
Abstract
Purpose:
The Gram-negative bacterium Aeromonas caviae is an important opportunistic facultative anaerobic pathogen. In the present study, we aimed to elucidate the whole-genome sequence of the multidrug-resistant (MDR) A. caviae strain AC1520, detailing its acquired antibiotic resistance genes (ARGs) and their genetic elements.
Patients and Methods:
The A. caviae strain AC1520 was isolated from a urine sample taken from a patient with urinary tract infection. Whole-genome sequencing was performed following strain identification and antimicrobial susceptibility testing. Overall, we identified ARGs, integrons, insertion sequences (IS), and transposons acquired by strain AC1520, systematically analyzing the genetic elements associated with these ARGs.
Results:
The A. caviae strain AC1520 contained a circular chromosome and a plasmid. Multilocus sequence typing revealed that this strain belonged to ST-1056. All ARGs within this strain were distributed on the circular chromosome. We identified two MDR regions: (1) IS common region 1 (ISCR1) and class 1 integron (IntI1) elements associated with aadA16, aac(6’)-Ib-cr, catB3, qacE (two copies), sul1 (two copies), and blaPER-3; one gene cluster structure (IS6100-mphR(A)-mph(A)-mrx(A)-IS26); (2) Two IntI1 elements, linked to ant(2’’)-Ia, blaOXA-10, aadA2b, aph(3′’)-Ib, aph(6)-Id, ARR-2, aac(6’)-Ib3, dfrA1, qacE, and sul1. Notably, these two MDR regions were not only present in A. caviae but also in other bacteria, such as Aeromonas hydrophila, Aeromonas media, and Edwardsiella tarda.
Conclusion:
The A. caviae strain AC1520 with two separate MDR regions and 20 ARGs, conferring resistance to aminoglycoside, fluoroquinolone, phenicol, sulfonamide, beta-lactam, macrolide, rifampicin, and trimethoprim, was identified in a hospital in China. Mobile genetic elements including TnAs1, ISCR1, ISAs25, IS6100, IS26, TnAs3, ISAs1, and Tn3, were found within the MDR region, which could play important roles in the global dissemination of these resistance genes.
Introduction
Aeromonas species, widespread bacteria found in aquatic environments, have been recognized as significant pathogens since their first isolation from a human blood sample in 1954. 1 As Gram-negative facultative anaerobic bacteria, they are easily found in nature and can be isolated from water, soil, and human feces. 2 Until 2023, 36 species within the Aeromonas genus had been described, with each capable of causing a wide range of infections. 3 However, recent studies suggest that clinical infections caused by the Aeromonas spp. are primarily linked to four species: Aeromonas caviae, Aeromonas dhakensis, Aeromonas hydrophila, and Aeromonas veronii. 3 Among these, A. caviae is the most commonly reported species 4 and is responsible for various infections, including gastroenteritis, wound infections, bloodstream infections, biliary tract infections, respiratory system infections, urinary tract infections, ocular infections, meningitis, peritonitis, and liver abscesses. 1
Aeromonas spp. are becoming increasingly resistant to cephalosporins and carbapenems.5–7 The resistance to carbapenem is primarily attributed to CphA metallo-β-lactamase, encoded by the cphA gene. 8 In 2007, French researchers reported the first clinical isolation of a carbapenem-resistant strain of A. caviae capable of producing the class B carbapenemase IMP-19. 9 Meanwhile, quinolone resistance in Aeromonas spp. is mediated by specific chromosomal and plasmid-mediated quinolone resistance genes; recent studies have identified an increasing trend in quinolone resistance.10,11
Antimicrobial resistance (AMR) has emerged as an increasingly significant threat to public health in recent years. 12 The sharp rise in the incidence of multidrug-resistant (MDR) Gram-negative bacteria infections has garnered increasing attention worldwide. Specifically, A. caviae is becoming increasingly prevalent in clinical settings, and MDR A. caviae has emerged as a major threat to public health.
Mobile genetic elements, such as integrons, insertion sequences (IS), transposons, plasmids, bacteriophages, and genomic islands, have been determined to play an important role in the spread of multidrug resistance genes. 13 Therefore, this study aimed to sequence the whole genome of an MDR strain of A. caviae (AC1520) isolated from a patient with urinary tract infection. In addition, we aimed to determine the genetic structures that contribute to the acquisition of antibiotic resistance genes (ARGs), with a particular focus on mobile genetic elements.
