
Abstract
Primary hyperoxaluria type 1 (PH1) is a rare autosomal recessive disorder that leads to kidney and liver failure. PH1 is caused by a mutation in the alanine glyoxylate aminotransferase (AGXT) gene, which encodes a key metabolic enzyme that converts glyoxylate to glycine in the liver. Inability to metabolize glyoxylate leads to oxalate overproduction, yielding insoluble calcium oxalate crystals; accumulation of these crystals leads to progressive organ failure. Here, we used a novel, minimally disruptive genome-editing approach to disrupt the mechanism of action of hydroxyacid oxidase 1 (HAO1), an upstream enzyme in the glyoxylate metabolic pathway. Successful gene editing and disruption of the HAO1 gene is expected to increase levels of glycolate, a harmless intermediate of the glycine metabolic pathway, thereby preventing the formation of calcium oxalate crystals. We intravenously administered an adeno-associated virus (AAV) vector expressing the M1HAO1 meganuclease to both wild-type and Agxt−/− mice, a mouse model of PH1. We observed >30% editing of HAO1 in Agxt−/− mice, correlating with a dose-dependent increase in serum glycolate levels. At the highest dose tested, urine glycolate levels increased by 79%, with a concomitant 75% decrease in urine oxalate levels. We also evaluated in vivo targeting in rhesus macaques injected with AAV expressing two different versions of the HAO1 meganuclease. Dose-dependent editing of hepatic DNA and RNA was achieved, and serum glycolate levels changed in a manner consistent with successful liver editing; additionally, the treatment was well tolerated. Our results indicate that AAV-delivered meganucleases can effectively target HAO1 in mice and nonhuman primates to achieve high levels of HAO1 gene editing. Moreover, increased glycolate levels in serum indicate that this intervention significantly impacts the HAO1-mediated glycolate-to-glyoxylate pathway. These data suggest that this approach may represent an effective treatment for PH1.
Keywords
INTRODUCTION
Primary hyperoxaluria type 1 (PH1) is a rare genetic disease caused by mutations in the alanine glyoxylate aminotransferase (AGXT) gene, 1 which encodes alanine-glyoxylate aminotransferase, a key enzyme responsible for converting glyoxylate to glycine within peroxisomes in the liver (Supplementary Fig. S1). The low level of oxalate produced by normal metabolism is continuously removed from the blood by the kidneys and excreted in the urine. However, the inability to metabolize glyoxylate leads to oxalate overproduction, which yields insoluble calcium oxalate crystals; the accumulation of these crystals leads to kidney stones, nephrocalcinosis, kidney failure, and systemic oxalosis.2–4
Supportive treatment for PH1 involves using crystallization inhibitors in combination with hyperhydration. 5 Dialysis can also be used to treat kidney dysfunction. As AGXT is primarily synthesized in the liver, liver transplantation can restore glyoxylate detoxification capabilities. Liver transplantation can be performed alone or concomitant with kidney transplantation (either sequentially or simultaneously), depending on the damage accrued in the kidney. However, organ transplantation requires immunosuppression to prevent rejection, where potential allograft dysfunction carries significant long-term risks. Due to a lack of treatment options, there is a high unmet need for the development of new therapeutic approaches for PH1.
A gene therapy approach involving expression of alanine-glyoxylate aminotransferase has been evaluated for the treatment of PH1. 6 However, this strategy would only achieve short-term effects due to the loss of vector genomes during hepatocyte proliferation, meaning it would be limited to the adult PH1 population and not infants with rapidly growing livers.
Two gene knockdown approaches have recently received approval or are in ongoing clinical trials for the treatment of PH1, lumasiran and nedosiran, respectively. These drugs target different genes through different mechanisms of action but attempt to reduce urinary oxalate in PH1 patients. However, these drugs require regular readministration. Lumasiran is an RNA interference (RNAi) therapy that binds to glycolate oxidase (also known as hydroxyacid oxidase, hydroxyacid oxidase 1 [HAO1]) RNA and targets it for degradation; this treatment requires monthly or quarterly subcutaneous injections following an initial loading phase involving three monthly doses. 7 Nedosiran is a small-interfering RNA (siRNA) that targets lactate dehydrogenase (LDH) RNA for degradation to reduce the production of oxalate and is administered monthly via subcutaneous injection. 8 Both therapies are associated with injection site reactions. In contrast to these treatments, a gene-editing approach may represent an effective one-time treatment.
Others have explored gene-editing approaches with clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9-based strategies. Like the gene knockdown approaches using RNAi or siRNA, these gene-editing approaches have targeted either HAO1 or LDH. Researchers have shown that a single systemic administration of an AAV8-CRISPR/Cas9 vector targeting either HAO1 9 or LDH 10 can prevent oxalate overproduction and kidney damage in Agxt1−/− mice. However, both of these approaches utilized Staphylococcus aureus Cas9, which has reduced translatability from mice to primates.
We previously evaluated an AAV vector-delivered, liver-directed meganuclease for in vivo gene editing of the pro-protein convertase subtilisin/kexin type 9 (PCSK9) gene11–13 and transthyretin (TTR) gene. 14 This approach enables the generation of a double-stranded break within a specific 22-bp sequence in the gene of interest, thereby allowing endogenous DNA repair mechanisms to generate insertions or deletions (indels) at the cleavage site that disrupt PCSK9 or TTR protein expression, respectively.
Here, we evaluated the ability of an AAV-delivered meganuclease targeted to HAO1 to disrupt the metabolic pathway leading to glyoxylate production. Successful gene editing and disruption of HAO1 were expected to result in increased levels of glycolate, a soluble intermediate of the glycine metabolic pathway,15,16 thereby preventing the formation of calcium oxalate crystals (Supplementary Fig. S1). We tested two engineered meganucleases, each having a unique target site within the HAO1 gene, in mice and rhesus macaques. Physiological analysis of a first-generation M1HAO nuclease in a mouse model of PH1 (Agxt−/− mice) produced significant differences in serum glycolate and urine oxalate levels, with reduced localization of HAO1 to the peroxisome. A second-generation nuclease, M2HAO, produced up to 72% indels in HAO1 RNA in rhesus macaques. This significant editing potential indicates that AAV delivery of an HAO1-specific nuclease may represent an effective one-time treatment strategy for PH1.
MATERIALS AND METHODS
Note that many techniques outlined below were previously described in Breton et al. 13 and involve standardized procedures and workflows that were modified for this specific study.
AAV vector production
All AAV vectors were produced by the Penn Vector Core at the University of Pennsylvania as previously described. 17 Briefly, plasmids expressing either M1HAO or M2HAO meganucleases from the thyroxine-binding globulin (TBG) promoter, including the WPRE sequence, were packaged within the AAV8 capsid. The M1HAO meganuclease targets a site in exon 8 within the HAO1 gene that is conserved in mice, nonhuman primates (NHPs), and humans, whereas the M2HAO nuclease targets a site in HAO1 exon 1 that is conserved in NHPs and humans but is not present in the mouse genome.
Mice
All animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. An Agxt−/− mouse strain was generated at The Jackson Laboratory (Bar Harbor, ME) by CRISPR/Cas9 gene editing of exon 1 of the mouse Agxt gene. A homozygous Agxt−/− breeding colony was maintained at the University of Pennsylvania under specific pathogen-free conditions. Adult male Agxt−/− mice received an intravenous (IV) injection with 3 × 1011–3 × 1013 genome copies (GC)/kg of AAV8.TBG.M1HAO via the tail vein or vehicle (phosphate-buffered saline, PBS) on day 0. While PH1 affects males and females equally, only male mice were evaluated in this study to enable maintenance of the Agxt−/− mouse breeding colony. We monitored the mice for serum and urine glycolate and oxalate levels throughout the in-life phase of the study. Mice were necropsied on day 63.
