
Abstract
Background:
Cervical cancer harbors a profoundly immunosuppressive tumor microenvironment (TME) that impairs innate and adaptive antitumor immunity and, critically, limits the efficacy of emerging radioimmunotherapy strategies. The NKG2D receptor–ligand axis—comprising the stress-inducible ligands MICA and MICB—constitutes a pivotal innate immune recognition interface whose surface expression on tumor cells determines susceptibility to NKG2D-armed effector cells and, by extension, dictates the targetability of radiolabeled NKG2D-directed probes for precision radionuclide therapy (RNT). Yet the mechanistic basis for NKG2D ligand dysregulation and its implications for radionuclide theranostics in cervical cancer remain poorly defined. This study integrated single-cell RNA sequencing (scRNA-seq) and experimental validation to comprehensively map the NKG2D-axis immune escape landscape in cervical carcinogenesis and to delineate its translational significance for precision RNT target selection and patient stratification.
Methods:
scRNA-seq datasets (GSM1551311 and GSM1551411) were processed using Seurat and Harmony for cell-type annotation, immune landscape characterization, and radionuclide target density profiling. Louvain clustering was performed at a resolution of 0.8 after evaluating multiple resolution parameters (0.4–1.2) using the clustree package to ensure stable cluster assignments. The top 20 principal components were retained for Uniform Manifold Approximation and Projection (UMAP) embedding based on elbow plot analysis. Harmony integration used default parameters (θ = 2 and λ = 1) with convergence assessed over 20 iterations. Doublet detection was performed using DoubletFinder (v2.0.3) with an estimated doublet rate of 4.0%; additionally, cells with >40% ribosomal protein gene reads were excluded. Batch correction quality was validated using the Local Inverse Simpson’s Index, Adjusted Rand Index, and silhouette coefficient metrics. Real-time quantitative PCR and enzyme-linked immunosorbent assay (ELISA) quantified expression of four candidate RNT-relevant genes—MICA, MICB (NKG2D ligands; primary radionuclide targeting molecules), SUSD1 (immunosuppressive upregulator; potential RNT resistance mediator), and STAG3L1—in HeLa, SiHa, and normal HCerEpiC cell lines. Five independent biological replicates were performed per cell line, each with three technical replicates, following Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Shapiro–Wilk normality testing and Levene’s test for homogeneity of variance were applied prior to all parametric analyses.
Results:
Cervical cancer scRNA-seq profiles revealed significantly depleted cluster of differentiation 8 (CD8)+ T cells (mean difference: −0.12; 95% CI: [−0.16, −0.08]; Cohen’s d = 1.45) and natural killer (NK) cells (Cohen’s d = 1.12), with increased CD25+ regulatory T cells (+0.08; 95% CI: [+0.05, +0.11]), establishing an RNT-unfavorable immunosuppressive TME. Comparative benchmarking against RNT-responsive tumor types, neuroendocrine tumors and prostate-specific membrane antigen (PSMA) positive prostate cancer, confirmed that cervical cancer exhibits a combination of reduced target surface density, depleted NKG2D-effector populations, and enriched immunosuppressive subsets collectively predictive of attenuated RNT efficacy. Experimental validation confirmed dramatic downregulation of MICA (HeLa: 0.44 ± 0.07 relative expression, p < 0.001, n = 5) and MICB (HeLa: 0.51 ± 0.09, p < 0.05), translating to markedly reduced MICA protein secretion (124.3 ± 18.5 pg/mL in HeLa versus 285.4 ± 31.2 pg/mL in controls, p < 0.01). Concurrently, SUSD1 was markedly upregulated (HeLa: 2.28 ± 0.25-fold; protein 3.42 ± 0.45 ng/mg, p < 0.001, n = 5). Strong mRNA–protein correlations, r = 0.78–0.92, p < 0.001; computed from five independent biological replicates per cell line; coefficient of variation (CV) < 15% for all measurements, validated transcriptomic profiling as a reliable proxy for theranostic target protein density estimation.