Material and Methods
Isolation and characterization of the MDR bacterial strain AC 1520
The A. caviae strain AC1520 was isolated from a urine sample of a 68-year-old person with a urinary tract infection at the Department of Urology of Zhuhai People’s Hospital on November 13, 2022. The present study complies with the Declaration of Helsinki. Subsequently, the strain was identified using VITEK matrix-assisted laser desorption ionization-time of flight mass spectrometry (bioMérieux, France). Identification of the AC1520 strain was confirmed through 16S rRNA gene sequencing. According to the Clinical and Laboratory Standards Institute (CLSI M45, 3rd Edition) guidelines, the broth microdilution method was used for initial susceptibility screening. MICs were confirmed using the VITEK 2 XL system. CLSI criteria (M45, 3rd Edition) were used to interpret the results. CLSI criteria (M45, 3rd Edition) defines an MDR organism: Non-susceptibility to at least one agent in three or more antimicrobial categories. Strain AC1520 resists three classes: quinolones, sulfonamides, and β-lactam/β-lactamase inhibitors (Table 1).
Minimum Inhibitory Concentration of Various Antibiotics for the Aeromonas caviae Strain AC1520
I, Intermediate; R, Resistant; S, Susceptible.
Whole-genome sequencing, assembly, and annotation
Whole-genome shotgun sequencing was performed using Novaseq 6000 (Illumina, 2 × 150 bp reads) and PacBio Sequel lle (10–15 Kb insert whole-genome shotgun libraries; Pacific Biosciences). PacBio reads were then assembled using Hifiasm (version 0.13-r308) and Canu (version 1.7). Meanwhile, assembly polishing of Illumina reads was conducted using Pilon (version 1.22). 14 The genome assemblies of the AC1520 strain were submitted to the GenBank database for annotation, 15 according to the NCBI Prokaryotic Annotation Pipeline. 16
Genome analysis of the A. caviae strain AC1520 using bioinformatics
Acquired ARGs were identified in the A. caviae strain AC1520 using ResFinder 4.1 software. 17 Subsequently, multilocus sequence typing (MLST) was performed based on six housekeeping genes (gltA, groL, gyrB, metG, ppsA, and recA), using MLST 2.0. 18 PlasmidFinder 2.1 was utilized to determine the replicon types of the plasmids in the AC1520 strain. 19 Meanwhile, IS elements were identified using ISfinder software. 20 Finally, sequence comparison was performed using BLAST+ (version 2.11.0), 21 and a genetic map was generated with Easyfig (version 2.2.5). 22
Nucleotide sequence accession numbers
The complete genome sequences of the strain AC1520 reported in the present study were deposited in the GenBank nucleotide database under the accession numbers CP120942-CP120943.
Results
Antibiotic resistance profiles of the A. caviae strain AC1520
Antimicrobial susceptibility testing revealed that the A. caviae strain AC1520 obtained in this study exhibited resistance to quinolones (levofloxacin and ciprofloxacin), piperacillin–tazobactam, and sulfamethoxazole/trimethoprim (Table 1).
Genomic analysis of the A. caviae strain AC1520
Whole-genome sequencing revealed that the AC1520 strain contained a circular chromosome consisting of 4,484,517 bp (GenBank accession number: CP120942) and a plasmid containing 253,471 bp (GenBank accession number: CP120943). MLST typing revealed that this strain belonged to ST-1056. Then, ResFinder results showed that the acquired ARGs within this strain were distributed on the circular chromosome. A total of 20 ARGs were identified in this genome: blaMOX-3, aadA16, aac(6’)-Ib-cr, catB3, sul1 (three copies), blaPER-3, mphR(A), mph(A), mrx(A), ant(2’’)-Ia, blaOXA-10, aadA2b, aph(3′’)-Ib, aph(6)-Id, ARR-2, aac(6’)-Ib3, dfrA1, and tet(A) (Figs. 1 and 2).