Male Rag1−/− mice were purchased from The Jackson Laboratory and received an IV injection of 3 × 1012 GC/kg of AAV8.TBG.hHAO1native or vehicle on day 0. On day 14, mice received an IV injection of 3 × 1010–3 × 1012 GC/kg of AAV8.TBG.M1HAO or AAV8.TBG.M2HAO. Mice were necropsied on day 42 post-meganuclease vector administration (day 56 of the study).
Rhesus macaques
Wild-type rhesus macaques aged 2–6 years old (n = 8 for four groups, one male and one female per group) were obtained from Covance (Princeton, NJ). NHP studies were conducted at the University of Pennsylvania within a facility that is registered with the United States Department of Agriculture, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and assured by the Public Health Service. As previously described, 18 animals were housed in stainless steel cages with perches. All cage sizes and housing conditions were in compliance with the Guide for the Care and Use of Laboratory Animals. A 12-h light/dark cycle was maintained and controlled via an Edstrom Watchdog system. Animals were fed Certified Primate Diet 5048 (PMI Feeds, Inc., Brentwood, MO, USA) two times per day (morning and evening). An additional variety of food treats that were fit for human consumption, including fruits, vegetables, nuts, and cereals, were given daily as part of the standard enrichment process. Manipulanda such as kongs, mirrors, a puzzle feeder, and raisin balls were provided daily. Animals also received visual enrichment along with human interaction on a daily basis. All interventions were performed during the light cycle, and animals were fasted overnight prior to being anesthetized.
On study day 0, macaques received 10 mL of vector into the saphenous vein at a rate of 1 mL/min via an infusion pump (Harvard Apparatus, Holliston, MA). Two macaques each received a dose of 6 × 1012 GC/kg or 3 × 1013 GC/kg of AAV8.TBG.M1HAO or AAV8.TBG.M2HAO (n = 2/dose/vector). All NHPs received oral prednisolone at a dose of 1 mg/kg/day from the day of vector administration through weeks 8–12, after which animals were tapered off prednisolone by a gradual reduction of the daily dose.
NHP analyses during the in-life phase
We anesthetized rhesus macaques and collected blood on selected days via the femoral vein. Antech (Irvine, CA) subsequently performed complete blood counts, clinical chemistries, and coagulation panels on the blood samples. Neutralizing antibody titers were determined on serum samples taken prior to initiation of the study, as previously described.19,20 We also screened the macaques for the presence of the meganuclease target sites prior to vector administration on DNA extracted from peripheral blood mononuclear cells.
NHP liver biopsy
We performed mini laparotomy procedures on days 18 and 128 post-vector administration to isolate liver tissue, as previously described. 14
Serum and urine analyses
Blood was collected from mice and NHPs in serum separator tubes and allowed to clot, and serum was isolated. Urine was also collected from mice and NHPs at certain time points. Both serum and urine were analyzed for glycolate and oxalate levels by gas chromatography–mass spectrometry (GC–MS) at the Children’s Hospital of Philadelphia Metabolomics Core. Briefly, samples were prepared as follows: 50 μL of serum or 25 μL of urine was spiked with a precise amount of 13C-labeled oxalic and glycolic acids. Samples were then applied to an AG1 column and washed extensively with water, and the oxalic and glycolic acids were eluted with 3 mL of 3 N HCl. The eluate was placed in 4-mL vials and completely dried at 60°C in a stream of air. Then, 50 μL of acetonitrile and 50 μL of MTBSTFA (derivative reagent) were added to a dried sample, which was heated for 30 min at 60°C.
Samples were analyzed via GC–MS (Agilent Intuvo 9000 GC system and Agilent 5977B mass spectrometer). Glycolic and oxalic acids were separated on an HP-5MS capillary column (30 m × 250 mm × 0.25 μm) with the following temperature regime: initial temperature of 60°C—hold for 3 min; ramp 1—increase by 5°C/min until 120°C is reached; ramp 2—increase by 30°C/min until 300°C is reached and hold at 300°C for 1 min. The total run time was 22 min. The retention time was 18.1 min for glycolic acid and 18.35 min for oxalic acid. Mass spectrometry was run in single-ion monitoring mode; the ions for following glycolic acid are 247, 248, 249, and 250, and the ions for following oxalic acid are 261, 262, 263, and 264.
The oxalic and glycolic levels in urine or serum samples were determined by a modification of the conventional isotope dilution technique. The initial enrichment of 13C (APE) is 99APE in oxalic or glycolic acid (I1); serum or urine samples were spiked with a known amount (pmol) of 13C-labeled oxalic or glycolic acid (D1). Samples were then passed through an ion exchange, dried, and derivatized as outlined above. The isotopic abundance (I2) was then measured for each sample. The concentration of oxalic or glycolic acid in the sample (Cx) was calculated as follows: Cx = D1 × (I1/I2 − 1).
Next-generation sequencing for on- and off-target analysis
For on-target analysis of DNA extracted from mouse liver samples, we evaluated the percentage of indels in the region of interest using amplicon sequencing (amplicon-seq) as previously described.11–14 Briefly, the region of interest within HAO1 exon 8 for M1HAO and exon 1 for M2HAO was amplified by polymerase chain reaction (PCR). We then generated next-generation sequencing libraries from the PCR product and sequenced them on a MiSeq instrument (Illumina, San Diego, CA). These sequences were then mapped to a reference genome (Assembly GRCm38.p6). Using a custom script, we quantified unedited reads and reads containing indels.11–13 We performed the same analysis for RNA, which was reverse-transcribed to complementary DNA using random primers.
For NHP samples, we quantified AAV integration and translocations in addition to indels by AMP-seq.11–14,21 We sequenced the libraries on a MiSeq instrument and mapped the resulting sequences to a reference genome (Mmul_8.0.1 for rhesus macaque) or the administered AAV vector genome to characterize and quantify edited alleles, as previously reported.11–14
To evaluate off-targets in NHP samples, we performed inverted terminal repeat (ITR) sequencing (ITR-seq) to identify off-target sites from the mapped reads, as previously described. 22
Vector GC and transgene RNA analysis
Liver samples were snap-frozen at the time of necropsy. We detected and quantified vector GCs in extracted DNA; relative meganuclease transcript expression in extracted RNA was assessed via real-time PCR, as previously described.18,23 Briefly, vector GC and RNA levels were quantified using primers/probes designed against a vector-specific sequence. We also determined the relative expression of HAO1 RNA using a TaqMan gene expression assay (Thermo Fisher Scientific). Samples from wild-type C57BL/6J mice (background strain of the Agxt−/− mouse) were included as a control.
