
Abstract
In this research, fluorine substituent influence on the electronic structure, 14NQR and electronic spectra of carbo-borazine molecule was explored at M062X/6-311G(d,p) level of theory. Energetic stability of possible isomers was examined. QTAIM results illustrated ring critical points (RCP) and the electrostatic potential of RCP reported. The curvature of electron density perpendicular to ring plane at RCP was considered to exploring of aromaticity of these molecules. The ELF map were used to description of chemical bonds (C-H, C-B, C-N, B-F, N-F and B-N) in the studied molecules. Natural transition orbital (NTO) analysis was employed to origin of corresponding transition of λmax value. Fluorine substituent influence on the computed λmax and 14N NQR parameters values was studied.
Introduction
Carbomers are extended molecules, which are attained by the insertion of -C≡C- unit in each bond of a molecule.1–3 For instance, carbo-benzene (C18H6, D6h) can be created with of six -C≡C- units in each bond of benzene. Preparation of this “bare” carbo-benzene has been unsuccessful. Computational studies have been illustrated electronic structure and properties of cabo-benzene.4–9 But, synthesis of carbo-benzene derivatives has been reported.10–12 Also, the inorganic analogues of carbo-benzene can be made. Carbo-borazine (B3C12N3H3, D3h) is obtained from the inclusion of six -C≡C- units in each bond of borazine (B3N3H, D3h). Preparation of this molecule was impossible, due to dimerization tendency of this molecule. The aromaticity of this molecule was examined, and it was illustrated that the electronegativity difference between B and N atoms disturbed the electronic delocalization. The computed induced magnetic field exhibited nonaromatic property in this molecule. 13
In this research, we attracted to explore fluorine substituent influence on the structure, vibration and 14N NQR spectra and electronic properties of carbo-borazine at M062X/6-311G(d,p) level of theory. The curvature of electron density perpendicular to ring plane at RCP was considered to exploring of aromaticity of these molecules.
Computational methods
Optimizations of the considered molecules were done using the 6-311G(d,p) basis set14–17 and hybrid functional of Truhlar and Zhao (M06-2X) 18 by software package of Gaussian 09. 19 Harmonic vibrational frequencies were verified that the optimized molecules have no imaginary frequency.
The Hirshfeld population analysis was made based on optimized molecules. This analysis is a very general atomic population way well-known on deformation density partition. 20
The computations of quantum theory of atoms in molecules analysis (QTAIM) and interacting quantum atoms (IQA) method21–24 were calculated with the AIMAll software package. 25 The “Promega” basin integration method using fine interatomic surface mesh was applied.
Atomic dipole corrected Hirshfeld atomic charge (ADCH), 26 the electron localization function (ELF) 27 and curvature of electron density perpendicular to ring plane at RCP 28 were attained by Multiwfn 3.8 package.29–31
Electronic transitions of the molecules were explored using TD-DFT 32 at M062X/6-311G(d,p) level of theory. 30 lowest singlet excited states were evaluated.
Natural Transition Orbital (NTO) analysis 33 was accomplished with TD-DFT calculations on optimized molecules. The NTO distributions of the most intensity transition of molecules were provided using Chemissian version 4.2.3 software. 34
Results and discussion
Energetic aspects
Figure 1 presents the structures of carbo-borazine (

The structures of carbo-borazine and fluorinated-carbo-borazine molecules.