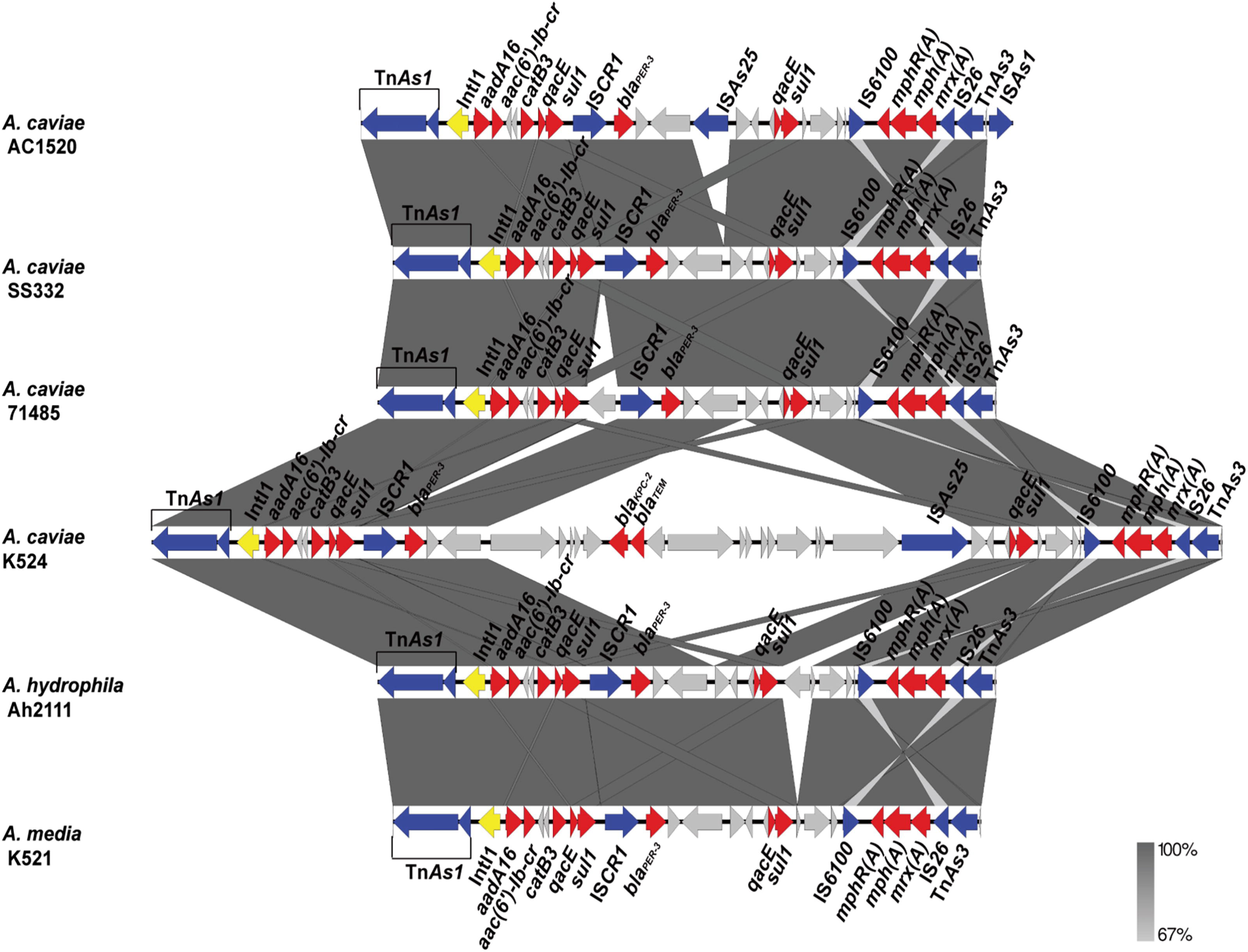
Comparative analysis between the MDR 1 region in the Aeromonas caviae strain AC1520 and MDR regions identified in other strains, including SS332 (A. caviae, CP071151), 71485 (A. caviae, CP085468), K524 (A. caviae, CP118940), Ah2111 (Aeromonas hydrophila, CP095280), and K521 (Aeromonas media, CP118993). Red, blue, and yellow represent resistance, transposase, and integrase genes, respectively. MDR, multidrug-resistant.

