
Abstract
Multi-arm multi-stage designs can improve the efficiency of the drug-development process by evaluating multiple experimental arms against a common control within one trial. This reduces the number of patients required compared to a series of trials testing each experimental arm separately against control. By allowing for multiple stages experimental treatments can be eliminated early from the study if they are unlikely to be significantly better than control. Using the TAILoR trial as a motivating example, we explore a broad range of statistical issues related to multi-arm multi-stage trials including a comparison of different ways to power a multi-arm multi-stage trial; choosing the allocation ratio to the control group compared to other experimental arms; the consequences of adding additional experimental arms during a multi-arm multi-stage trial, and how one might control the type-I error rate when this is necessary; and modifying the stopping boundaries of a multi-arm multi-stage design to account for unknown variance in the treatment outcome. Multi-arm multi-stage trials represent a large financial investment, and so considering their design carefully is important to ensure efficiency and that they have a good chance of succeeding.
Keywords
1 Introduction
Bringing a drug from the laboratory to the market is a long and expensive process often ending in failure. 1 Typically, a novel medicinal product will take 10–15 years to develop and validate, at the cost of hundreds of millions of dollars. 2 Any improvements in design that potentially increase the efficiency of the development process are therefore of great practical interest.
One class of trial designs that have been proposed to improve the efficiency of the drug development process as a whole are multi-arm multi-stage (MAMS) designs. MAMS designs are a rich class of designs but fundamentally consist of simultaneously testing several experimental treatments against a common control. Interim analyses are used in order to decide which treatments should continue. Using MAMS designs provides several advantages over running separate controlled trials for each experimental treatment:
a shared control group can be used, instead of a separate control group for each treatment; a direct head-to-head comparison of treatments is conducted, minimising biases that can be introduced from making comparisons between treatments tested in separate trials; the use of interim analyses allows ineffective treatments to be dropped early, or early stopping of the trial if one treatment is clearly superior (although this advantage applies also in the case of separate trials of each treatment through use of group-sequential designs).
Within the class of MAMS studies a variety of different designs are available that differ mainly in the treatment selection at the interim analyses. A ‘Pick-the-winner’ design selects the most promising experimental treatment at the first interim analysis and compares it to control in the subsequent stages. 3 – 5 Stallard and Friede 6 allow more than one treatment to continue beyond the first stage, where the number of treatment arms within each stage is pre-specified while Kelly et al.7 prefer using a rule that allows all treatments that are close to the best performing treatment to be selected. Flexible adaptive two-stage multi-arm designs utilising p-value combination ideas together with closed testing have been discussed in, for example.8,9 These designs do not require pre-specification of a treatment selection rule and hence flexible decision making that takes other information from the first stage of the trial into consideration is possible. Study designs with two or more stages in which all treatments are continued at each stage, provided they are sufficiently promising, are discussed in Royston et al. 10 and Magirr et al. 11 This class, which we refer to as a group-sequential MAMS design, will be considered throughout the rest of the manuscript, although most statements will hold true irrespective of the selection rule used.
In this article, we discuss a range of statistical issues faced in the design of group-sequential MAMS trials and use the TAILoR trial, in which the same normally distributed endpoint is used at each analysis, as a motivating example. Much of our discussion will also apply to more complex MAMS designs in which endpoints are not necessarily normally distributed or the same at each analysis. We consider aspects of controlling the type-I error rate and power in a MAMS trial; choice of stopping boundaries; how to adjust boundaries when the variance of the normally distributed endpoint is unknown; the impact of adding a treatment arm during a MAMS trial; and whether additional patients should be allocated to the control group.
2 Motivating trial and notation
At present there are only a few examples of MAMS designs being used in practice, which include the MRC STAMPEDE trial 12 and the TAILoR trial, discussed in Magirr et al. 11 At the time of writing, additional MAMS trials are in various stages of being set up. To provide a case-study to frame discussion in this article, we consider the TAILoR (TelmisArtan and InsuLin Resistance in HIV) trial. This trial initially was planned to test four experimental arms corresponding to four different doses of Telmisartan. Although the final protocol of the study only uses three experimental arms we will use four experimental arms in our examples for consistency with previous publications. Telmisartan is thought to reduce insulin resistance in HIV-positive individuals on combination antiretroviral therapy (cART). The primary endpoint is reduction in insulin resistance in the telmisartan-treated groups in comparison with the control group as measured by HOMA-IR at 24 weeks. The assumption of monotonicity of dose–response relationship was thought to not be valid based on experimentation of the treatment in a different indication. As a consequence, a design that made no assumption of a dose–response relationship was used.