Immunohistochemistry and immunofluorescence
Liver samples were fixed in 10% neutral buffered formalin and paraffin embedded for the determination of meganuclease and HAO1 expression via immunohistochemistry; HAO1 colocalization with a peroxisome marker, catalase, was determined via immunofluorescence. Sections were deparaffinized through a xylene and ethanol series followed by antigen retrieval (boiling for 6 min in 10 mM citrate buffer, pH 6.0). To detect the meganuclease and human HAO1 via immunohistochemistry, we further treated the sections sequentially with 2% H2O2 (15 min; Sigma), avidin/biotin blocking reagents (15 min each; Vector Labs), and blocking buffer (1% donkey serum in PBS + 0.2% Triton for 10 min) followed by incubation with the primary (1 h) and biotinylated secondary antibodies (45 min; donkey antibodies, Jackson ImmunoResearch) diluted in blocking buffer. The primary rabbit antibody against the meganuclease (
For immunofluorescence, sections were blocked after antigen retrieval with 1% donkey serum in PBS containing 0.2% Triton for 15 min, followed by sequential incubation with primary (overnight at 4°C) and fluorescence-labeled secondary antibodies (Jackson ImmunoResearch, 45 min) diluted in blocking buffer. The rabbit antibody against human HAO1 (Thermo Fisher Scientific, PA5-62006) was diluted at 1:400. We utilized a mouse antibody against catalase (LS-Bio LS-B2554, 1:100) for costaining. Both primary antibodies were mixed, and the HAO1 antibody was detected via secondary antibody, whereas the catalase antibody was sequentially detected with a VectaFluor™ Excel Amplified DyLight® 488 Anti-Mouse IgG kit according to the manufacturer’s protocol (Vector Labs).
Statistical analysis
We used linear mixed-effect modeling to evaluate significant differences in serum glycolate and urine oxalate levels over time in mice. All values are presented as mean ± standard error of the mean (SEM). A p-value of <0.05 was considered significant.
RESULTS
Increased serum glycolate and reduced urine oxalate levels following gene editing in a mouse model of PH1
We initially evaluated the M1HAO meganuclease, which targets a site conserved in the mouse, NHP, and human genomes. To determine the potential of the M1HAO meganuclease to edit the mouse genomic HAO1 sequence, we treated male Agxt−/− mice with 3 × 1011, 3 × 1012, or 3 × 1013 GC/kg of AAV8.TBG.M1HAO or vehicle (PBS) via IV injection. Similar to another previously described Agxt−/− mouse strain, 15 the Agxt−/− mice had a very low probability of developing crystals of oxalate within the kidney, and so this was not utilized as an endpoint for these studies. We collected blood and urine throughout the in-life phase to evaluate serum glycolate (Fig. 1A) and urine oxalate levels (Fig. 1B).
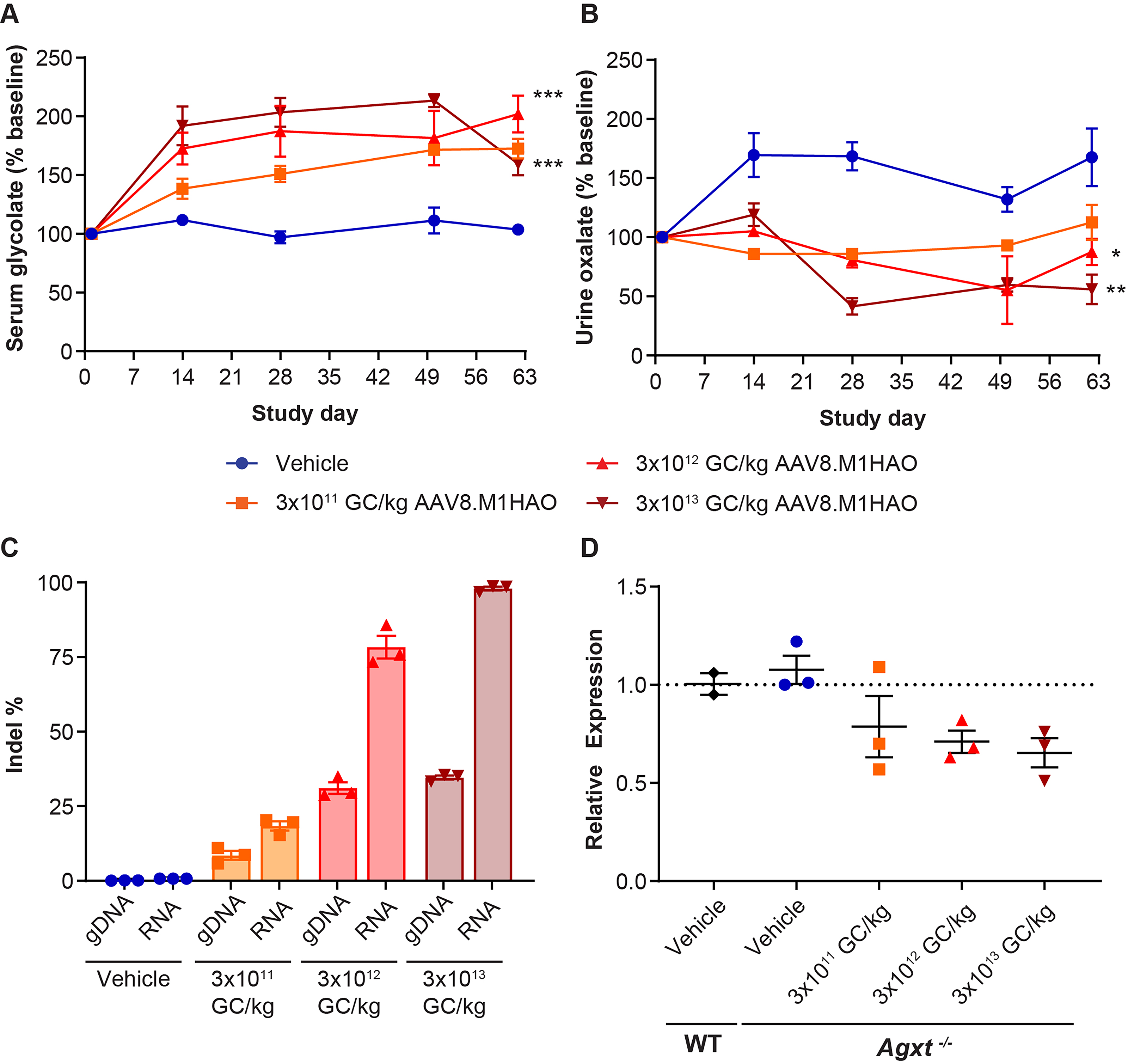
HAO1 gene editing reduces serum glycolate and urine oxalate levels in Agxt−/− mice. Agxt−/− mice received IV injections of 3 × 1011, 3 × 1012, or 3 × 1013 GC/kg AAV8.TBG.M1HAO or vehicle control on day 0. Blood and urine were collected at selected time points for
Mice that received the highest vector dose (3 × 1013 GC/kg) of meganuclease-expressing vector exhibited an 81% increase in serum glycolate levels (Fig. 1A) and a concomitant 56% decrease in urine oxalate levels compared with vehicle-administered Agxt−/− mice (Fig. 1B).
We also evaluated the editing potential of the M1HAO meganuclease for the mouse HAO1 gene (Fig. 1C). We observed a dose-dependent increase in the indel percentage in both mouse genomic DNA (gDNA) and mouse HAO1 RNA (RNA); 35% and 98% editing in gDNA and RNA, respectively, was achieved with the highest vector dose (3 × 1013 GC/kg) (Fig. 1C). Of the indels observed, approximately 3% were insertions and 97% were deletions, with most of the edits observed being 3-bp deletions. These indels resulted in the following effects on the amino acid sequence of the mouse HAO1 genomic sequence: 54% original parental sequence (unedited), 28% deletion, 10% stop codon, 6% frameshift, and 2% substitution. There were no insertions in the amino acid sequence detected. The resulting mutations in the HAO1 RNA sequence could lead to instability in the RNA; thus, we determined the relative expression levels of mouse HAO1 RNA (Fig. 1D). Vehicle-administered wild-type and Agxt−/− mice exhibited similar levels of HAO1 RNA expression. A dose-dependent decrease in HAO1 RNA expression was observed following AAV8.TBG.M1HAO vector administration.
