
Abstract
The Platform Vector-Gene Therapy (PaVe-GT) program is a National Institutes of Health (NIH) initiative that aims to develop adeno-associated virus (AAV) gene therapies for four monogenic rare diseases, two organic acidemias and two congenital myasthenic syndromes. PaVe-GT’s platform-based approach identifies and diminishes redundancies and applies efficiencies in preclinical, clinical, and regulatory activities. The program’s hypothesis is that implementing these efficiencies can accelerate clinical trial initiation. Based on its platform-centric experience and public-serving mission, the PaVe-GT program actively shares its scientific and regulatory learnings with the public to benefit the development of similar gene therapy products for rare diseases. PaVe-GT’s first investigational AAV gene therapy candidate is AAV serotype 9 human propionyl-CoA carboxylase alpha subunit (AAV9-hPCCA) for propionic acidemia caused by PCCA deficiency, which received initial feedback from the Food and Drug Administration (FDA) in an
Keywords
INTRODUCTION
Recombinant adeno-associated virus (AAV)-based gene therapy is a gene replacement strategy in which a functional copy of the mutated gene is introduced into living cells using an AAV vector. In the United States (U.S.), a milestone was achieved in 2017 with the U.S. Food and Drug Administration (FDA) approval of the first AAV gene therapy product, LUXTURNA®, for confirmed biallelic RPE65 mutation-associated retinal dystrophy. 1 Six more AAV-based gene therapies were approved by the FDA for rare diseases by the end of 2024, and more than 200 AAV gene therapy trials for multiple indications are recorded in ClinicalTrials.gov.2,3 Still, with greater than 10,000 rare diseases 4 and only ∼5% of them having an FDA-approved treatment, 5 there is a clear need for accelerated gene therapy development.
The Platform Vector-Gene Therapy (PaVe-GT) program is a National Institutes of Health (NIH) initiative to support the translation of AAV gene therapies for four rare diseases, two organic acidemias and two congenital myasthenic syndromes.
6
The program’s goals are: (1) to determine whether a platform approach can define efficiencies and apply them across preclinical and clinical development to accelerate clinical trial startup and (2) to share with the public the lessons learned from the translation of the four AAV gene therapies. The PaVe-GT shared scientific and regulatory knowledge can also enable and de-risk the development of other gene therapy programs. The first investigational PaVe-GT candidate is AAV9-hPCCA (or NCATS BL-0746), an AAV9 vector carrying a human codon-optimized cDNA of PCCA for the treatment of propionyl-CoA carboxylase alpha subunit (PCCA)-related propionic acidemia (PA). PA is a rare monogenic disease with an estimated incidence of 0.13–1.2 infants per 100,000 births and no causal treatment available.7–9 AAV9-hPCCA received early feedback from the FDA in an
To test an investigational product in humans, a sponsor must, at a minimum, file an IND application with the FDA and obtain a “study may proceed” notification. The pre-IND is a Type B meeting with the FDA that occurs before the submission of an original IND and, compared with an INTERACT meeting, is intended for a more mature stage of translation of the investigational product. 12 A pre-IND meeting is most appropriate when the sponsor has defined the manufacturing process, completed proof-of-concept (POC) pharmacological studies, has an IND-enabling toxicology study plan, and developed a draft clinical study design for the investigational product. In the development of AAV9-hPCCA, we continued the pharmacology, chemistry, manufacturing, and controls (CMC) and clinical protocol design activities, incorporating INTERACT feedback, and then proceeded to a pre-IND meeting. The FDA reviewed the overall preclinical development plans for AAV9-hPCCA at this meeting and responded to our specific Pharmacology/Toxicology, CMC, and clinical questions.
In addition to a pre-IND meeting, a sponsor can have other formal regulatory meetings with the FDA, such as a Type C or Type D meeting for early consultation on targeted topics. 12 In contrast to a pre-IND meeting, multiple Type C or Type D (more narrowly focused) meetings can be requested by the sponsor to gain clarification on post-pre-IND development questions that arise, e.g., preclinical, clinical, and manufacturing. After the pre-IND meeting, we continued the development of AAV9-hPCCA and subsequently obtained valuable clarifications from the FDA on surrogate biomarker endpoints and CMC issues through a Type C meeting.
The goal of this article is to share regulatory lessons learned during the pre-IND and Type C meetings for AAV9-hPCCA, which can be applied to other AAV gene therapy translational programs. Here, we have summarized the process, our questions, and the FDA’s feedback for each of the two regulatory meetings. The meeting requests (MRs), meeting packages (MPs), preliminary responses, and FDA meeting feedback are provided on the PaVe-GT website (https://pave-gt.ncats.nih.gov/). 13
PRE-IND MEETING FOR AAV9-hPCCA
Strategy
After the INTERACT meeting in July 2021,
10
we applied the FDA’s feedback and recommendations to continue development of the investigational product AAV9-hPCCA for PCCA-related PA. We focused on the following IND-enabling activities:
Pharmacology: POC studies using the research-grade or early process development AAV9-hPCCA batches were completed, and an additional non-Good Laboratory Practices (GLP) efficacy and safety study in the PA animal model was planned; CMC: a pilot-scale (50 L) lot of AAV9-hPCCA was manufactured to define the manufacturing process and release specifications for the product; Toxicology: the GLP toxicology study protocol was drafted; Clinical: the clinical protocol synopsis draft for the first-in-human (FIH) Phase 1/2 study was updated based on INTERACT feedback.