Comparative analysis between the MDR 2 region in the Aeromonas caviae strain AC1520 and MDR regions identified in other strains GT88 (Edwardsiella tarda, CP099739), vAh ST251 (Aeromonas hydrophila, LR963135), W3 (A. caviae, CP097209), and WP5-W18-ESBL-02 (Aeromonas media. AP022110). Red, blue, and yellow represent resistance, transposase, and integrase genes, respectively. MDR, multidrug-resistant.
Comparative analysis of the MDR regions of the A. caviae strain AC1520
Next, we divided the AC1520 ARGs into two separate MDR regions based on their genetic locations. Following BLAST analysis and comparison with the GenBank database, sequences similar or identical to these MDR regions were identified in other bacterial strains.
MDR region 1 of AC1520 was determined to be associated with the following resistance genes: aadA16 (aminoglycoside resistance), aac(6’)-Ib-cr (fluoroquinolone and aminoglycoside resistance), catB3 (phenicol resistance), sul1 (sulfonamide resistance), blaPER-3 (beta-lactam resistance), mphR(A), mph(A), and mrx(A) (macrolide resistance) (Fig. 1 and Table 2). QacE is a disinfectant resistance gene (not antibiotic). MDR region 1 was determined to be flanked by TnAs1 and ISAs1 elements; overall, this region is composed of a 29,669 bp fragment that is similar to regions identified in strains SS332 and 71485, with the exception of two insertions (ISAs25 and ISAs1) in the AC1520 strain, supported by the presence of 1,615 and 1,097 bp direct repeats (Fig. 1). IntI1 was identified at the 5′ end of this MDR sequence, followed by the ARGs aadA16, aac(6’)-Ib-cr, catB3, qacE, and sul1 (Fig. 1). Meanwhile, insertion common sequence region 1 (ISCR1) was located downstream of sul1 and upstream of blaPER-3 (Fig. 1). In addition, BlaPER-3 was flanked by ISCR1 and ISAs25, which were adjacent to two direct repeat genes (qacE-sul1) (Fig. 1). Moreover, AC1520 sequencing revealed that the mphR(A)-mph(A)-mrx(A) gene cluster was embedded in IS6100 and IS26, forming IS6100-mphR(A)-mph(A)-mrx(A)-IS26 (Fig. 1). MDR region 1 was almost identical (identity >99%, coverage = 99%) to corresponding regions in the chromosomes of A. caviae strain SS332 (GenBank accession number: CP071151) and A. caviae strain 71485 (GenBank accession number: CP085468) (Fig. 1). This region also possessed an almost identical sequence (identity >99%, coverage >90%) to regions in the pK524-KPC plasmid of A. caviae strain K524 (GenBank accession number: CP118940) and the chromosomes of Aeromonas hydrophila strain Ah2111 (GenBank accession number: CP095280), and Aeromonas media strain K521 (GenBank accession number: CP118993) (Fig. 1 and Table 3). But samples of the aforementioned other strain were isolated from various sources, including bile and ascites.
Resistance Genes and Antibiotics Resistance for the Aeromonas caviae Strain AC1520
Genomic Similarities of MDR Region 1 in AC1520 to Other Bacterial Sequences
Coverage: % alignment length relative to AC1520 MDR region.
Identities: % nucleotide similarity.
Length: bp of aligned region.
MDR, multidrug-resistant.