We necropsied the Agxt−/− mice treated with 3 × 1011, 3 × 1012, or 3 × 1013 GC/kg AAV8.TBG.M1HAO or vehicle control on study day 63 and harvested livers to evaluate meganuclease and mouse HAO1 expression via immunohistochemistry (Fig. 2). The dose-dependent increase in liver vector GCs and M1HAO transgene RNA levels (Supplementary Fig. S2) corresponded to a dose-dependent increase in nuclease expression and a concomitant reduction in HAO1 levels. We also evaluated liver sections for the localization of HAO1 protein in relation to a peroxisome marker, catalase (Fig. 2), providing both functional and physiological analyses for this mouse model of PH1. In Agxt−/− mice treated with the vehicle control, HAO1 (red) colocalized with catalase (green) to generate mostly yellow staining throughout the liver. As the AAV8.TBG.M1HAO dose increased, we observed reduced peroxisomal localization of HAO1, as indicated by the reduction in yellow signal (Fig. 2).
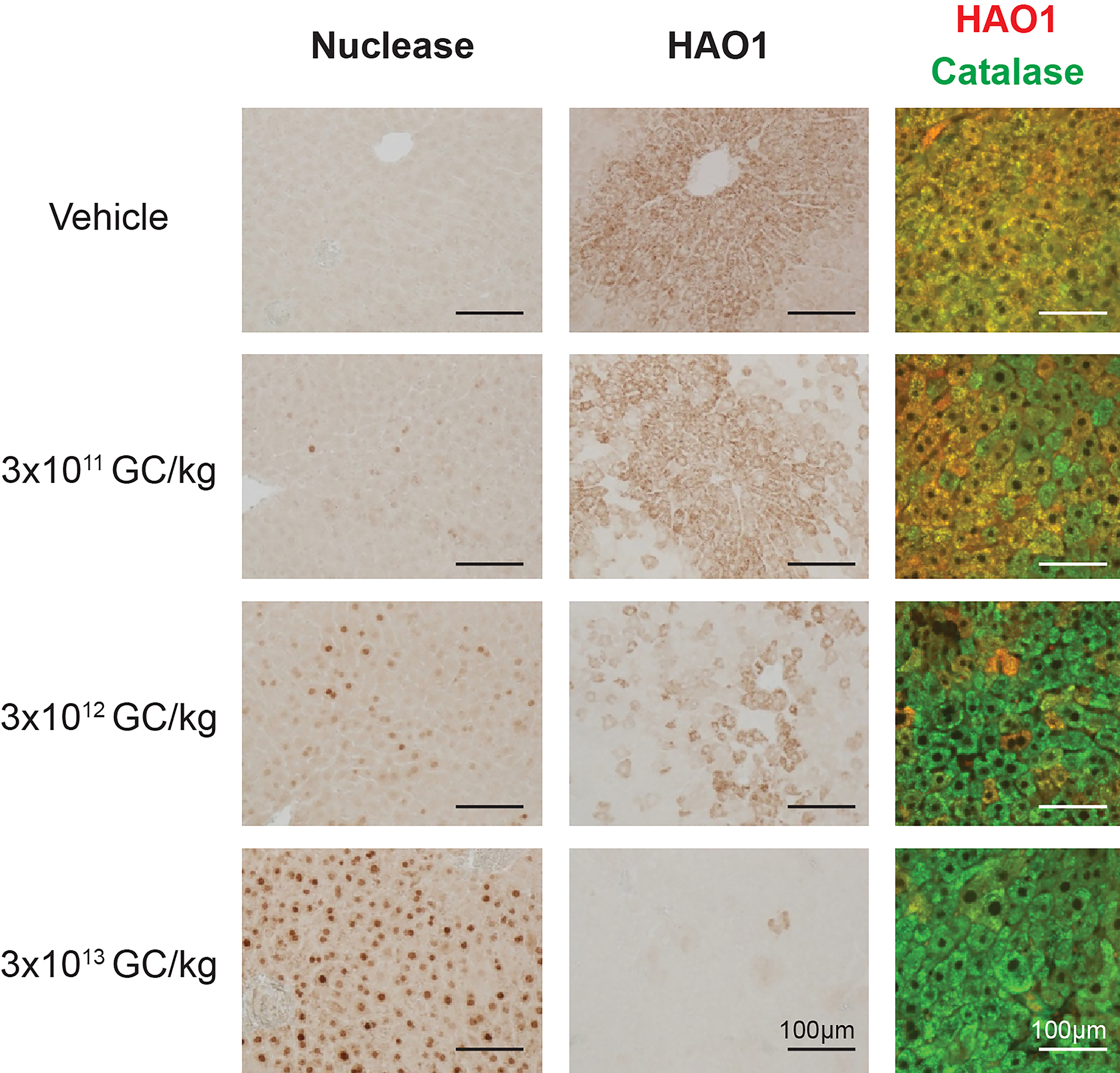
Gene editing reduces peroxisome-localized HAO1 in Agxt−/− mouse liver. Agxt−/− mice received IV injections of 3 × 1011, 3 × 1012, or 3 × 1013 GC/kg AAV8.TBG.M1HAO or vehicle (phosphate-buffered saline) on day 0. Mice were necropsied on day 63. Livers were harvested at necropsy to evaluate meganuclease and mouse HAO1 expression by immunohistochemistry. The localization of HAO1 (red) was also evaluated by immunofluorescence compared with a peroxisome marker, catalase (green).
We further evaluated the potential of the (human) HAO1 sequence to undergo meganuclease-mediated editing by administering an AAV8 vector expressing human HAO1 gene as the transgene (AAV8.TBG.hHAO1native). A similar strategy has been employed to evaluate gene editing of the human PCSK911,13 and TTR 14 genes in mice. The M1HAO meganuclease targets a sequence that is conserved in mice, NHPs, and humans (meaning it is present even in the absence of hHAO1 vector), but hHAO1 vector administration was included in M1HAO evaluation (Fig. 3A) to provide experimental conditions consistent with those used in the evaluation of M2HAO, whose target site is not present in mice (Fig. 3B). To evaluate the in vivo editing ability of M1HAO, we pretreated immunodeficient male Rag1−/− mice with 3 × 1012 GC/kg of AAV8.TBG.hHAO1native to supply the human HAO1 sequence on day 0 (or vehicle control). On day 14, mice received 3 × 1010, 3 × 1011, or 3 × 1012 GC/kg of AAV8.TBG.M1HAO or vehicle. On day 56 (42 days post-nuclease administration), mice were necropsied, and livers were harvested for evaluation of indels in (1) mouse gDNA (Fig. 3A), (2) the AAV vector genome (AAV DNA), and (3) HAO1 RNA derived from endogenous mouse expression and the AAV8.TBG.hHAO1native expression vector (RNA, Fig. 3A). We observed only 0.6–1.8% indels in gDNA, AAV DNA, and RNA in mice that received the lowest dose of meganuclease vector (3 × 1010 GC/kg). We detected 12–27% indels across the mouse gDNA, AAV DNA, and RNA for mice that received 3 × 1011 GC/kg. By contrast, mice that received 3 × 1012 GC/kg exhibited indels that ranged from 45% to 87% across the DNA and RNA analyses (Fig. 3A).