We consider a trial testing K experimental treatments against a control treatment, we define
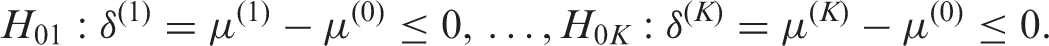
For a multi-stage design, the above set of null hypothesis is tested at up to J analysis time points (stages). After stage j, standard z-test statistics are calculated to compare each remaining experimental arm to control. The test statistic comparing experimental arm k to the control group is labelled
The TAILoR trial follows this setting and uses two-stages with futility boundaries (0, 2.18) and efficacy boundaries (2.91, 2.18). These boundaries are found to give a family-wise error rate of 5%. Note that the boundaries are similar to the popular O'Brien-Fleming boundary shape. 13 The sample size required to obtain a power of 90% is found to be n = 44 patients per arm per stage if a standardised effect (i.e. σ = 1) of 0.544 is considered interesting while an effect of 0.178 is considered too small to warrant further study. The maximum total sample size of the study is therefore 440.
3 Error control
Controlling the type-I and type-II error in multi-arm trials is more complicated than in traditional randomised controlled trials (RCT) due to the simultaneous testing of several hypothesis.
3.1 Type-I error considerations
For a set (or family) of hypotheses, a type-I error is defined as rejecting any true null hypothesis. Controlling the family-wise error rate (FWER) in the strong sense means that the probability of rejecting any true null hypothesis is controlled at a pre-specified level for any possible values of (δ(1),…, δ(K)). The guidance on multiplicity issues in clinical trials from the European Medicines Agency 14 states that controlling the familywise type-I error in the strong sense is required for confirmatory trials.
Magirr et al. 11 extend the multiple-testing procedure of Dunnett 15 to multiple stages. They show that the probability of rejecting any true null hypothesis is maximised when δ(1) = … = δ(K) = 0, and so controlling this probability provides strong control of the FWER. The authors derive an analytic formula for this probability which contains multi-dimensional integration, with the number of integrations being equal to the number of stages in the trial. Thus evaluating the formula becomes more computationally intensive as the number of stages increases. A simulation approach using a large number of independent replicates is an alternative method to evaluate the maximum FWER, and may be necessary when there are more than three stages. This approach is described in Wason and Jaki. 16 The probability of rejecting any null hypothesis at δ(1) = … = δ(K) = 0 is determined only by the stopping boundaries, and not the group size used as the mean of each test statistic is 0 under the null hypothesis, regardless of n. Similarly the covariance between the test-statistics is not dependent on n which implies that one can find a MAMS design by first choosing stopping boundaries that give the correct FWER, and then subsequently choose a group size to power the trial.
Although we recommend that the FWER of the design should be specified and controlled in confirmatory trials, there are contrary opinions. Freidlin et al. 17 advocate not adjusting multi-arm trials for multiple testing at all when the different arms correspond to different treatments. The argument for this position is that if the treatments were compared in separate trials, they would not be subjected to multiple testing adjustment. Although this argument has merit, we feel that the situation of conducting a MAMS trial is conceptually quite different to running a series of separate trials. As an analogy, consider testing multiple primary outcomes in a confirmatory trial. In this case, regulatory bodies would encourage (or require) that a multiple testing correction is made. However, one could test each primary endpoint in a separate trial without requiring multiple testing.
The MRC STAMPEDE trial, 12 does not explicitly control or specify the FWER, but instead controls the pairwise type-I error rate, i.e. the type-I error rate of a test of one experimental treatment against the control treatment. Since this pairwise type-I error rate is low (0.013) and early stopping for efficacy is not allowed, it is likely that the overall FWER is low.