Upon completion of these activities, the PaVe-GT drug development team worked with a regulatory Contract Research Organization (CRO) to review all available AAV9-hPCCA information and study results and to strategize the next steps in product translation. The PaVe-GT team included more than 20 scientists with expertise in genomics, AAV vector design, disease animal models, biologics manufacturing, assay development, and Pharmacology/Toxicology studies, clinicians/geneticists specializing in organic acidemias natural history studies (NHS), and project managers (PMs) experienced in translational research and regulatory strategy. The team determined that the FDA’s feedback was needed on Pharmacology/Toxicology, CMC, and clinical issues, especially in the context of rare diseases and creating platform efficiencies. Specifically, we needed FDA’s feedback on: a) our proposed additional non-GLP efficacy and safety protocol, which was a Pharmacology/Toxicology study planned to be performed in the same PA mouse model used in the POC studies and neonate wild-type (WT) controls, and a GLP toxicology study in adult WT mice; b) our proposed manufacturing process for AAV9-hPCCA, product quality attributes, and stability testing plans; and c) the clinical protocol synopsis for testing AAV9-hPCCA in children and adults with PCCA-related PA. Overall, regulatory strategy and the details on goals, readiness, and process are shown in Figure 1.
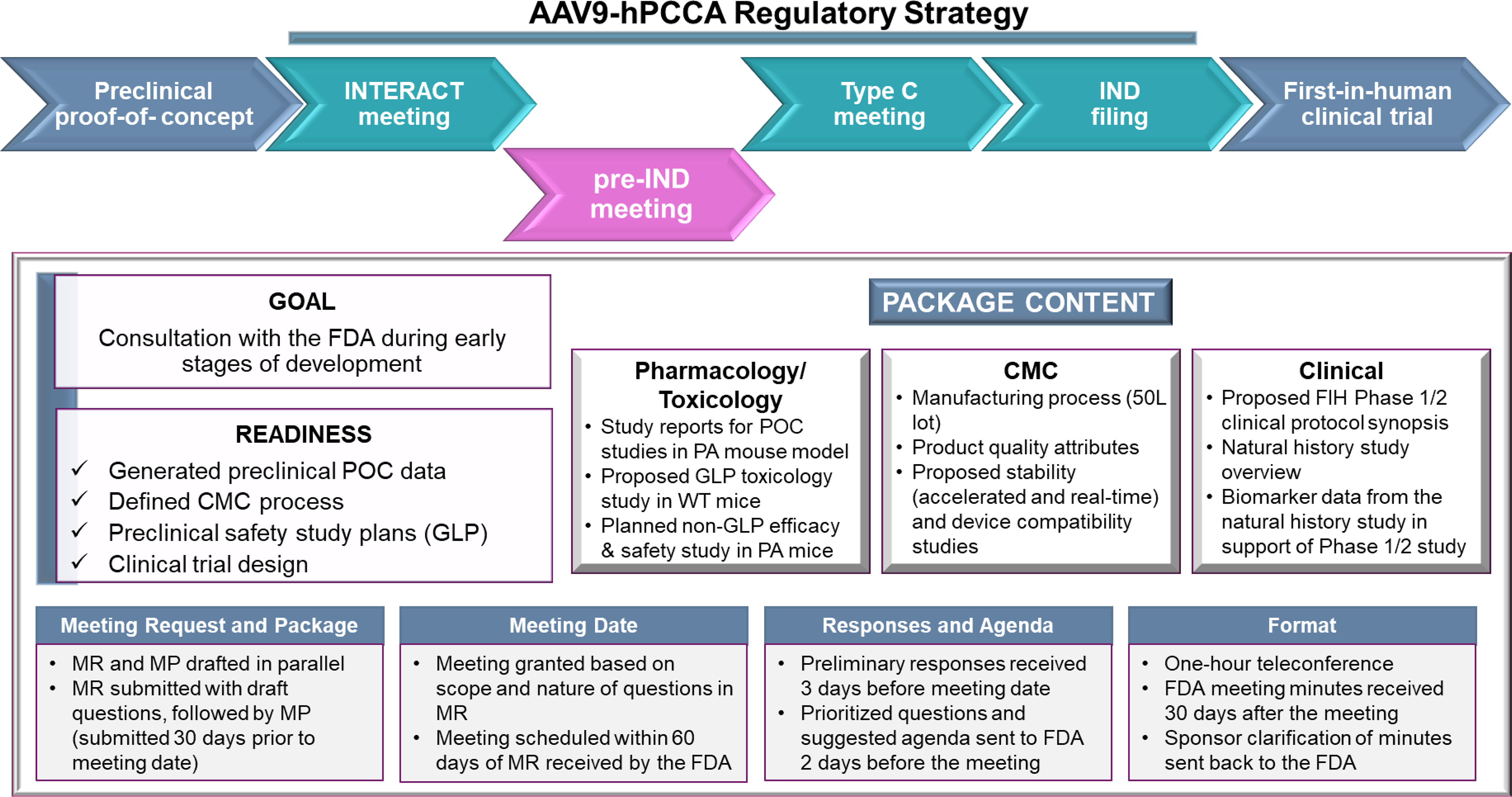
AAV9-hPCCA regulatory strategy, with a focus on the pre-IND meeting goal, readiness, and package content. The regulatory strategy for the first product of PaVe-GT, AAV9-hPCCA, included INTERACT, pre-IND, and Type C meetings prior to IND filing and executing the FIH clinical trial. The pre-IND meeting goal, readiness, and meeting package content are summarized, along with highlights of the pre-IND meeting logistics, process, and timeline. AAV, adeno-associated virus; AAV9-hPCCA, AAV serotype 9 human propionyl-CoA carboxylase alpha subunit; IND, investigational new drug; INTERACT,
Meeting and materials preparation
A small team led by the PMs kicked off the pre-IND meeting preparation by collecting and organizing the documentation on the current AAV9-hPCCA development, followed by data evaluation to determine readiness for the meeting. We then drafted the questions for the FDA based on the needed feedback and created a target schedule for pre-IND–related activities, including MR and MP preparation. Milestones of the pre-IND meeting and documentation preparation process, as they integrate with the AAV9-hPCCA development process, are shown in Figure 2A.