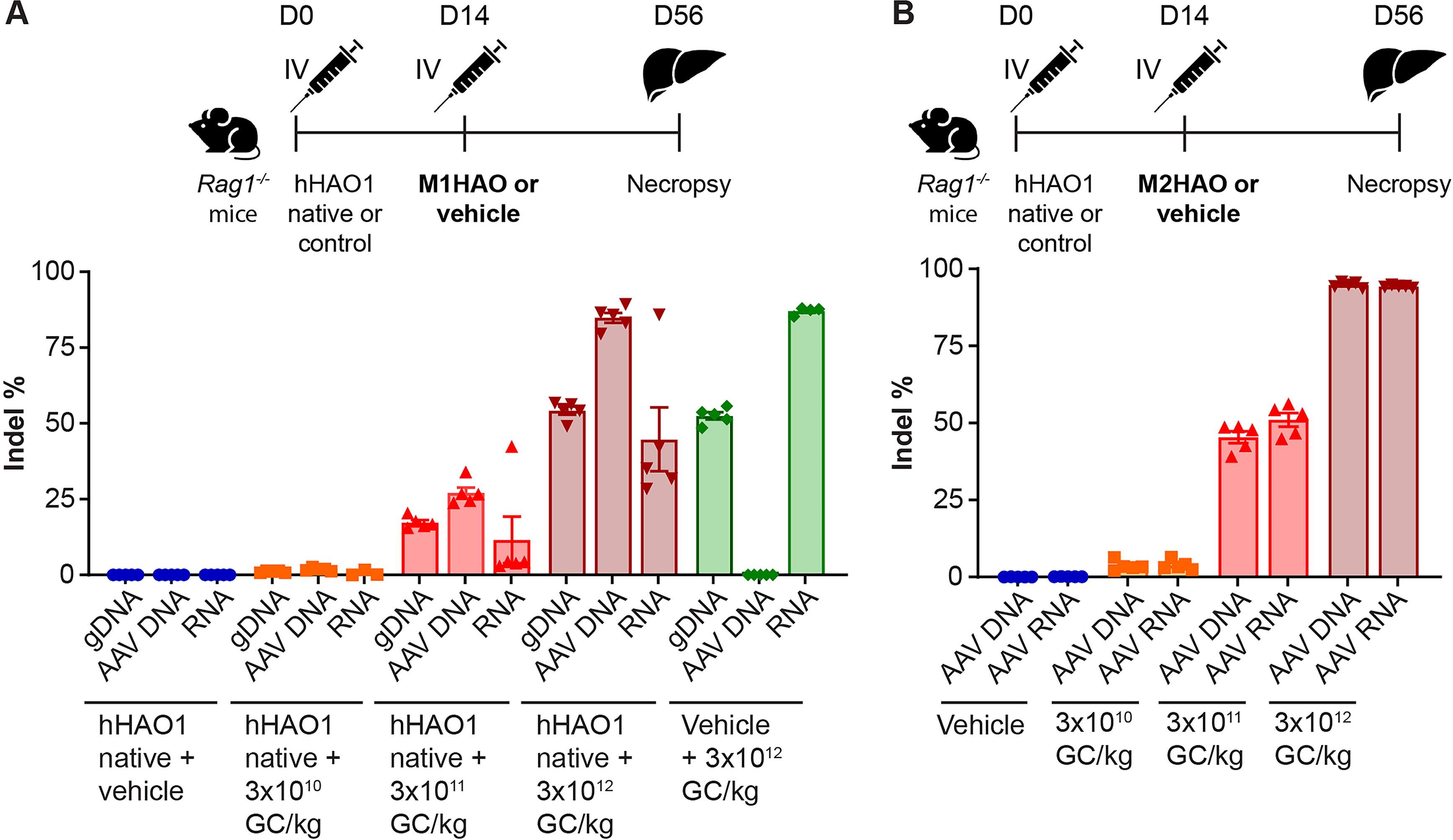
Enhanced in vivo gene editing is observed with a meganuclease that targets an alternative site in HAO1. Rag1−/− mice received an IV injection of 3 × 1012 GC/kg AAV8.TBG.hHAO1native or control vehicle on day 0.
Enhanced in vivo gene editing with a meganuclease that targets an alternative site in HAO1
To target a different sequence within the HAO1 gene compared with that of the M1HAO meganuclease, we designed an alternative engineered meganuclease, M2HAO. The target site for the M2HAO meganuclease is conserved between NHPs and humans but is not present in the mouse genome. To evaluate the in vivo efficacy of this meganuclease, we pretreated immunodeficient male Rag1−/− mice with 3 × 1012 GC/kg of AAV8.TBG.hHAO1native to supply the human HAO1 sequence on day 0 (or vehicle control). On day 14, mice received 3 × 1010, 3 × 1011, or 3 × 1012 GC/kg of AAV8.TBG.M2HAO or vehicle. On day 56 (42 days post-nuclease administration), mice were necropsied, and livers were harvested for evaluation of indel percentage in the AAV genome (AAV DNA) and in the HAO1 RNA derived from the AAV8.TBG.hHAO1native expression vector (AAV RNA, Fig. 3B). We observed a dose-dependent increase in indels in both AAV DNA and AAV RNA, with the highest vector dose (3 × 1012 GC/kg) resulting in 95% and 94% editing in AAV DNA and AAV RNA, respectively (Fig. 3B). Of the indels observed, 6% resulted in the original parental amino acid or an amino acid substitution, 8% caused a small deletion (≤4 amino acids), 61% resulted in a stop codon, 4% were large deletions (≥5 amino acids), and 21% gave rise to a frame shift in the HAO1 sequence.
Translation of gene editing potential from mice to NHPs
To determine the translatability of gene-editing efficacy, we evaluated the same engineered meganucleases (M1HAO and M2HAO) in rhesus macaques. AAV8 vectors expressing either the M1HAO or M2HAO meganuclease were IV administered to rhesus macaques at doses of 6 × 1012 GC/kg or 3 × 1013 GC/kg. All NHPs received prednisolone at a dose of 1 mg/kg/day orally for 8–12 weeks post-vector administration, and prednisolone was then tapered off by a gradual reduction in the daily dose. NHPs were routinely monitored by blood draws for complete blood cell counts and serum chemistries during the in-life phase of the study. Some NHPs receiving either vector exhibited peaks in aspartate transaminase or alanine aminotransferase that primarily occurred around the time of the first liver biopsy procedure (day 18, see Supplementary Figs. S3 and S4 for NHPs treated with AAV8.TBG.M1HAO or AAV8.TBG.M2HAO, respectively).
Liver biopsies were performed on days 18 and ∼128 post-vector administration, and the NHPs were necropsied at 1-year post-vector administration. We evaluated the indel frequency (insertions, deletions, vector ITR integrations) in the HAO1 locus of NHP gDNA and RNA via AMP-seq and amplicon-seq, respectively, as well as off-targets via ITR-seq in liver samples (Fig. 4). We observed an average of 17% and 26% editing in gDNA in macaques that received the M1HAO meganuclease on day 18 following administration of 6 × 1012 GC/kg or 3 × 1013 GC/kg vector, respectively (Fig. 4A). This DNA editing level was sustained through to 1 year post-vector administration, when the NHPs were necropsied. Similar to our observation in mice, the indels generated by the M1HAO meganuclease in gDNA predominantly comprised 3-bp deletions (Supplementary Fig. S5). The editing level in RNA determined by amplicon-seq at day 18 was higher, at 21% and 67% following the low and high vector doses, respectively, but these levels decreased to 13% and 33% by 1 year, respectively (Fig. 4B). We observed a similar reduction over time in the number of off-target sites determined via ITR-seq (Fig. 4C). The average number of off-target sites decreased from 791 at day 18 to 55 at 1 year post-vector administration following a dose of 6 × 1012 GC/kg AAV8.TBG.M1HAO and from 1,776 at day 18 to 152 one year after the high vector dose (3 × 1013 GC/kg).