For exploratory MAMS trials (for example in phase II), controlling the FWER would not be required by regulatory bodies. However, we believe that the FWER is a more relevant quantity than the pairwise type-I error rate associated with each experimental treatment. The FWER provides the maximum probability of recommending an ineffective treatment, which is important if a phase III trial is to be carried out subsequently. An additional reason to consider designing such trials with FWER control is due to the increased use of phase II studies as the second pivotal study when making a confirmatory claim.
3.2 Powering a MAMS trial
If the objective of the trial is to detect the truly best treatment, then the power to do so depends on both the mean effect of the best treatment, and also the mean effects of all the other experimental treatments. 18
The TAILoR trial was powered to detect the best treatment using what is known as the least favourable configuration (LFC). The LFC requires specification of a clinically relevant difference, δ1, and an uninteresting treatment difference threshold, δ0. The uninteresting treatment difference threshold is the smallest mean difference between an experimental treatment and the control treatment that would make that experimental treatment clinically interesting. Given δ1 and δ0, the LFC is the probability of recommending experimental treatment 1 when δ(1) = δ1 and δ(2) = … = δ(K) = δ0. It is referred to as the least favourable configuration because out of all scenarios where treatment 1 has the clinically relevant treatment effect and treatments 2, … , K are uninteresting, it provides the lowest probability of recommending treatment 1. 4
Group size and power of designs 1-3 at different power scenarios. Design 1 has sample size chosen so that power at the LFC with δ1 = 0.545 and δ0 = 0.178 is 0.9; design 2 has sample size chosen so that power at the LFC with δ1 = 0.545 and δ0 = 0 is 0.9; design 3 has sample size chosen so that power to recommend any treatment when all have effect δ = 0.545

Table 1 shows that the choice of δ0 for the LFC does not affect the power greatly provided that δ0 is not too close to δ1. For example design 2, powered for the LFC with δ0 = 0, still has 87.2% power at the LFC with δ0 = 0.178. On the other hand design 3, powered to recommend any experimental treatment when they are all effective, does not adequately power the trial at either LFC considered. It would be unusual for all experimental treatments in a trial to be highly effective in comparison to the control treatment. Thus powering the trial for this situation would be highly optimistic and will often result in under-powered trials in practice.
3.3 Choosing stopping boundaries
As for group-sequential trials, the choice of stopping boundaries influences the operating characteristics of a MAMS trial. One approach to setting stopping boundaries is to specify a function that determines the shape, such as those of Pocock, 20 O'Brien and Flemming, 13 or the triangular stopping boundaries of Whitehead and Stratton. 19 As discussed in Section 3.1, with a given stopping boundary shape it is conceptually straightforward, although computationally demanding, to find the MAMS design with required FWER and power. Even more complex, though achievable, is the use of the more flexible alpha-spending approach. 21 The disadvantage of using set stopping boundaries (or alpha-spending) is that the expected sample size properties may not be to ones liking. Wason and Jaki 16 show that the triangular design performs well in terms of expected sample size, so is a good choice if a pre-specified design is desirable.
An alternative is to search for an optimal design. This is an extremely computationally demanding procedure, but does produce designs which have desirable expected sample size properties. Of particular interest is a generalisation of the δ-minimax design,22,23 which is described in Wason and Jaki. 16 The generalised δ-minimax design has very good expected sample size characteristics, generally improving over the triangular design when the experimental treatments are not much better than control. It does not perform as well as the triangular test when some experimental treatments are considerably better than control.
Due to the computational complexity of finding optimal designs, a compromise between the fixed boundary approach and the optimal design approach may be useful. The power family of group-sequential tests24,25 specifies a family of stopping boundaries indexed by a parameter, Δ which determines the shape of the futility and efficacy stopping boundaries. By increasing Δ, more weight is put on the expected sample size, and less on the maximum sample size. An extension to allow the shape parameter for the futility boundaries to differ to that of the efficacy boundaries was proposed for group-sequential RCTs in Wason. 26 It was found that the boundaries of optimal designs were well approximated by boundaries within the extended power-family. Investigating whether this result holds for MAMS trials is an area for future research.