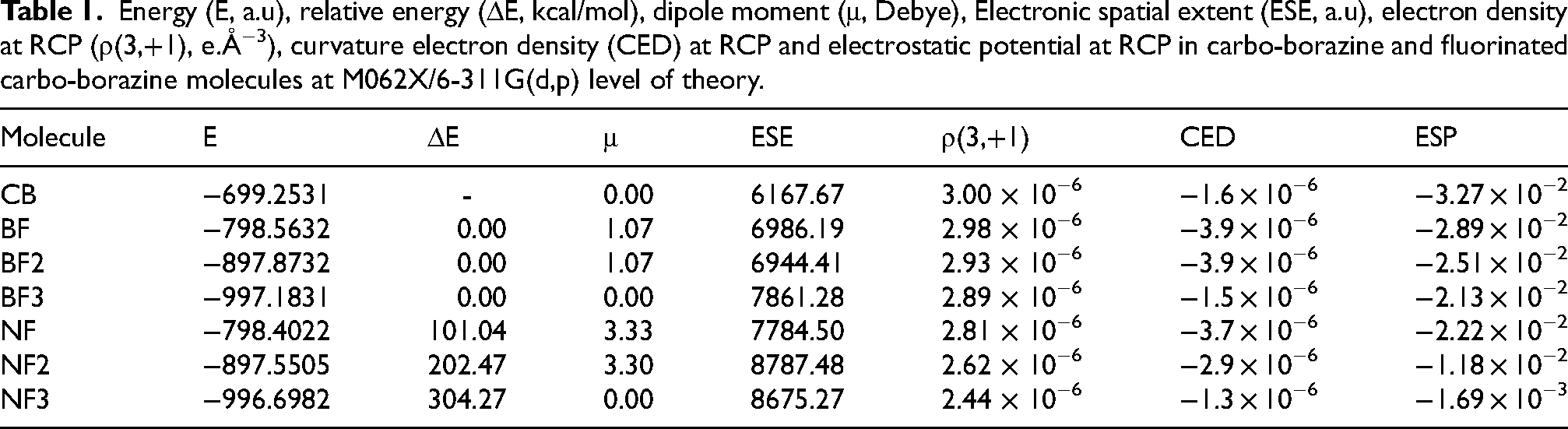
Energy (E, a.u), relative energy (ΔE, kcal/mol), dipole moment (μ, Debye), Electronic spatial extent (ESE, a.u), electron density at RCP (ρ(3,+1), e.Å−3), curvature electron density (CED) at RCP and electrostatic potential at RCP in carbo-borazine and fluorinated carbo-borazine molecules at M062X/6-311G(d,p) level of theory.
Interacting Quantum Atoms (IQA) approach
The interacting quantum atoms (IQA) method is used to computation of the repulsion energy values of between of B and N nucleuses with F atom (Vnn(A,B)) in the fluorinated-carbo-borazine systems (Table 2). It can be detected greater Vnn(N,F) values in compared to Vnn(B,F) values. This consequence is well-matched with existence of LP/LP electron repulsion in the N-F bond. This impact is negligent in the B-F bond. Hence, the
Repulsion energy between nucleus of atom B and nucleus of atom F, also atom N and nucleus of atom F (Vnn(A,B), a.u), in fluorinated molecules at M062X/6-311G(d,p) level of theory.

Polarity
Computed dipole moment values of the considered systems are given in Table 1. It can be deduced greater polarity in the N-fluorinated molecules than B-fluorinated molecules. This tendency is well-matched with larger polarity of N-F bond than B-F bond. Computed atomic dipole corrected Hirshfeld atomic charge (ADCH) of B and N atoms of the considered molecules are recorded in Table 3. It can be realized more positive ADCH values for N atoms of in the N-fluorinated molecules respect to B atoms in the B-fluorinated systems.
Computed atomic dipole corrected Hirshfeld atomic charge (ADCH) of B and N atoms in carbo-borazine and fluorinated carbo-borazine molecules at M062X/6-311G(d,p) level of theory.

Electronic spatial extent (ESE)
ESE is estimated as the probability value of the electron density times the distance from the center of mass of a molecule. More it is features physical property of the electron density volume round the molecule. Greater defused electron cloud is provided with increasing of the range. Computed ESE values of the investigated systems are recorded in Table 1.
Computed ESE values reveal larger ESE values in fluorinated systems than
It can be deduced larger ESE values in the B-fluorinated molecules than N-fluorinated molecules. In the N-fluorinated molecules, both N and F atoms are electron-withdrawing, pulling electron density inward toward the N–F bond. There is a more polarized B–F bond in the
QTAIM
Results of QTAIM computations displays bond critical points (BCP) between CC, CB, and CN units. It can be found the BCP is nearer to C in CN units, nearer to B in CB units, and nearer to B in BN units. The shift of BCP adopts the atomic radii pattern, BCP is place at middle in CC units.