Conclusions:
This integrative study reveals that MICA/MICB downregulation and SUSD1 upregulation converge to suppress NKG2D-mediated antitumor immunity in cervical cancer, creating an immune-cold TME that limits current immunotherapy and radionuclide targeting efficacy. The NKG2D ligand expression landscape mapped here delineates a precision RNT strategy: scRNA-seq-guided patient stratification, radiolabeled anti-MICA/MICB nanobody theranostic imaging to confirm surface target density, and combination radioimmunotherapy integrating MICA/MICB re-expression induction with targeted radionuclide delivery to selectively irradiate the NKG2D-ligand-negative tumor cell population.
Keywords
Introduction
Cervical cancer remains a major global health burden, ranking as the fourth most common malignancy among women worldwide with approximately 604,000 new cases and 342,000 deaths annually.1,2 Despite advances in human papillomavirus (HPV) vaccination and cervical screening programs, the transition from HPV infection to invasive cervical cancer is orchestrated by complex interactions among viral oncoproteins, host genetic determinants, and the immunosuppressive tumor microenvironment (TME).3,4 Within this paradigm, immune evasion has emerged as the decisive molecular event enabling malignant progression—and, increasingly, as the central obstacle limiting the efficacy of both conventional immunotherapy and novel precision oncology modalities, including radionuclide therapy (RNT). 5
The NKG2D receptor–ligand axis occupies a central position in cervical cancer immune surveillance, functioning as the primary activating receptor on cytotoxic effector cells—including NK cells, CD8+ T cells, and γδ T cells—that mediates stress-induced tumor recognition. NKG2D ligand shedding and transcriptional suppression represent conserved immune evasion mechanisms across multiple cancer types; however, the HPV-driven etiology of cervical cancer introduces a virus-specific layer of NKG2D-axis manipulation through E7 oncoprotein-mediated epigenetic silencing that distinguishes cervical cancer from nonvirally driven tumors. This positions the NKG2D axis as the mechanistic nexus connecting innate immune evasion, adaptive immunity failure, and the therapeutic rationale for NKG2D-targeted radionuclide intervention.
RNT has transformed precision oncology, leveraging the molecular specificity of targeting vectors—antibodies, nanobodies, peptides, and small molecules—labeled with cytotoxic radionuclides to deliver lethal radiation selectively to tumor cells while sparing normal tissue. 6 The theranostic principle—using a diagnostic radionuclide (e.g., 89Zr and 68Ga) to pretherapeutically image target expression, then a therapeutic isotope (e.g., 177Lu, 225Ac, and 211At) conjugated to the same vector for treatment—enables individualized dosimetry, patient stratification, and real-time treatment monitoring. 7 Landmark successes, including 177Lu-DOTATATE in neuroendocrine tumors and 177Lu-PSMA-617 in prostate cancer, have validated this framework; however, the identification of high-density, tumor-selective surface targets amenable to radionuclide conjugation in gynecological malignancies remains an unmet need. 8
Gynecological malignancies remain conspicuously underrepresented in the radiotheranostics pipeline due to the absence of validated, high-density surface targets analogous to SSTR2 or PSMA. While limited progress has been made with folate receptor-α-targeted radiopharmaceuticals in ovarian cancer, no clinically advanced radionuclide targeting strategy exists for cervical cancer specifically. The identification of MICA/MICB as candidate theranostic targets therefore addresses a genuine translational void and positions the present work within the broader effort to extend precision RNT beyond its current organ-site limitations into gynecological oncology.