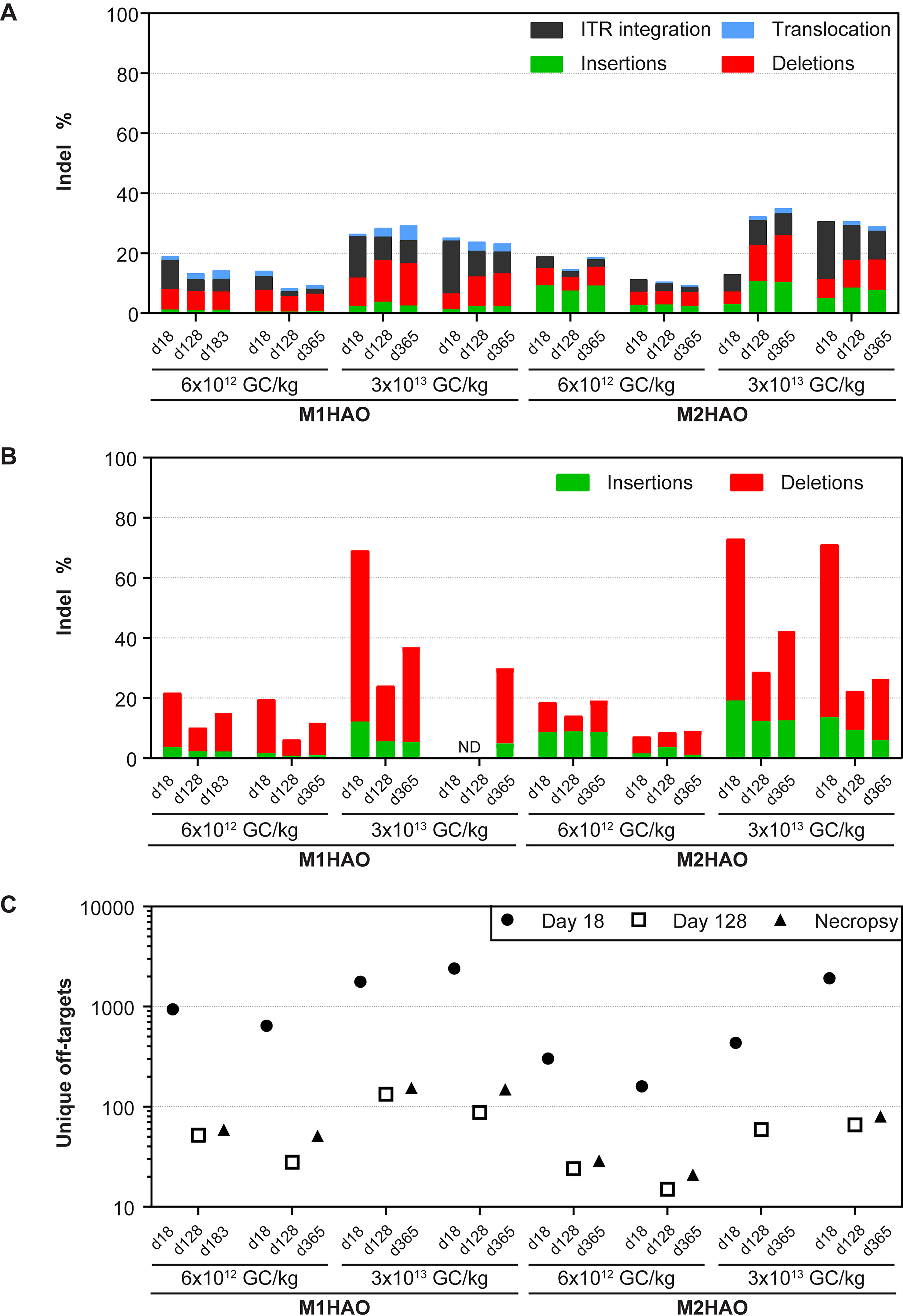
HAO1 gene editing translates from mice to NHPs. Rhesus macaques received an IV injection of 6 × 1012 or 3 × 1013 GC/kg AAV8.TBG.M1HAO or AAV8.TBG.M2HAO. Liver biopsies were performed on days 18 and 128 post-vector administration. Animals were necropsied 1 year post-vector administration. We extracted DNA and RNA and performed next-generation sequencing analysis, including
In comparison, macaques that received the low dose (6 × 1012 GC/kg) of vector expressing the M2HAO meganuclease exhibited an efficacy similar to that observed for NHPs treated with the M1HAO vector, whereas increased editing was observed at the high dose of 3 × 1013 GC/kg (Fig. 4A). The M2HAO meganuclease generated an indel pattern in gDNA comprising a mixture of 1-bp insertions, 2-bp insertions, and 2-bp deletions (Supplementary Fig. S6). NHPs that received 6 × 1012 GC/kg of AAV8.TBG.M2HAO displayed an average of 15% indels on day 18, whereas 13–35% editing was observed for a dose of 3 × 1013 GC/kg across the three time points evaluated. This higher level of DNA editing resulted in a slight increase in RNA editing (increasing from 67% for the M1HAO nuclease to 72% for the M2HAO nuclease), as detected via amplicon-seq (Fig. 4B). Similar to the M1HAO meganuclease, the number of off-target sites detected upon administration of the M2HAO nuclease was high at day 18 post-vector administration (an average of 231 and 1,177 sites for the low and high vector doses, respectively), which decreased to <80 sites by 1-year post-vector administration (Fig. 4C). The M1HAO1 and M2HAO1 meganucleases used here were not optimized for specificity in the mouse or NHP genomes, and validation of the off-target site by editing frequency analysis within a given off-target site was not performed, thereby precluding further analysis.
We also evaluated serum glycolate levels in NHPs administered the M1HAO and M2HAO meganuclease-expressing vectors (Fig. 5). Both macaques that received the low vector dose (6 × 1012 GC/kg) of M1HAO exhibited minor fluctuations from baseline serum glycolate levels over the course of the study from 11 to 23 µM (Fig. 5A). Following the high vector dose (3 × 1013 GC/kg) of M1HAO, there was a distinct peak in serum glycolate of 61 and 83 µM at day 28 post-vector administration in the two NHPs evaluated, an increase of 3.6- and 4.9-fold, respectively. These levels then plateaued at approximately 40 µM, more than double the baseline levels (Fig. 5A). Although a dose-dependent effect was still observed in macaques treated with the M2HAO meganuclease-expressing vector, the effect on serum glycolate levels was weaker following the high vector dose (Fig. 5B). A dose of 3 × 1013 GC/kg of AAV8.TBG.M2HAO resulted in peak serum glycolate levels of 34 µM and 57 µM, an increase of 2.6- and 3.6-fold, respectively. The subsequent plateau in serum glycolate levels was also lower at approximately 30 µM (Fig. 5B). Vector GCs and meganuclease transcript levels did not differ between the two nucleases evaluated, with both displaying vast reductions in DNA and RNA levels over time (Table 1).
