linkage. 31 This could explain why individuals who were sequenced shortly after diagnosis were more likely to cluster in close proximity to one another compared to those who were sequenced later (OR 2.3 [1.7–3.0]). Participants with more recent HIV infections and shorter clinical follow-up had less STI episode information available (data not shown) and this could have contributed to a lower misclassification rate for those without (reported) STI episodes. Moreover, the STI episode rate for the MSM participants in this study could be underestimated since the mean follow-up time of the MSM group was significantly shorter (P < 0.001) than the heterosexual transmission group.
Our phylogenetic tree is only based on HIV transmission. Since compared to other STIs, HIV is relatively inefficiently transmitted sexually, 32 our tree likely underestimates the sexual network connectivity of individuals therein. Phylogenetic analyses are limited by the fact that they often cannot assign directionality, thus it cannot be ruled out that the one female clustered with the possible misclassified transmission individuals could have been the transmission link instead of a MSM in the same cluster. The number of individuals defined as possibly misclassified was very small; a Penalized Multinomial Logistic Regression analysis was performed in an attempt to compensate for rare event biases28,29; however, the sample size remains small and multivariate analytical approaches were not feasible.
Increased use of phylogenetic analysis brings with it important ethical, security and privacy considerations that need to be addressed. 9 With the further refinement of accessible bioinformatics tools, individual smaller centres will be able to use these methods to define and characterize their cohorts. Furthermore, patient care would benefit from standardized and optimized approaches to sexual behaviour reporting. A recent study by Haider et al. 33 investigating patient-centred approaches to sexual behaviour data collection found that patients were willing to be asked about their sexual behaviour, were likely not to take offense at such questioning and both patients and clinicians prefer nonverbal data collection methods.
Conclusions
In our setting the superimposition of STIs on the HIV phylogram provided insights into how sexual behaviour could have been misclassified for some individuals. This has important implications for patient care and understanding local STI transmission characteristics since these individuals could operate as a bridge population into otherwise low-risk groups. With regards to patient care, it is crucial to know if individuals are MSM as they require different anal neoplasia and STI screening 19 than other populations.
Footnotes
Acknowledgements
The authors gratefully acknowledge the participation of the individuals in this study. Thanks to Virginie Maes from the Scientific Institute of Public Health (WIV-ISP) for providing a summary of the ITM’s STI data. We appreciate Tania Crucitti’s (ITM) constructive comments during the manuscript preparation and for supervision of the STI testing.
Authors’ contributions
Study design: CRK, CJM, AT, KKO; Cohort study and sample collection EF, ME; Sequence analysis CJM; Statistical analysis: KKO, CRK, AT; Draft manuscript preparation: KKO, CJM; CRK; Final manuscript edit and approval: CRK, CJM, EF, ME, KKO, LH, KKA, KF, SGR, AT.
Availability of data and material
The datasets generated and analysed during the current study are not publicly available due to them containing information that could compromise research participant privacy but are available from the corresponding author (CRK) on reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
The Institutional Review Board of the ITM approved this study (901/13).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was part of Project ID: 757003 funded by the Flemish Government-Department of Economy, Science & Innovation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.