On the other hand, QTAIM reveals the position of ring critical point (RCP) is changed. The CB, BF3 and NF3′s RCPs are located at (00,0), RCP of BF and NF are located at (0.00, 0.00, −0.67) and (0.23,-0.58, 0.00), respectively.
RCP of BF2 and NF2 are located at (0.00, 0.00, −0.63) and (0.00, 0.00, 0.58), respectively. It can be seen 1.21 Å displacement along z-axis. RCP of BF and NF are placed at (0.00, 0.00, −0.67) and (0.23, −0.58, 0.00), respectively.it can be found, −0.23, 0.58, and 0.67 Å displacement along x, y, and z-axes, respectively.
On the other hand, the RCP electrostatic potential of ring critical point (RCP) is altered (Table 1). It can be deducing more negative ESP values for B-fluorinated structures than N-fluorinated structures.
Aromaticity
Electron density at RCP is faithfully correlated to aromaticity of resultant ring. 28 Computed ρ(3,+1) values of the studied systems are given in Table 1. The larger the density, the stronger the aromaticity. As a result, B-fluorinated molecules have a stronger aromaticity than N-fluorinated molecules.
Now, we estimate the curvature of electron density perpendicular to ring plane at RCP. This parameter explores noteworthy relationship with the aromaticity of ring. More negative curvature suggests greater aromaticity. Comparison between the two curvatures again displays that B-fluorinated molecules have a stronger aromaticity than N-fluorinated molecules.
Two provided results are compatible with more stability energetically of B-fluorinated molecules respect to N-fluorinated molecules.
ELF
The ELF function explores the amount of electron delocalization in chemical systems, facilitating covalent bonding analysis by poor in electrons and red for rich-electron regions. ELF maps of carbo-borazine and fluorinated carbo-borazine molecules in the defined plane by three atoms are presented in Figure 2. Located hydrogen atoms in red areas, representative the electron deficiency distinctive of protons. But, B, C, and N atoms are surrounded by blue areas, suggesting the presence of electron density.

ELF maps of carbo-borazine and fluorinated carbo-borazine molecules in the defined plane
The ELF map similarly describes chemical bonds (N-F, B-F, C-B, C-H, B-N, and C-N) colored between red and orange. Furthermore, the electron pairs round fluorine atoms seem as orange areas.
14NQR parameters
Electric field gradient tensor
Table 4 represents EFG tensors (qzz, qyy and qxx) of 14N in studied molecules. Computed qyy, qxx values of N atoms of B-fluorinated molecules are smaller than
Electric field gradient tensor (qii, a.u), Nuclear quadrupole coupling constant (χii, MHz), asymmetry parameter (η) and nuclear quadrupole resonance frequencies (MHz) of, 14NQR spectra of carbo-borazine and fluorinated carbo-borazine molecules at M062X/6-311G(d,p) level of theory.

Computed qxx and qzz values of N atoms bonded to F in N-fluorinated molecules are larger than
Nuclear quadrupole coupling constant
Nuclear quadrupole coupling constants (χii) of 14N in considered molecules are collected in Table 4. χzz, χyy and χxx values of N atom of B-fluorinated molecules are larger than
Computed χzz and χyy values of N atoms bonded to F in N-fluorinated molecules are larger than
Asymmetry parameter
Computed asymmetry parameters (η) of 14N atom in considered molecules are collected in Table 4. These value demonstrate altered distribution of electric charge around the N nucleus respect to cylindrical symmetry in nitrogen molecules.
No significance changes are observed in the η values of N atoms of B-fluorinated molecules respect to
On the other hand, the computed η values of N atoms bonded to F are larger than N atoms bonded to H in N-fluorinated molecules. Also, these values are larger than the corresponding values in
NQR frequencies
Calculated NQR frequencies of 14N in considered molecules are given in Table 4. The N atoms of these molecules are located in the electric field of the other nuclei, hence, the symmetry of the EFG surrounding them alters. This alteration results in the splitting of the energy levels of 14N nuclei, accordingly, three NQR frequencies for 14N will be seen. The variances between the frequencies of nitrogen in these molecules can be associated to the direct participation of the electron pairs of N atom through chemical bond realization or diatropic current in the cycle.