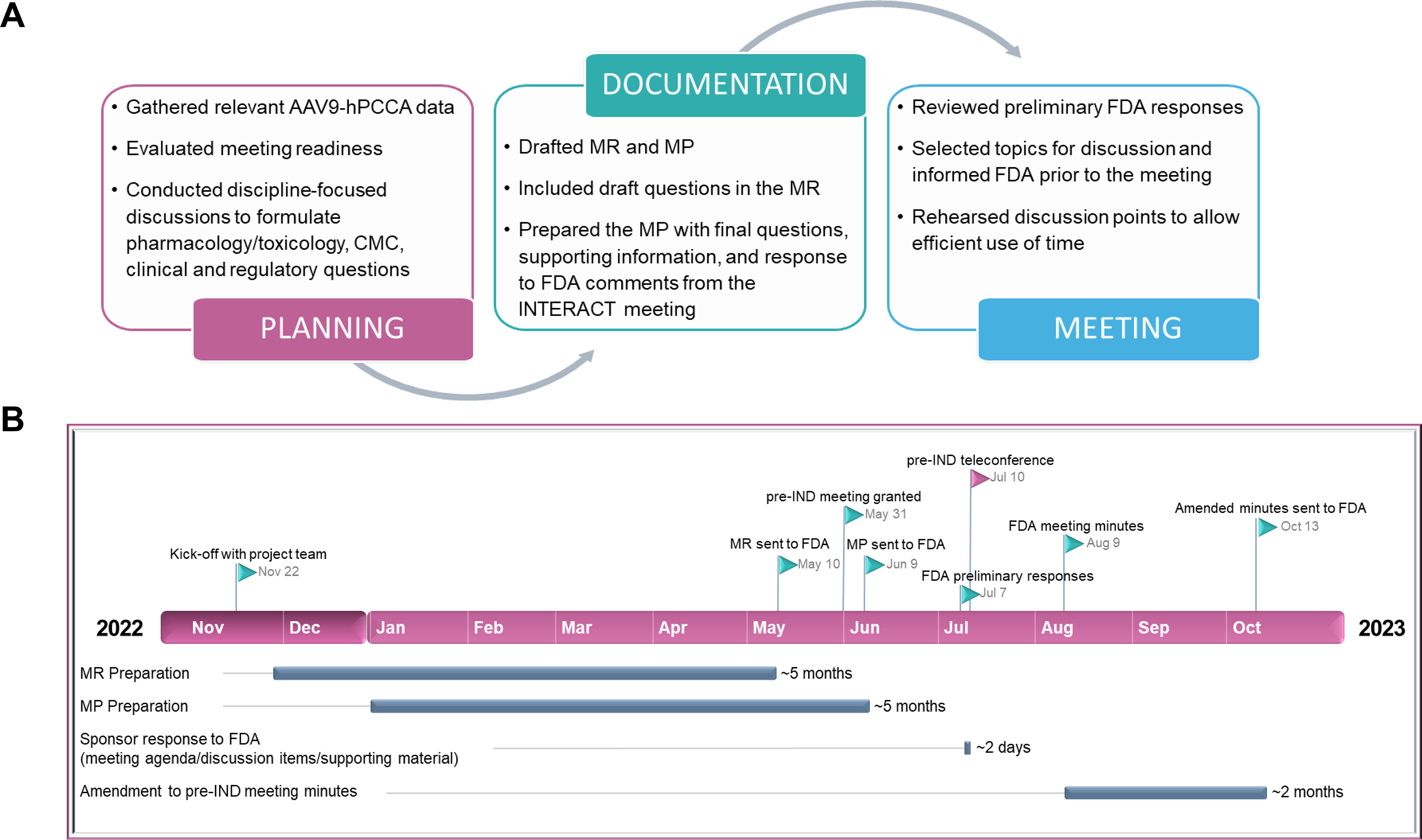
Overview of the pre-IND meeting for AAV9-hPCCA.
To work in parallel on the MR and MP, the PM team was crucial in leading the overall pre-IND preparation process, setting up digital communications, ensuring efficient information sharing across subteams, and organizing all documentation using a team collaboration platform. In the MR for AAV9-hPCCA, we included a brief description of the product, the proposed indication, and planned Pharmacology/Toxicology, CMC, and clinical questions for the FDA. This request (∼10–15 pages) helped the FDA assemble reviewers with subject matter expertise in AAV gene therapies and rare metabolic diseases. Approximately 30 days after the FDA received our MR, they responded, informing us that a meeting (teleconference) was scheduled. The MP prepared was an extensive document (∼300 pages in length), which included comprehensive Pharmacology/Toxicology, CMC, and clinical information (see description in Fig. 1 and summary of MP preparation in Table 1). The MP was submitted on time via the Electronic Submission Gateway to meet the FDA’s deadline of at least 30 days prior to the pre-IND meeting date (see timeline of pre-IND activities in Fig. 2B).