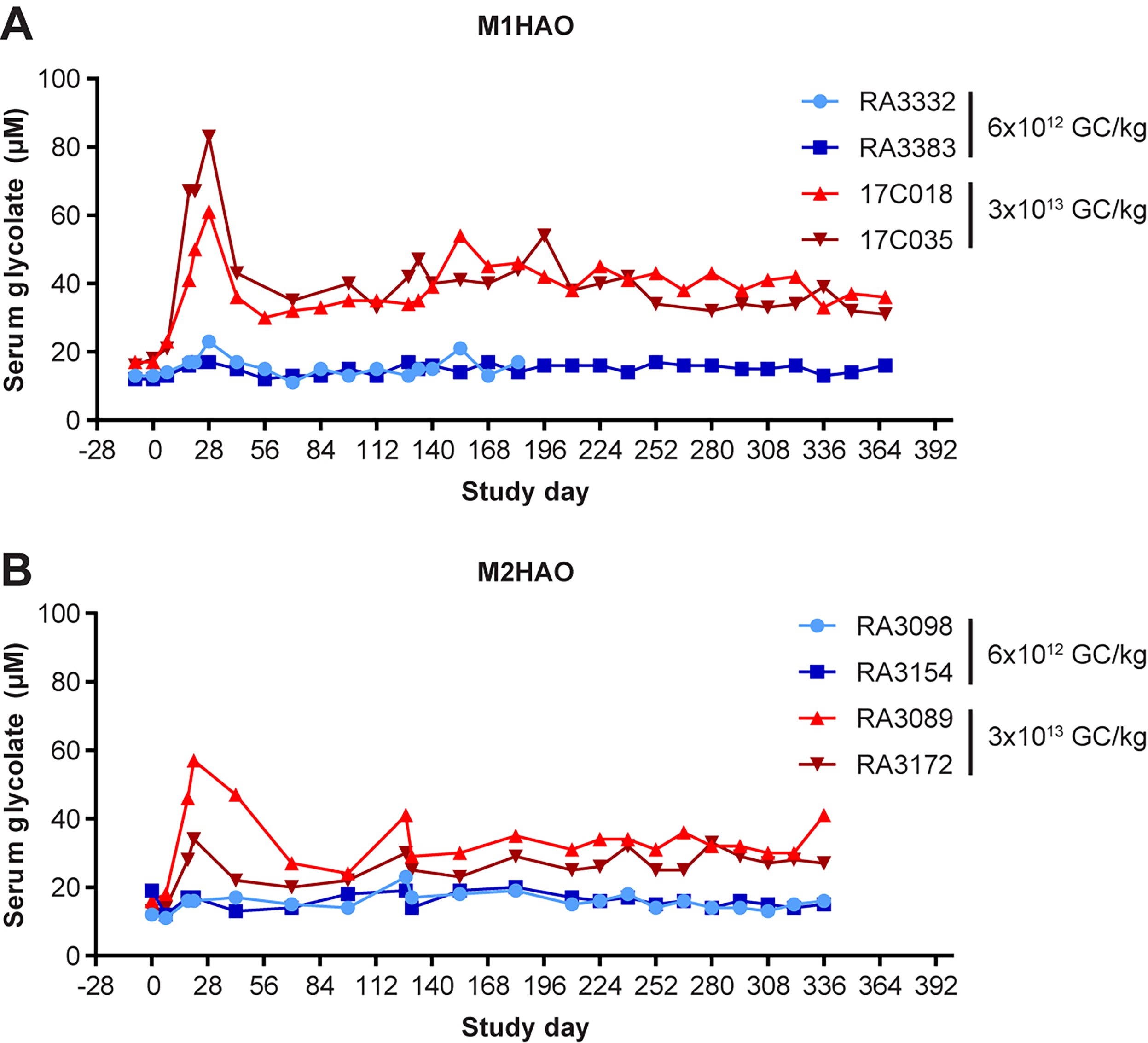
NHPs exhibit a substantial increase in serum glycolate levels following systemic administration of AAV8.TBG.M1HAO. Rhesus macaques received an IV injection of 6 × 1012 or 3 × 1013 GC/kg of
Liver Vector Genome Copies and Meganuclease RNA Levels in the Liver Following Systemic Administration of AAV8.TBG.M1HAO or AAV8.TBG.M2HAO
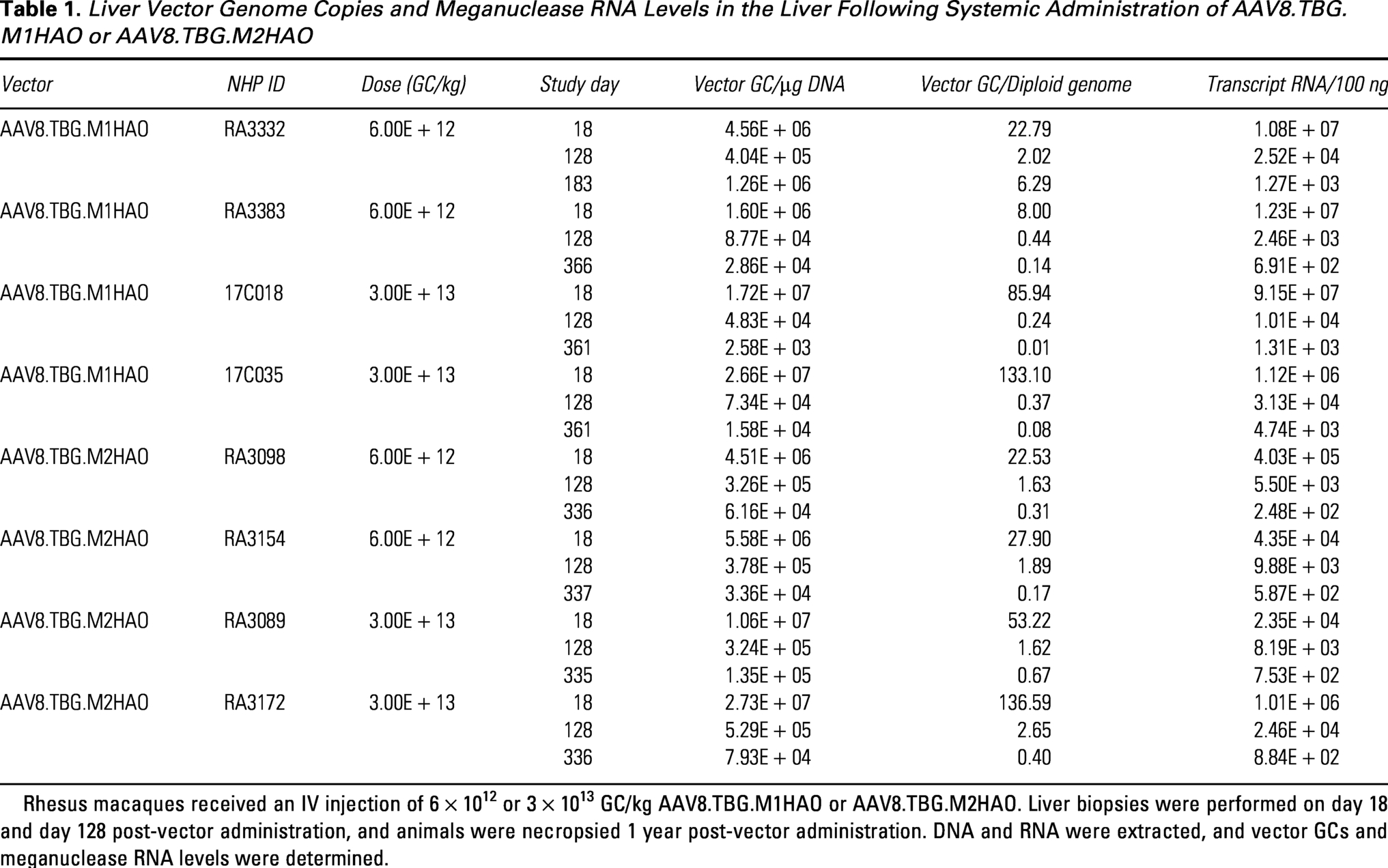
Rhesus macaques received an IV injection of 6 × 1012 or 3 × 1013 GC/kg AAV8.TBG.M1HAO or AAV8.TBG.M2HAO. Liver biopsies were performed on day 18 and day 128 post-vector administration, and animals were necropsied 1 year post-vector administration. DNA and RNA were extracted, and vector GCs and meganuclease RNA levels were determined.