MDR region 2 of AC1520 also contained genes associated with resistance to aminoglycosides [ant(2’’)-Ia, aadA2b, aph(3′’)-Ib, and aph(6)-Id], beta-lactam (blaOXA-10), rifampicin (ARR-2), fluoroquinolone and aminoglycoside [aac(6’)-Ib3], trimethoprim (dfrA1), and sulfonamide (sul1) (Fig. 2 and Table 2). MDR region 2 was determined to consist of an 11,440 bp fragment that is nearly identical to a region identified in A. caviae DNA (WP5-W18-ESBL-02) (Fig. 2). The first IntI1 element was identified at the 5′ of this sequence, followed by the ARGs ant(2’’)-Ia, blaOXA-10, aadA2b, aph(3′’)-Ib, and aph(6)-Id. Meanwhile, the second IntI1 was identified downstream of aph(6)-Id, initiating the sequence followed by the ARGs ARR-2, aac(6’)-Ib3, dfrA1, qacE, and sul1 (Fig. 2). Notably, MDR region 2 was determined to be highly similar to corresponding regions in the WP5-W18-ESBL-02 DNA of A. caviae (GenBank accession number: AP022110), the chromosome of Edwardsiella tarda strain GT88 (GenBank accession number: CP099739), the chromosome of A. hydrophila strain vAh ST251 (GenBank accession number: LR963135), and the chromosome of A. caviae strain W3 (GenBank accession number: CP097209) (Fig. 2 and Table 4). But samples of the aforementioned other strain were isolated from various sources, including frog farm sediment, activated sludge, and wastewater treatment plant effluent.
Genomic Similarities of MDR Region 2 in AC1520 to Other Bacterial Sequences
Coverage: % alignment length relative to AC1520 MDR region.
Identities: % nucleotide similarity.
Length: bp of aligned region.
MDR, multidrug-resistant.
Discussion
This study employed whole-genome sequencing to elucidate the chromosomal and plasmid sequences of the MDR A. caviae strain AC1520, isolated from a urine sample of a patient with urinary tract infection.
In this study, we established that the AC1520 strain contains a typical IntI1 gene. Integrons, genetic platforms that can be mobilized by transposons or plasmids, 23 are classified into two types: chromosomal and mobile integrons. Class 1 mobile integrons are the most common type of integron. These integrons serve as major reservoirs of AMR genes and facilitate the exchange of resistance genes between Gram-negative bacteria.24,25
Class 1 mobile integrons contain 5′ conserved segments (5′CS) and 3′ conserved segments (3′CS). 26 In the present study, the AC1520 strain was found to contain a partial IntI1 lacking the 5′CS within MDR region 1. This MDR region was found to contain four ARGs: aadA16, aac(6’)-Ib-cr, catB3, and sul1. Meanwhile, two IntI1 elements were identified in MDR region 2; these IntI1 elements were associated with four separate ARGs. Notably, our findings revealed that IntI1 is widely expressed not only in A. caviae but also in A. hydrophila, A. media, and E. tarda. Thus, our findings suggest that “IntI1” may facilitate horizontal ARG transfer in A. caviae and related pathogens.
A subgroup termed ISCR elements is now recognized as a system involved in the capture and transfer of major ARG, playing a key role in the evolution and dissemination of these genes. 27 Currently, 23 groups of the ISCR family (ISCR1–23) have been identified. Similar to IS91, ISCR1 operates on a rolling-circle mechanism and conducts sustained transposition. 28 This element plays a crucial role in the dissemination of resistance genes that encode various enzymes, ultimately providing a powerful capture system for ARGs.
Therefore, we investigated the structure of ISCR1 adjacent to the 3′CS of integrons. 29 Known as a “complex IntI1” or “unusual IntI1,” this structure involves interactions between class 1 integrons and ISCR1. 30 As its frequency increases, this structure can become mobile, resulting in the transpositions of large sections of DNA. 29 Since ISCR1 inserts DNA into the 3′ conserved regions of class 1 integrons, complex class 1 integrons harboring sul1, resX, and trIb resistance genes may coexist with IntI1.31–35
In the present study, ISCR1 was found to coexist with IntI1, forming a complex class 1 integron carrying aadA16, aac(6’)-Ib-cr, catB3, qacE, sul1, and BlaPER-3 resistance genes. This structure was not only observed in the AC1520 strain but also identified in the chromosomes and plasmids of other Aeromonas spp., including A. hydrophila and A. media.