Key Highlights of the Pre-IND MP Preparation for AAV9-hPCCA
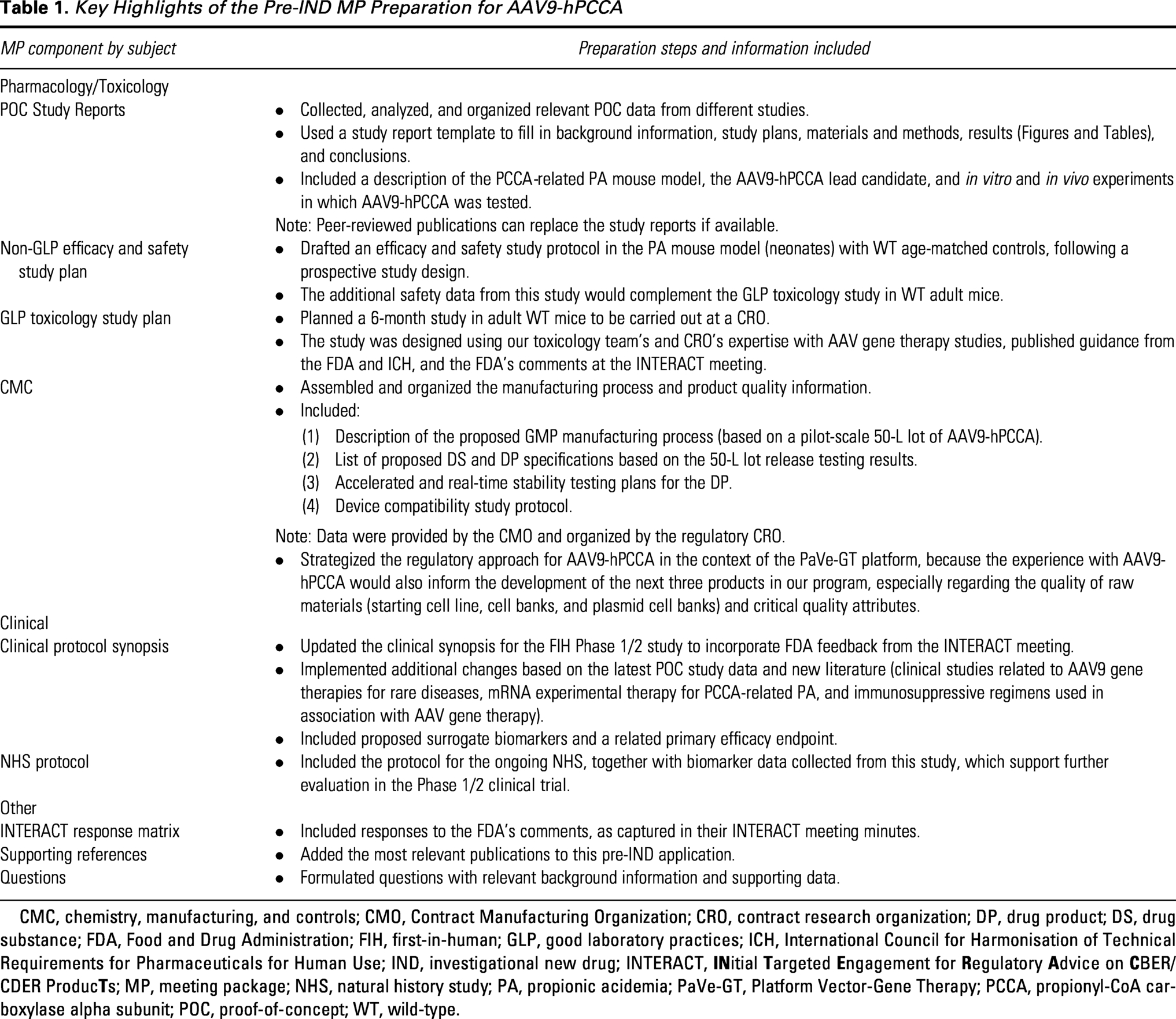
CMC, chemistry, manufacturing, and controls; CMO, Contract Manufacturing Organization; CRO, contract research organization; DP, drug product; DS, drug substance; FDA, Food and Drug Administration; FIH, first-in-human; GLP, good laboratory practices; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; IND, investigational new drug; INTERACT,
Meeting with the FDA
Three days prior to the meeting date, the Agency provided us with preliminary responses to our questions and a sample agenda for the pre-IND meeting. In their responses, the FDA reviewers, who included more than 10 experts in viral vectors, immunology, Pharmacology/Toxicology, and CMC/manufacturing of gene therapies, and clinicians, provided valuable feedback for some aspects of AAV9-hPCCA development not targeted by us in the questions to the Agency. Within hours of receiving these responses, the team reviewed all the feedback and prioritized the top questions for discussion at the meeting. In addition, we identified preliminary responses that could benefit from added clarification from the FDA. For these items, our subject matter experts (SMEs) prepared a slide presentation to present at the actual meeting, ensuring there were no new data or findings that had not been previously included in the MP. Our slide presentation and proposed meeting agenda were then submitted to the FDA at least 24 h prior to the meeting.
The SMEs, two patient advocates, and the PM team participated in the meeting with the FDA. In their opening remarks, the patient advocates shared their experience living with family members with PA and urged the FDA to expedite new therapies for this devastating rare disease. The meeting discussions covered: (1) the suitability of the PA mouse model to support the proposed clinical trial assessments; (2) CMC/drug product (DP) potency assay; (3) residual host cell DNA (rHCD); d) the planned use of data from the proposed non-GLP efficacy and safety study and the GLP toxicology study to inform clinical trial dosing; and (4) the target population for the Phase 1/2 clinical trial (details of preliminary FDA responses and meeting discussions organized into Pharmacology/Toxicology, CMC, and clinical feedback are captured in Table 2A–C). While the pre-IND meeting cannot be recorded, according to FDA rules, we captured meeting minutes that we later compared with the official minutes received from the Agency to ensure sponsor and Agency alignment on all points discussed. The discussions with the FDA reviewers were helpful and led to the clarification of issues raised in the MP and preliminary responses (see highlights in Fig. 3A). The FDA’s position was supportive of the development of AAV9-hPCCA with a line of sight toward longer-term regulatory strategy and the possibility of leveraging the Phase 1/2 study data to support a license application.