No significance changes are observed in the υ+, υ−, and υ0 values of N atoms of B-fluorinated molecules respect to
On the other hand, the υ+, υ−, and υ0 values of N atoms bonded to F are larger than N atoms bonded to H in N-fluorinated molecules. These values are larger than
Electronic spectra
The carbo-borazine molecule fits to the D3h point group. The strongest peak (fmax) is the S0→S2 transition. This transition appears at 270.20 nm (f = 0.8424). TD-DFT consequences expose no one transition evidently governs, and probing the appropriate canonical orbitals is not predominantly useful in this example: HOMO-9 → LUMO HOMO-2 → LUMO+1 HOMO-1 → LUMO+1 HOMO-1 → LUMO+2 HOMO → LUMO
Percentages of transitions contributions are 2.50%, 5.23%,41.35%, 3.39% and 41.45%, respectively.
Accordingly, we accomplished natural transition orbital (NTO) analysis on the most intense singlet-singlet transition for the carbo-borazine molecule. NTO analysis permits a direct clarification of electronic transitions in terms of a hole-particle representation. It can be inferred, this excitation implicates mostly the promotion of one π→π* electronic transition (Figure 3).
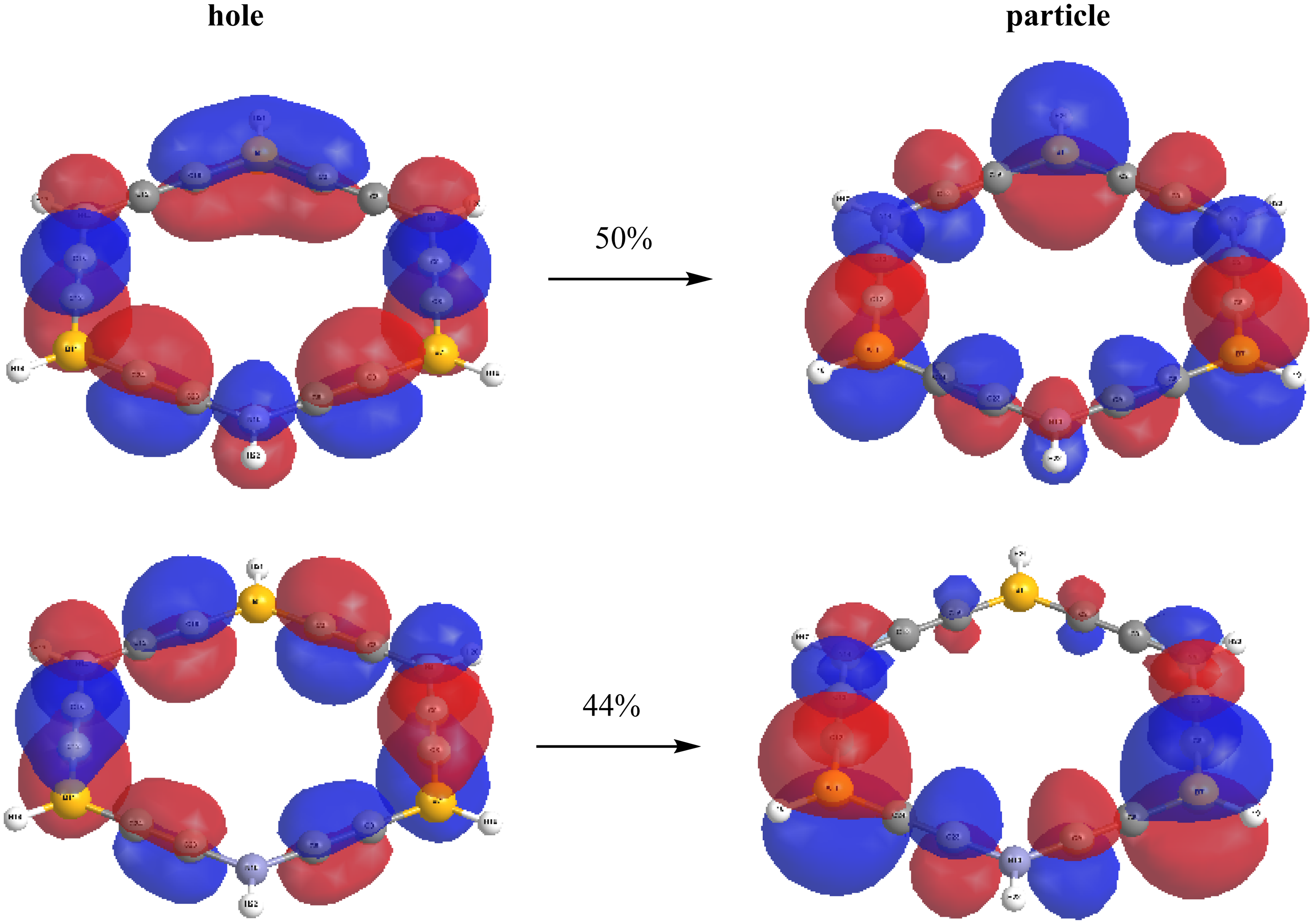
The NTO distributions of the most intensity transition of carbo-borazine molecule.