4 Control group allocation
In a traditional RCT in which the endpoint measured for both the control and experimental treatments have the same variance, the optimal allocation between arms, in terms of maximising the power, is 1:1. However, when there are multiple experimental arms all being compared against a control arm, the optimal allocation is no longer 1:1. If there were no early stopping, then the optimal allocation to the control group has been shown to be approximately
Changing the allocation ratio affects both the expected sample size and maximum sample size of the trial. Wason and Jaki
16
investigate the optimal allocation ratio as part of searching for an optimal design. For three stages and four experimental arms, the optimal allocation ratio to controls was found to be approximately 1.33:1. The optimal allocation ratio increases when there are six experimental arms, but is still considerably below 2:1. The optimal allocation ratio based on expected sample size is thus substantially below the
Allocation ratio giving lowest maximum sample size as J (number of stages) and K (number of experimental arms) varies

Although efficiency (in terms of maximum sample size) can be gained by deviating from an equal allocation to each arm, the gain is generally fairly small (as also shown by Wassmer
27
). Figure 1(a) shows the maximum sample size for the three-stage triangular design with the TAILoR design parameters across a range of allocation ratios. By choosing the optimal allocation ratio, the maximum sample size is reduced by only 2.5% compared to an equal allocation. Interestingly, one has to increase the allocation to controls considerably in order to noticeably increase the maximum sample size. Put conversely this implies that a large number of patients can be put on the control treatment without inflating the maximum sample size considerably. This may, for example, be of interest if the control treatment is considerably cheaper than the experimental treatments or thought to have a better safety profile than the experimental treatments. This effect is shown in Figure 1(b), where the total cost of allocating patients is shown as the ratio of the cost of the control treatment and experimental treatments varies. If the cost of the control treatment is very low, then a high allocation to control patients would be optimal.
Maximum sample size and maximum cost (arbitrary units) of treatment as allocation ratio changes. Designs are chosen using triangular stopping boundaries such that they give 5% type-I error and 90% power. Maximum cost assumes that the cost of allocating a patient to the control group is c, and the cost of allocating a patient to an experimental treatment is 1 where c ∈ {1, 0.5, 0.25, 0.1}.
The downside of allocating additional patients to the control treatment is that it may reduce recruitment to the trial. There is some evidence that in placebo controlled trials, patient willingness to take part in the trial is reduced as the allocation to the control group increases. 28
5 Unknown variance
For trials with a normally distributed endpoint, a common assumption made at the design stage is that the variance, σ2, is known. Of course this is not generally the case, and even if a prior estimate of the variance is available, it is usually subject to considerable uncertainty. Using a test statistic that assumes a known variance will lead to incorrect operating characteristics if the actual variance differs from the quantity assumed in the test statistic. For group-sequential trials, several papers have suggested approaches to modifying stopping boundaries to allow for unknown variance including Monte Carlo simulation, 29 a recursive algorithm 30 and quantile substitution, i.e. replacing the stopping boundaries, which are quantiles of the standard normal distribution, with the equivalent quantiles of Student's t-distribution, as described in Jennison and Turnbull. 31 Currently there is no work on extending the recursive algorithm to group-sequential MAMS trials; instead we examine the third method, which is straightforward and not computationally intensive.
Recall that l
j
and u
j
are the stopping boundaries for analysis j, and jn is the number of patients per arm that are randomised by the time of the analysis. Then the thresholds for stopping in terms of p-values are attained from the respective quantiles of the normal distribution, i.e. 1 − Φ(u
j
) and 1 − Φ(l
j
) respectively. With unknown variance, when δ = 0, the test-statistics would be marginally distributed as a Student's t-distribution with 2jn − 2 degrees of freedom. A natural approach to take the unknown variance into consideration is to find new stopping boundaries as
To evaluate whether the quantile-substitution method works adequately for MAMS trials, we compare the FWER and power for three different approaches. The first is to use the known variance test statistic with presumed value of σ; the second is to use a t-test without modifying the stopping boundaries; and the third approach is to use the t-test together with using quantile substitution to change the stopping boundaries. The following two designs are considered:
n = 35, f = (0, 1.44, 2.34), e = (2.71, 2.39, 2.34) a three-stage four experimental arm triangular design when δ0 = 0.178, δ1 = 0.545, σ = 1, α = 0.05, 1 − β = 0.9; n = 10, f = (0, 1.43, 2.34), e = (2.70, 2.39, 2.34) a three-stage four experimental arm triangular design for δ0 = 0, δ1 = 1, σ = 1, α = 0.05, 1 − β = 0.9.