DISCUSSION
We evaluated an AAV-delivered, meganuclease-based gene-editing approach to reduce HAO1 as a potential treatment for PH1. We performed initial experiments in Agxt−/− mice, a mouse model of PH1, in which we observed significant improvements in serum glycolate and urine oxalate levels and detected a dose-dependent increase in indel percentages for gDNA and RNA from the liver. We conducted follow-up experiments in NHPs in which dose-dependent editing of hepatic gDNA and RNA was achieved and serum glycolate levels changed in a manner consistent with successful liver editing; additionally, the treatment was well tolerated. Together, these data suggest that using our engineered meganuclease to edit the HAO1 gene may represent a viable approach for treating PH1 with a single AAV dose.
The M1HAO meganuclease targets a site in exon 8 within the HAO1 gene that is conserved in mice, NHPs, and humans. At the two highest vector doses evaluated (3 × 1012 and 3 × 1013 GC/kg), we detected significant differences in serum glycolate and urine oxalate consistent with successful disruption of HAO1 enzyme activity and a dose-dependent increase in indel percentage in both mouse gDNA and mouse HAO1 RNA.
The efficacy of the AAV-expressed M1HAO meganuclease was similar to that of CRISPR-Cas9-based strategies within the same dose range.9,10 Like gene knockdown approaches using RNAi or siRNA, CRISPR-Cas9-based editing approaches have been reported that target either HAO1 9 or LDH. 10 A single systemic administration of an AAV8-CRISPR/Cas9 vector has been shown to prevent oxalate overproduction and kidney damage in Agxt1−/− mice. However, these approaches both utilize S. aureus Cas9, which has reduced translatability from mice to primates. Thus, we chose to use the engineered M1HAO and M2HAO meganucleases, whose coding sequences are small enough to be easily packaged within a single AAV genome and expressed from a single transcript.
At the highest vector dose evaluated in Agxt1−/− mice (3 × 1013 GC/kg), 35% editing was achieved in gDNA, with 98% editing in RNA. The resulting mutations in the HAO1 RNA sequence likely led to instability in the RNA, which probably resulted in the very high indel percentage in RNA compared with gDNA. This may mean that the RNA data were skewed, as only intact RNA (which likely only contains small indels) could be measured. We assessed the function and physiology of Agxt−/− mice treated with increasing doses of AAV8.TBG.M1HAO by performing costaining via immunohistochemistry for HAO1 protein and catalase (a peroxisome marker) in hepatocytes; we determined that meganuclease administration resulted in reduced peroxisomal localization of HAO1.
The HAO1 exon 1 target site for the M2HAO nuclease is not present in the mouse genome but is conserved in NHPs and humans, so we could not perform the same functional analysis in Agxt−/− mice. However, we were able to evaluate its editing potential by using our previously described strategy in which human genes are introduced to mice.11,13,14 By pretreating immunodeficient male Rag1−/− mice with 3 × 1012 GC/kg of AAV8.TBG.hHAO1native to supply the human HAO1 sequence on day 0 (or vehicle control), we evaluated the editing of this (episomal) human gene following administration of the meganuclease-expressing vector on day 14. In these mice, we observed a dose-dependent increase in indels, with the highest vector dose (3 × 1012 GC/kg) resulting in 95% and 94% editing in DNA and RNA, respectively.
The editing levels seen for both the M1HAO and M2HAO meganucleases in these mouse studies translated well into NHPs. Following expression of the M1HAO meganuclease from an AAV vector in NHPs, we detected a high level of HAO1 gene editing, corresponding to increased serum glycolate levels that plateaued at approximately 40 µM, more than double baseline levels. These increased serum glycolate levels are indicative of significantly disrupted HAO1-mediated glycolate-to-glyoxylate metabolism and highlight how this approach could be effective for treating PH1. While the M2HAO meganuclease had a higher level of HAO1 gene editing than M1HAO in mice, there was a lesser increase in serum glycolate levels in NHPs. This may be due to the nature of the mutations generated by these meganucleases and their relative impact on HAO1-mediated metabolism. However, elucidating the mechanism(s) underpinning such differences across species is complicated significantly by the fact that the M2HAO nuclease HAO1 target site is not present in the mouse genome.
Although there is now an approved RNAi therapy targeting HAO1 (Lumasiran), this treatment requires monthly or quarterly subcutaneous injections following three monthly doses as a loading phase. 7 Therefore, there is a potential risk of nonadherence with this treatment, which would be avoided with a one-time genome-editing approach. In addition, there are no known detrimental effects from knocking down HAO1 expression, and a few published cases have indicated that glycolic aciduria is asymptomatic.24,25 This bodes well when considering potential long-term consequences associated with therapeutic HAO1 disruption. Overall, our data suggest that editing the HAO1 gene with an engineered meganuclease delivered in a single AAV dose may represent a viable approach for treating PH1.
AUTHORS’ CONTRIBUTIONS
J.A.G.: Conceptualization, data curation, formal analysis, investigation, methodology, supervision, visualization, writing—original draft preparation, and writing—review and editing; C.B.: Data curation, formal analysis, investigation, methodology, supervision, visualization, and writing—review and editing; M.K.S.: Data curation, investigation, methodology, project administration, and writing—review and editing; T.F.: Data curation, investigation, methodology, project administration, and writing—review and editing; P.B.: Investigation, methodology, and writing—review and editing; P.C.: Funding acquisition, investigation, methodology, visualization, and writing—review and editing; J.M.W.: Conceptualization, methodology, supervision, validation, and writing—review and editing.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank the Penn Vector Core, Program in Comparative Medicine, Nucleic Acid Technologies Core, Immunology Core, and Histology Core at the Gene Therapy Program. They thank the Metabolomic Core at the Children’s Hospital of Philadelphia for performing metabolite measurements. In addition, the authors would like to thank Scott N. Ashley, Joanna K. Chorazeczewski, Matthew Jennis, and Jayme M. L. Nordin for their continued support of this project. The authors would like to thank Hanying Yan for performing the statistical analysis.
AUTHOR DISCLOSURE
J.M.W. is an employee and shareholder of GEMMA Biotherapeutics, Inc., a professor emeritus at the University of Pennsylvania, and an advisor to and holds equity in iECURE, Inc. and Passage Bio, Inc. He also holds equity in the Center for Breakthrough Medicines/SK Pharmteco. J.M.W. is the chair and a shareholder of Franklin Biolabs. This article includes work conducted at the Gene Therapy Program at the University of Pennsylvania, which has since transitioned its activities to GEMMA Biotherapeutics, Inc. The University of Pennsylvania previously had sponsored research agreements with Alexion Pharmaceuticals, Amicus Therapeutics, CBM, Ceva Santé Animale, Elaaj Bio, FA212, Foundation for Angelman Syndrome Therapeutics, former G2 Bio asset companies, iECURE, Inc., and Passage Bio, Inc., which are licensees of GEMMABio and Penn technology. J.M.W. and J.A.G. are inventors on patents that have been licensed to various biopharmaceutical companies and for which they may receive payments.
FUNDING INFORMATION
This research was supported by Precision BioSciences.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.