Summary of the FDA Feedback and Recommendations from Both the Preliminary Responses and Teleconference Discussions, as Provided in the Final Meeting Minutes

CBER, Center for Biologics Evaluation and Research; CFR, Code of Federal Regulations; cGMP, current good manufacturing practices; CTCAE, Common Terminology Criteria for Adverse Events; FOB, functional observational battery; ICH, International Conference on Harmonisation; IP, investigational product; MC, methylcitrate; NGS, next-generation sequencing; NHS, natural history study; OTAT, Office of Tissues and Advanced Therapies; OTP, Office of Therapeutic Products; PCR, polymerase chain reaction; PDUFA, Prescription Drug User Fee Act; PICC, peripherally inserted central catheter; POBT, 1-13C-propionate oxidation breath test; rHCD, residual host cell DNA; SEND, Standard for the Exchange of Nonclinical Data; WHO, World Health Organization.

FDA highlights provided during early AAV9-hPCCA regulatory review.
Meeting minutes and post-pre-IND activities
Thirty days after the pre-IND meeting, we received the official meeting minutes from the FDA, which also included a list of templated information applicable to the Pharmacology/Toxicology and clinical sections, with generic regulatory advice and references to applicable FDA guidance (captured in Table 2A–C). In comparing our minutes with the FDA’s minutes, we identified an area related to the target population for clinical testing that required an amendment. We informed the FDA about our understanding of the final discussions regarding the first patient enrolled in each clinical trial cohort, which differed from what was captured in the Agency’s minutes. While this is not a compulsory action, our goal was to ensure that the clinical enrollment aspect was clear, as it has an impact on the performance of the clinical trial. The meeting minutes will guide the rest of the IND-enabling studies for AAV9-hPCCA, the regulatory strategy beyond the Phase 1/2 clinical trial, and the development of the other AAV gene therapies in the PaVe-GT platform.
TYPE C MEETING FOR AAV9-HPCCA
IND-enabling activities undertaken after the pre-IND meeting and continuing review of the recommendations made by the FDA led us to uncover CMC and clinical topics for which we needed further input from the Agency prior to IND filing. Type C meetings enable sponsors to have early consultation with the Agency on a few specific topics related to product review or development. Thus, we decided to engage the FDA in a Type C meeting as part of our regulatory strategy for AAV9-hPCCA.
Strategy
During the pre-IND meeting, the level of rHCD in the 50-L pilot-scale lot of AAV9-hPCCA was determined to be high relative to the World Health Organization (WHO)-recommended limit of 10 ng/dose. 23 Thus, we made adjustments to the manufacturing process to lower the rHCD level in the 200-L engineering lot of AAV9-hPCCA. In addition, we prepared a risk mitigation strategy for addressing the rHCD levels relative to the WHO limit. The assay to evaluate product potency was also discussed at the pre-IND meeting, and based on FDA recommendations, we planned a matrix potency-testing approach that would include the quantification of PCCA mRNA by PCR and of PCCA mutant variants via Next Generation Sequencing (NGS).
After the pre-IND meeting, the clinical team, which included clinical research investigators, geneticists, and nursing staff, continued to collect data on several biomarkers as part of the NHS of PA (NCT02890342) conducted at the NIH Clinical Center. 24 Data analysis showed a statistically significant correlation between the 1-13C-propionate oxidation breath test (POBT), metabolite levels, and clinical outcome measures. This finding led to proposed changes in the Phase 1/2 study design in which dose escalation would be dictated by safety measurements, with changes in POBT and metabolite levels representing secondary endpoints, not primary endpoints, as initially proposed prior to receiving pre-IND feedback. Their use as surrogate biomarkers for efficacy endpoints in a future clinical trial may be possible based on cumulative data from the natural history and Phase 1/2 studies.
To further de-risk AAV9-hPCCA development, we wanted to obtain FDA’s feedback on the risk assessment and approach to lower rHCD in the DP, the plans for testing and qualifying the planned matrix potency assay for AAV9-hPCCA, and the utilization of changes in POBT and metabolite levels in the Phase 1/2 clinical trial (Fig. 4A). The team decided that this could be done through a Type C meeting with the FDA. After evaluating readiness for the Type C meeting, the PM team led the CMC, clinical, and regulatory subteams to collect, organize, and analyze the data supporting the topics and to formulate the questions for the FDA.