At present, two other azithromycin resistance mechanisms have been reported. The first involves an efflux system that expels antibiotics from the bacterial cytoplasm. The second method involves the modification of macrolides or bacterial ribosomes by esterases, rRNA methylases, and phosphorylases encoded by the ere(A)/ere(B), erm, and mph(A)/mph(B) genes. 36 Notably, mph(A), a gene encoding macrolide 29 phosphotransferase, is the predominant genetic determinant for azithromycin resistance, proven through its identification in numerous bacterial species.37,38 In addition, research on 40 azithromycin-resistant Salmonella strains from 11 Salmonella serovars revealed that mph(A) is the most prevalent azithromycin resistance gene in this genus; this gene was identified in various incompatible plasmids and in chromosomes. 39 Meanwhile, the insertion sequence IS26 has been identified as a crucial element for the capture and spread of resistance genes,40–42 often forming complex resistance gene loci via associations with IntI1 and diverse transposons. Further research has demonstrated that azithromycin resistance in some strains is encoded by IncI1 and IncFIC/IncFIB type conjugative plasmids harboring the erm(B) gene and the IS26-mph(A)-mrx-mphR-IS6100 gene cluster. 43 Similarly, our study revealed an association between IntI1, IS6100, and IS26, forming a gene cluster encoding phosphorylase with the structure IS6100-mphR(A)-mph(A)-mrx(A)-IS26. Notably, this gene cluster is widespread, found in A. caviae, A. hydrophila, and A. media, residing in both chromosomes and plasmids. Thus, we postulate that the high level of macrolide resistance observed in the AC1520 strain is linked to the mphR(A)-mph(A)-mrx(A) gene cluster and its upstream promoter found in IS6100.
According to WGS, there is a contradiction between the phenotype and genotype of resistance. The possible reason is that the lack of drug resistance phenotypes in some ARGs(such as aadA16, aac(6’)-IB-CR, ant(2’)-Ia, aadA2b, aph(3′)-Ib, and aph(6)-Id) may reflect gene silencing, low expression, or compensatory mutations. 44
While single-strain studies are common for characterizing novel MDR mechanisms, we emphasize that the presence of two MDR regions from A. caviae strain AC1520 in a urine sample was less reported in China. The MDR regions share >99% identity with sequences from A. hydrophila (bile/ascites) and wastewater isolates (Tables 3 and 4), suggesting cross-species transmission. Our findings highlight clinical relevance and potential environmental dissemination routes.
But limitations include analysis of a single isolate, which precludes epidemiological conclusions. Future studies should screen larger clinical and environmental collections to quantify the prevalence of these MDR regions.
Conclusion
The A. caviae strain AC1520 with two separate MDR regions and 20 ARGs, conferring resistance to aminoglycoside, fluoroquinolone, phenicol, sulfonamide, beta-lactam, macrolide, rifampicin, and trimethoprim, was identified in a hospital in China. Mobile genetic elements, including TnAs1, ISCR1, ISAs25, IS6100, IS26, TnAs3, ISAs1, and Tn3, were found within the MDR region, which could play important roles in the global dissemination of these resistance genes. This study alerts clinicians to MDR A. caviae in Urinary Tract Infections and provides genomic evidence of mobile ARG spread across human/environmental bacterial populations.
Authors’ Contributions
T.X.: Acquisition of data: laboratory or clinical, analysis of data, drafting of article and/or critical revision, and final approval of article. J.C.: Acquisition of data: laboratory or clinical, analysis of data, and drafting of article and/or critical revision. S.W.: Acquisition of data: laboratory or clinical and analysis of data. Q.Z.: Acquisition of data: laboratory or clinical. W.X., S.S., X.L., F.Y., and J.D.: Analysis of data. X.L. and W.S.: Conception and design of study, drafting of article, and/or critical revision. W.N.: Conception and design of study, drafting of article and/or critical revision, and final approval of article.
Ethical Approval
It was a study that focuses on bacteria and does not contain any sensitive personal information. The current study was approved by the Ethics Committee of the Zhuhai People’s Hospital. Thus, informed consent was not required in accordance with “Measures for the Ethical Review of Biomedical Research Involving Humans” (https://www.gov.cn/gongbao/content/2017/content_5227817.htm).
Footnotes
Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
This study was supported financially by the grants from the Natural Science Foundation of Guizhou Province, China (Grant No. Guizhou Province-ZK [2023] General 244), the Zhuhai Industry-University-Research Cooperation Project (ZH22017001210008PWC), the Science and Technology Projects of Social Development in Zhuhai (Grant No. ZH22036201210111PWC), and the Xiangshan Talent Project of Zhuhai People’s Hospital (Grant No. 2020XSYC-02).