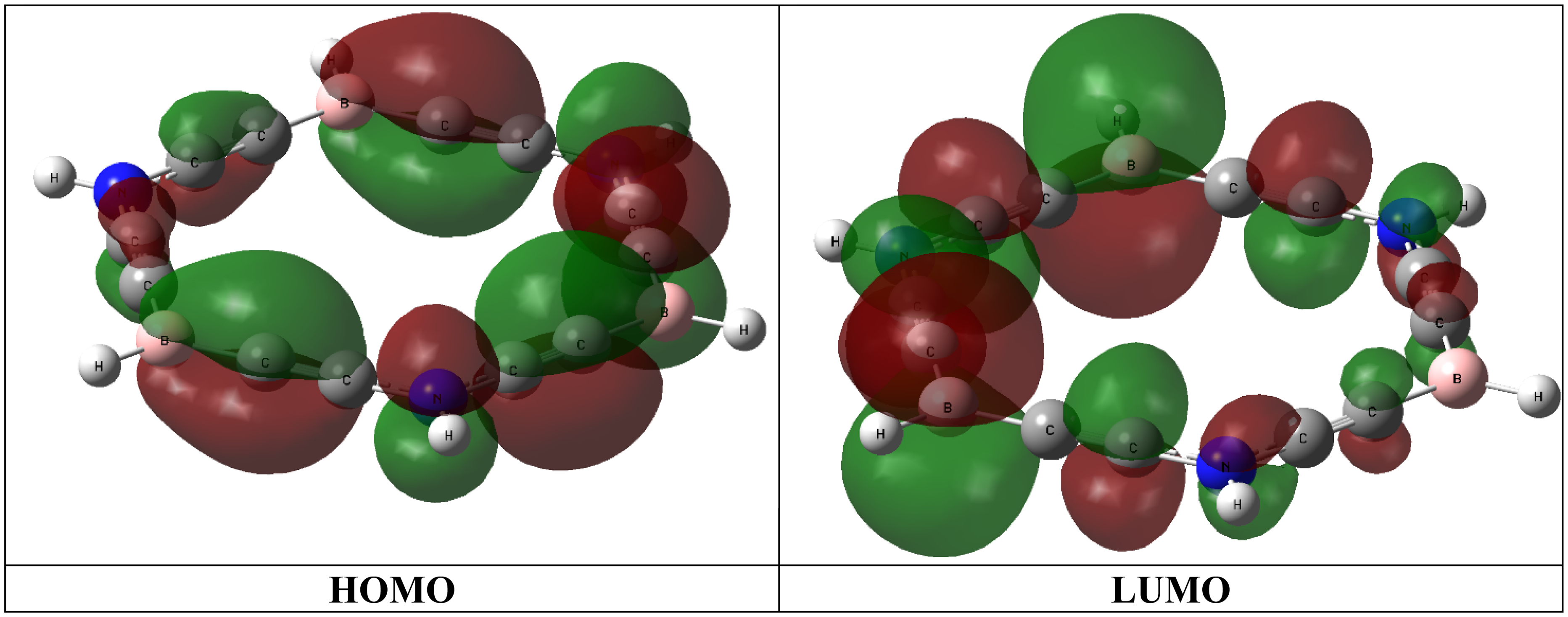
The HOMO and LUMO plots of carbo-borazine.