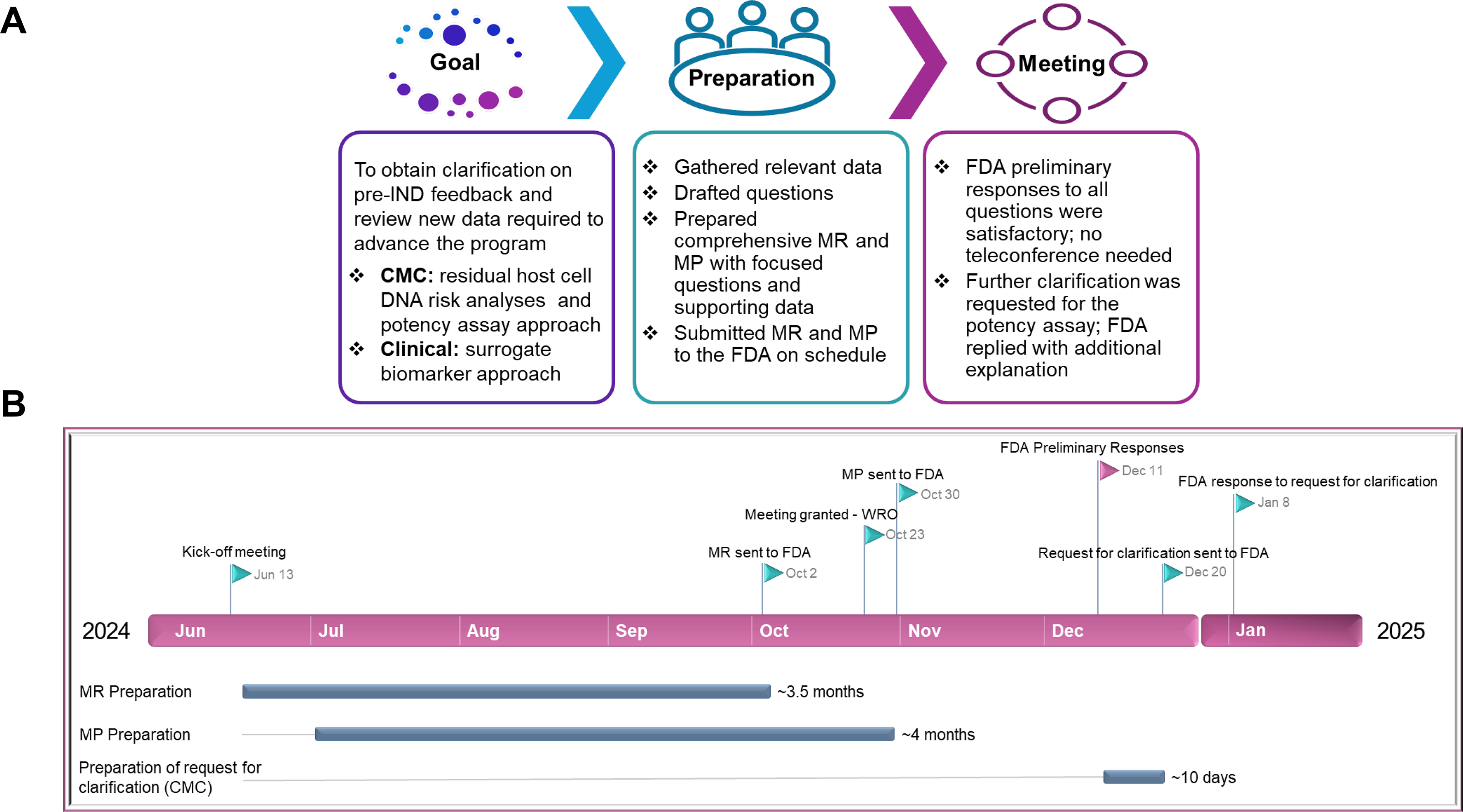
Overview of the Type C meeting for AAV9-hPCCA.
Meeting and materials preparation
Similar to the pre-IND meeting, the Type C meeting was requested by submitting an MR (10 pages), which included background and related questions for the FDA. The regulatory SMEs ensured that adequate data were shared with the FDA to allow the Agency to answer our questions. After the FDA responded to the MR in 21 days by initially granting us written responses only (WRO) (the timeline for all Type C meeting steps is shown in Fig. 4B), we submitted the MP (∼105 pages) 47 days prior to the meeting date and requested conversion of the WRO to a meeting to allow a more definitive dialogue about the surrogate biomarkers. The FDA approved our request.
Preliminary responses and meeting-turned WRO
Five days prior to the meeting date, the FDA provided preliminary responses to our questions. The reviewers agreed with our risk assessment and reporting of rHCD levels for AAV9-hPCCA lots and concurred with our proposed matrix approach to assess product potency using quantification methods for PCCA mRNA expression and PCCA mutant variants. The FDA reiterated its recommendation from the pre-IND meeting to demonstrate the suitability of the NGS method for quantitative analysis of sequence variants (Fig. 3B; details in Table 3) and to set up the assay by spiking mutated sequences in the test sample. With respect to biomarkers, the FDA agreed with our: a) proposed approach to evaluate changes in metabolite levels and POBT in the Phase 1/2 study to assess pharmacodynamic responses to AAV9-hPCCA treatment; and b) updated clinical study design. The FDA offered to cancel the virtual Type C meeting if we found their responses and advice clear and complete, thereby obviating the need for further discussion. We agreed and informed the Agency of our decision to cancel the meeting. However, subsequently, we found it challenging to implement the FDA’s advice to “spike” the test sample with mutant sequences to develop a quantitative NGS assay that can accurately and reproducibly measure the levels of variants in the drug substance (DS) as part of the potency assay. Thus, we submitted an official written request for clarification (two pages) of the spiking method, and the Agency responded in writing within 3 weeks (four pages) restating the need for a spiking study to assess the sensitivity of the NGS assay. We found the communication with the FDA valuable for preparing our IND application for AAV9-hPCCA and planning of the for the remaining three AAV gene therapy products in the PaVe-GT program.
Type C Meeting Questions and FDA Feedback

After submission of the Type C meeting package, the FDA shared preliminary responses, which the sponsor found satisfactory, and thus, the teleconference was canceled. However, we requested clarification regarding the potency assay from the FDA (refer to Fig. 3B). The following information is a summary of the FDA’s feedback and recommendations from both the preliminary responses and follow-up conversations.
AAV, adeno-associated virus; HCD, host cell DNA; PD, pharmacodynamic.
DISCUSSION
For the first product of the PaVe-GT program, AAV9-hPCCA, we published the experience and materials from the first regulatory interaction with the FDA at the INTERACT meeting 10 and committed to sharing the experience from subsequent FDA interactions, including future IND documentation. By sharing the approach and blueprint of our regulatory experience and application packages, we hope to assist other stakeholders in developing AAV gene therapies for rare diseases. Therefore, we summarize here our pre-IND and Type C meeting experience for AAV9-hPCCA, including goals, process, key feedback, and “dos” and “don’ts” (Fig. 5).