FWER and power estimates as the true standard deviation varies from the assumed value of 1 for three-stage design with four experimental arms, n = 35, f = (0, 1.44, 2.34), e = (2.71, 2.39, 2.34). 100,000 independent replicates used to estimate type-I error and power. Z-test is using the original boundaries with a Z-statistic, t-test the original boundaries with a t-statistic while t-testcorr uses a t-statistic with corrected boundaries. Monte Carlo standard error for estimated type-I error ≈ 0.0007. Maximum Monte Carlo standard for power estimate ≈0.0015
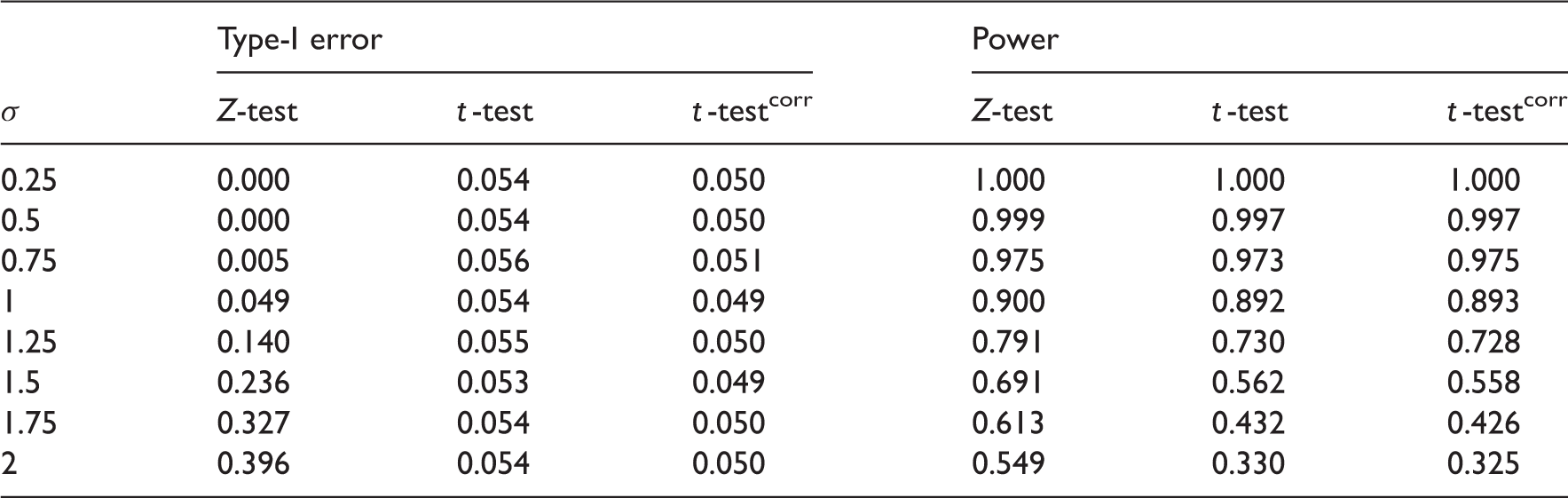
FWER and power estimates as the true standard deviation varies from the assumed value of 1 for three-stage design with four experimental treatments, n = 10, f = (0, 1.43, 2.34), e = (2.70, 2.39, 2.34). 100,000 independent replicates used to estimate type-I error and power. Z-test is using the original boundaries with a Z-statistic, t-test the original boundaries with a t-statistic while t-testcorr uses a t-statistic with corrected boundaries. Monte Carlo standard error for estimated type-I error ≈0.0007. Maximum Monte Carlo standard for power estimate ≈0.0015

Modifying the stopping boundaries is not sufficient to control both the FWER and power as σ varies from its design value. In confirmatory trials, the priority should be placed on controlling the FWER, which appears to be possible using quantile-substitution. If one wishes to simultaneously control the FWER and power, a sample-size reestimation technique could be applied as better estimates of σ are gathered throughout the trial. An alternative approach is to use a p-value combination test design,8,9 in which case an exact solution for unknown variance is available. 27
6 Adding treatment arms
Error rates when treatment is added at interim, keeping the original boundaries. Based on 100,000 simulations

We start by considering a, somewhat unrealistic, scenario in which one additional experimental treatment arm is always added at the interim. An additional 2n patients are recruited to treatment k = 5 in the second stage and an additional test statistic,
Since the fifth treatment can never stop early, the power is no longer independent of the treatment labels so that it is of interest to also investigate the power to select treatment 5 under the LFC. The corresponding Monte Carlo estimate,
Error rates when treatment is added at interim, adjusting the upper boundary at the second stage. Based on 100,000 simulations
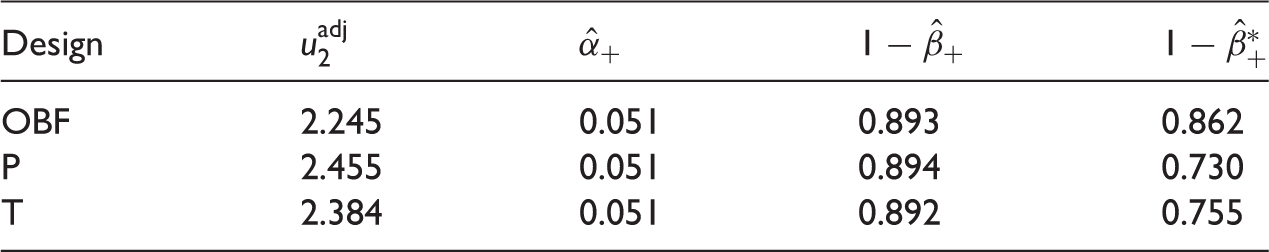