Calculated λmax values in the fluorinated systems are listed in Table 5. Evaluation of fluorine substituent on the λmax value exposes that red-shift in the N-fluorinated systems. It can be initiate red-shift in the λmax values with increasing of fluorine atom numbers. Then, blue-shift is deduced in the B-fluorinated molecules. A blue-shift in the λmax values with growing of fluorine atom numbers.
Computed λmax, oscillator strength (f), frontier orbital energies (eV) and HOMO-LUMO gap (eV) values of carbo-borazine and fluorinated carbo-borazine molecules at M062X/6-311G(d,p) level of theory.
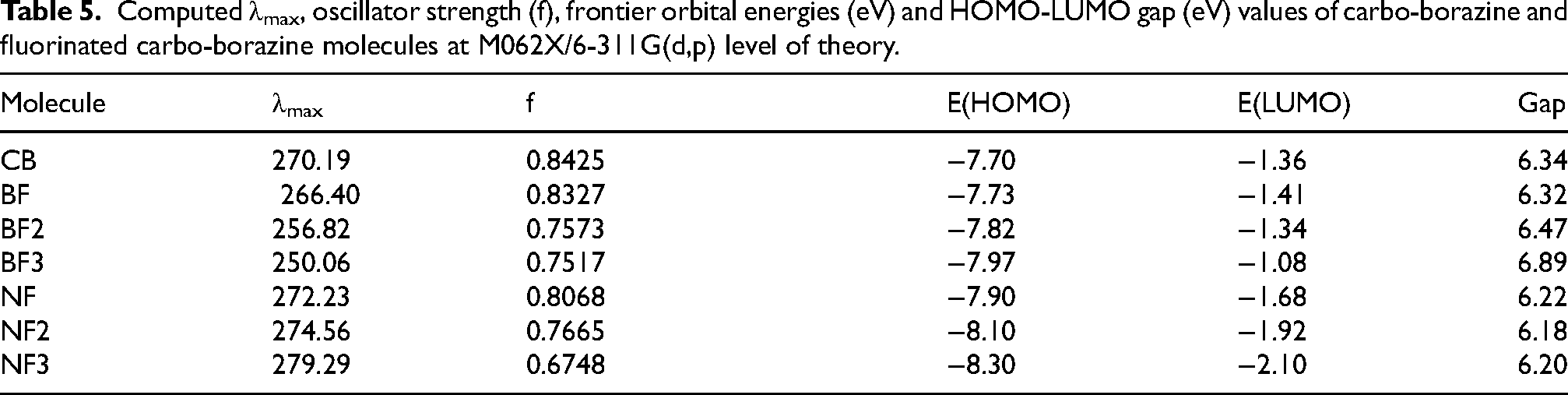
HOMO → LUMO transition has most involvement in S0→S2 transition. Figure 4 shows plots of HOMO and LUMO of carbo-borazine molecule. Molecular orbital analysis exposes that energy gap between two orbitals are increased with increasing of number of F atoms in the B-fluorinated systems. Contrary trend is detected the N-fluorinated systems (Table 5). Hence, λmax values drop with growing of fluorine atom numbers in the B-fluorinated systems. However, higher λmax values are realized with growing of fluorine atom numbers in the N-fluorinated systems.
Conclusion
Computational investigation of fluorine substituent influence on the electronic structure, spectroscopic properties of carbo-borazine molecule at M062X/6-311G(d,p) level of theory indicated B-fluorinated systemswere more stable than N-fluorinated systems, energetically. Larger polarity was established in the N-fluorinated molecules are more stable than
Footnotes
Consent to participate
All the co-authors consent to participate.
Consent to publish
All the co-authors consent to publish.
Author's contribution
Reza Ghiasi: Conceptualization, Data curation, investigation, Formal analysis, writing, review and editing.
Nahid Shajari: methodology, Data curation, writing, review and editing.
Funding
No funds or grants were received.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data generated or analyzed during this study are included in this published article.
Clinical trial number
Not applicable