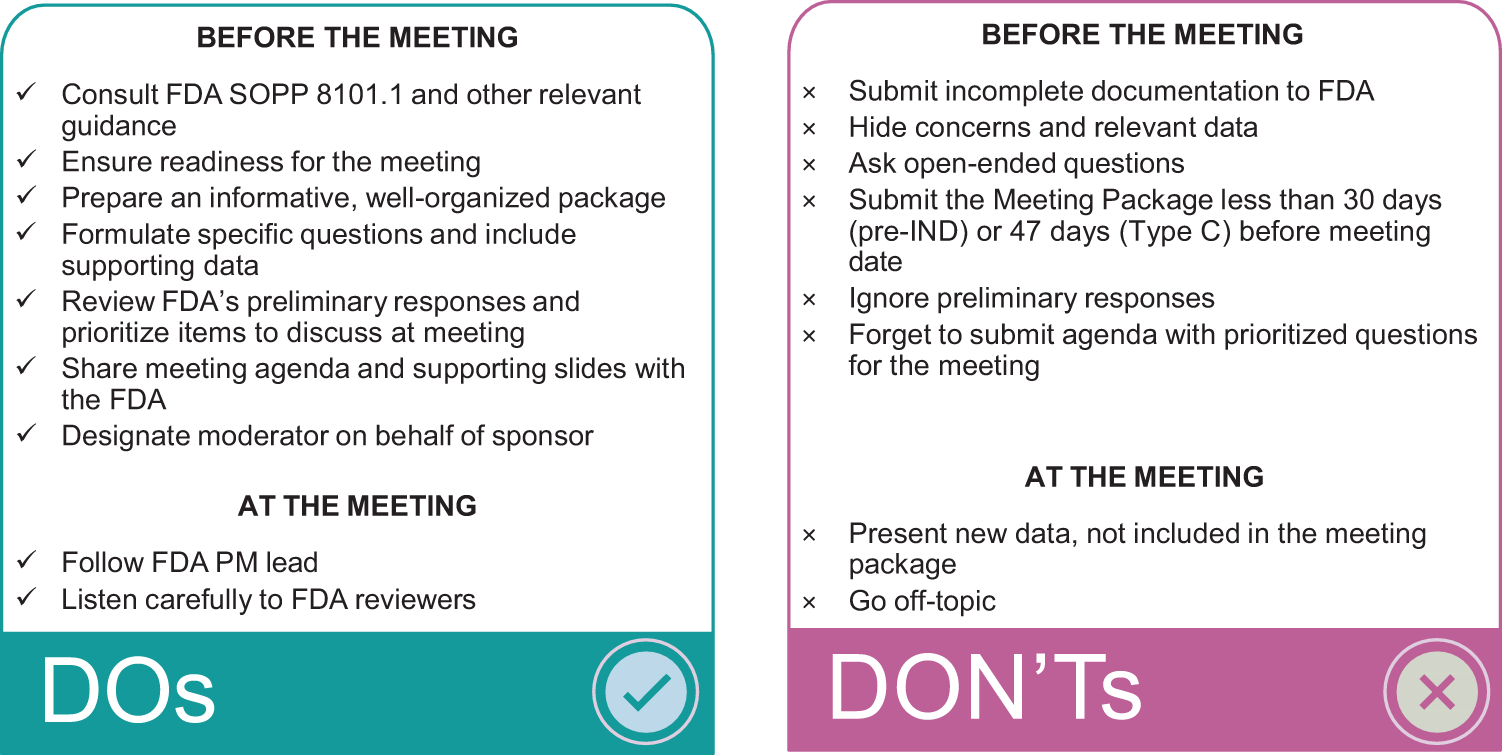
“Dos” and “Don’ts” to consider in preparing for pre-IND and Type C meetings with the FDA.
At the pre-IND meeting, the FDA reviewers provided advice on the Pharmacology/Toxicology aspects of AAV9-hPCCA development (Fig. 3A). We were initially encouraged to use a less severe disease model of PCCA-related PA in the additional non-GLP safety and efficacy study to enable better assessments of changes in biomarkers compared with untreated PA mice. However, after further discussions, we were able to convince the FDA reviewers that a severe disease mouse model was closer to the clinical scenario, since the risk:benefit ratio did not favor testing of AAV9-hPCCA in patients with mild or intermediate PA. Ideal candidates for participation in our Phase 1/2 study of AAV9-hPCCA are moderate-to-severe patients with PCCA-related PA who do not yet have irreversible organ damage (e.g., cardiac, renal). With respect to Pharmacology/Toxicology studies, the FDA agreed that for our program, safety data from a well-designed GLP toxicology study in WT adult mice and the non-GLP safety and efficacy study in the neonatal lethal PA mouse model could satisfy the requirements for preclinical safety data in support of a FIH Phase 1/2 study.
Regarding CMC aspects of AAV9-hPCCA development, some FDA-provided feedback focused on rHCD content and potency testing. After adapting the CMC process to reduce rHCD and drafting a risk mitigation plan, we shared this information with the FDA at the Type C meeting and received FDA concurrence on our approach. Based on the pre-IND and Type C meeting interactions, we plan to include in the IND: a) a justification for the proposed limit of rHCD content in the DP, which is anticipated to be higher than the WHO-recommended limit of 10 ng per dose, 23 b) a comprehensive rHCD risk assessment plan, and c) plans for manufacturing process optimization to reduce the level of rHCD in the investigational product.
A major lesson learned from the two regulatory meetings with the FDA was that we had to strategize early clinical development with the final goal in mind, i.e., to leverage the proposed Phase 1/2 clinical study data to support a marketing application. Recognizing the regulatory challenges of drug development for very small patient populations, the FDA advised us that CMC information submitted to support the clinical study should be appropriate for late-stage clinical development. This includes the early development of a potency assay applicable up to the submission of a marketing application, collecting quality data to tighten acceptance criteria for quality attributes, and ensuring that product quality data are appropriate for late-stage clinical development. Thus, we plan to develop a quantitative potency assay that best enables lot comparison throughout the lifecycle of AAV9-hPCCA development. The recommendation offered by the FDA at the pre-IND meeting and later clarified at the Type C meeting was to use a matrix approach in which an NGS assay would quantify mutant variants of PCCA in the DS and a quantitative mRNA assay for PCCA in the DP.