A more realistic setting than the one described above is when a treatment is added only with probability p+. In this case the original boundaries are used when no treatment is added while adjusted boundaries are used otherwise.
Monte Carlo estimates of familywise error rate (target α = α+ = 0.05) when Knew new treatments are added independently or on the basis of disappointing first stage results. Based on original OBF design, l1 = 0, u1 = 3.068, n = 44 and 100,000 simulations

7 Discussion
MAMS trials have an important role to play in improving the efficiency of the drug development process when several experimental treatments are awaiting testing. Parmar et al. 32 propose MAMS trials as a way of achieving more reliable results more quickly when evaluating new agents in cancer. A number of recent papers have discussed design of MAMS trials8,6,9,11,12,16,33 using a variety of different approaches.
In this article we have considered a multitude of issues in the design of MAMS trials. Our recommendations are as follows:
Strong control of the FWER should be considered a priority in the design of confirmatory MAMS trials. A MAMS trial should be powered to recommend a clearly superior treatment, with the value of δ1, the clinically relevant difference, being important; the value of δ0 (i.e. the mean effect of the other treatments) is less important. The efficiency benefits of a higher allocation of patients to control are low, and may be damaging to recruitment. However, if the control treatment is considerably cheaper than other treatments, then a higher allocation may lead to large cost reduction without compromising the design characteristics. If the group size is low (below 20), stopping boundaries should be adjusted using quantile substitution to account for unknown variance when considering normally distributed endpoints. For confirmatory MAMS trials, we do not recommend adding treatment arms on the basis of interim results. In the case of experimental treatment arms being added for other reasons, subsequent stopping boundaries should be adjusted to maintain the FWER at the level specified at the design stage.
Footnotes
Acknowledgements
The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. We thank the two anonymous reviewers for their useful comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Medical Research Council (grant numbers G0800860 and MR/J004979/1). This report is independent research arising from Dr Jaki’s Career Development Fellowship (NIHR-CDF-2010-03-32) supported by the National Institute for Health Research.