Regarding the clinical aspects of AAV9-hPCCA development, we learned from the pre-IND and Type C meetings that our preliminary data on biomarker response (POBT and metabolite levels) from participants in our NHS were insufficient to support their use as surrogate endpoints for efficacy at this early translational stage. However, we hope that ongoing data collection from the NHS and the Phase 1/2 study will support the use of POBT and other metabolite levels as surrogate biomarkers for efficacy in a future clinical trial. Other valuable clinical feedback obtained from these early regulatory interactions included the FDA’s alignment that: (1) a randomized, placebo-controlled clinical trial is not feasible for a very limited patient population such as that of PCCA-related PA and for a high-risk intervention such as gene therapy; instead, the use of an external control for the Phase 1/2 study, based on the NHS, was recommended; (2) the investigational product must be tested first in an adult or adolescent patient before it is tested in children, but if no such adult/adolescent participant is identified, the study can proceed with enrollment of children; and (3) patients with moderate-to-severe disease are the most suitable candidates for enrollment in the Phase 1/2 study. We consider the FDA’s feedback to be specific to the product and based on the sponsor’s submitted data and its unique challenges. Therefore, we ensured that all relevant information and questions on AAV9-hPCCA were included in the meeting packages.
For AAV9-hPCCA, the FDA’s feedback during this early regulatory process (Fig. 3 and Table 2), including feedback related to Pharmacology/Toxicology testing in a single species, product quality improvements, and FIH Phase 1/2 clinical study design, is leading to more efficient development of the other three PaVe-GT candidates and their regulatory strategies. For example, we plan to use a similar manufacturing process, critical quality attributes (with some improvements, as applicable), and an analogous matrix-based potency assay for the next PaVe-GT candidate, AAV9-metabolism of cobalamin associated B. This product will be tested in cblB-type methylmalonic acidemia, an indication similar to PA. Although intellectual property, inventions, and similar protections prevent the free circulation of information among potential gene therapy developers for rare diseases, and gene therapy development is an expensive and high-risk venture, our mission of disseminating PaVe-GT learnings and materials may help overcome some of the scientific and regulatory barriers. The public’s free use of our templates and examples could save resources, educate gene therapy developers, and improve access to information and healthy competition. Upcoming dissemination plans include publication of a PaVe-GT roadmap consisting of the preclinical and regulatory process, ODD, RPDD, and INTERACT templates and examples (already individually published and shared on the PaVe-GT website), as well as the pre-IND and Type C meeting templates for MR and MP, and examples of PaVe-GT regulatory documents. In the future, the PaVe-GT roadmap will also include the IND documentation. To complete the package of PaVe-GT dissemination resources, we plan to publish POC, CMC, and clinical development experiences, and the overall efficiencies from the AAV platform. This effort can also fulfill the broader goal of PaVe-GT to deliver a standardized, platformed, and scalable vector approach that could lead to more efficient AAV-based gene therapies across rare diseases.
AUTHORS’ CONTRIBUTIONS
R.S., R.M.L., and E.A.O. contributed to the conceptualization, methodology, and writing of the initial draft, reviewing, creating figures, and editing the article. The pre-IND and Type C meeting packages and requests used in this article are based on the original research of E.-Y.C., L.L., R.J.C., O.A.S., I.M., C.I.G.-A., J.L.S., and C.P.V., including design and conceptualization of AAV9-hPCCA, creation of the mouse model of PA, demonstration of preclinical efficacy in animal models, clinical research, and biomarker analysis. R.S., R.M.L., O.A.S., I.M., C.I.G.-A., V.M., E.-Y.C., R.J.C., L.L., J.L.S., P.T., X.X., J.D., P.J.B., C.G.B., C.P.V., and E.A.O. contributed to the gene therapy development activities as part of the PaVe-GT team and reviewed and edited the article.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to the patient advocates from the Organic Acidemia Association and the Propionic Acidemia Foundation for their contribution to the pre-IND meeting. The authors would like to thank Anjali Sarkar for her support in preparing the templates and examples shared on the PaVe-GT website. The authors also thank past and present members of the entire NIH PaVe-GT Team (in alphabetical order): Krishna Balakrishnan, Eggerton Campbell, Catherine Chen, Oksana Dukhanina, Malar Durai, Susan Ferry, Rahul Nandre, Asvelt Nduwumwami, London Toney, Carol Van Ryzin, Sury Vepa, Erik Wagner, and Amy Wang.
DISCLAIMER
This research was supported in part by the Intramural Research Program of the National Institutes of Health (NIH). The contributions of the NIH authors were made as part of their official duties as NIH federal employees, are in compliance with agency policy requirements, and are considered Works of the United States Government. However, the findings and conclusions presented in this article are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services.
AUTHOR DISCLOSURE STATEMENT
The authors have no competing interests relevant to this article.
FUNDING INFORMATION
PaVe-GT is funded by the NCATS Cures Acceleration Network and the Intramural Research Programs of NCATS, NHGRI, and NINDS. R.S., R.M.L., V.M., P.T., X.X., and E.A.O. are supported by the NCATS Intramural Research Program (1ZIATR00281). E.-Y.C., R.J.C., O.A.S., L.L., I.M., J.L.S., C.I.G.-A., and C.P.V. were supported by the Intramural Research Program of the NHGRI (1ZIAHG200318-21). C.G.B. is supported by the NINDS Intramural Research Program.