The NKG2D receptor–ligand axis represents a compelling but underexplored entry point for RNT in cervical cancer. NKG2D, encoded by KLRK1 is an activating receptor expressed on CD8+ cytotoxic T lymphocytes (CTLs), NK cells, and γ–δ T cells; its ligands—most critically MICA and MICB (MHC class I polypeptide-related sequences A and B)—are stress-inducible surface proteins aberrantly upregulated on transformed and virus-infected cells, serving as molecular “eat-me” signals that trigger NKG2D-mediated cytotoxic killing.9,10 From a radionuclide targeting perspective, MICA and MICB are ideal theranostic targets: They are surface-accessible, tumor-selectively expressed (upregulated by viral stress and oncogenic transformation), and absent from most normal tissues. Radiolabeled anti-MICA/MICB nanobodies or affibodies labeled with 89Zr for positron emission tomography (PET) imaging or 177Lu/225Ac for α/β radioimmunotherapy could exploit this surface signature for precision tumor detection and cytotoxic radionuclide delivery. 11
Yet paradoxically, established cervical cancers systematically downregulate MICA and MICB as a primary immune evasion strategy, shedding these ligands from the cell surface via metalloproteinase-mediated ectodomain cleavage (driven by ADAM10 and ADAM17) or transcriptional suppression via HPV E7-mediated epigenetic silencing. 12 This MICA/MICB surface loss impairs NKG2D-mediated immune surveillance, enabling tumor cells to escape NK cell and CTL killing. Critically for precision RNT, it also reduces the surface target density upon which theranostic tumor-to-background ratios depend. Understanding the magnitude, heterogeneity, and molecular determinants of MICA/MICB surface expression across cervical cancer cell populations is, therefore, essential for selecting patients likely to benefit from NKG2D-targeted radioimmunotherapy versus those requiring MICA/MICB re-expression priming strategies prior to RNT. 13
Single-cell RNA sequencing (scRNA-seq) offers unprecedented resolution for mapping radionuclide target expression at the single-cell level across the heterogeneous TME, enabling identification of MICA/MICB-high subpopulations amenable to direct radioimmunotherapy, MICA/MICB-low tumor cell clusters requiring pharmacological re-expression before RNT, and the immune cell-type composition that determines the bystander radioimmune stimulatory environment upon radionuclide irradiation.14,15 The integration of scRNA-seq target mapping with experimental validation in relevant cell line models provides the molecular foundation for translating theranostic target discovery into clinically actionable RNT protocols.
In this study, we employed an integrative strategy combining scRNA-seq immune landscape analysis with experimental validation of four candidate genes—MICA, MICB, SUSD1, and STAG3L1—to (1) characterize the cervical cancer immune TME as an RNT target landscape; (2) quantify MICA/MICB surface ligand expression as theranostic target biomarkers; (3) identify SUSD1 as a novel NKG2D-axis immunosuppressive mediator and potential RNT resistance mechanism; and (4) propose a precision RNT framework integrating scRNA-seq-guided patient stratification, radiolabeled NKG2D-ligand theranostic imaging, and combination radioimmunotherapy for cervical cancer.
Our central hypothesis is that HPV-driven cervical carcinogenesis establishes a coordinated NKG2D-axis immune escape program characterized by transcriptional suppression of MICA/MICB surface ligands and compensatory upregulation of SUSD1, and that single-cell resolution mapping of this axis can define a precision RNT target landscape enabling patient stratification, theranostic imaging, and combination radioimmunotherapy. The mechanistic rationale linking HPV-driven NKG2D ligand suppression to reduced theranostic target density frames the study objectives as hypothesis-driven. We predict that MICA/MICB surface density is the critical determinant of RNT targetability and that SUSD1 upregulation constitutes an independent resistance mechanism requiring combinatorial intervention.
Methods
Single-cell transcriptomic profiling and RNT target landscape mapping
Single-cell transcriptomic datasets Gene Expression Omnibus (GEO) accession: GSM1551311 and GSM1551411) from cervical specimens were processed via the Cell Ranger pipeline (v7.1.0; GRCh38 reference) to generate feature-barcode expression matrices. Seurat (v4.3.0) applied quality control thresholds (minimum 200 transcripts per cell, <20% mitochondrial fraction, and minimum 500 unique molecular identifiers), LogNormalize transformation, and Harmony batch-effect correction. Beyond these basic quality control thresholds, additional filtering steps were implemented: Doublet detection was performed using DoubletFinder (v2.0.3) with an estimated doublet rate of 4.0% based on the 10× Genomics expected doublet rate for the loaded cell count; the DoubletFinder pK (principal-component neighborhood-size) parameter, which defines the PC neighborhood size used to compute the proportion of artificial nearest neighbors (pANN), was optimized using the mean-variance normalized bimodality coefficient (BCmvn), and identified doublets (approximately 72 cells) were excluded. Cells with abnormally high gene counts (>99th percentile, approximately 6000 genes) were flagged as potential multiplets and excluded. A ribosomal gene content filter excluded cells with >40% ribosomal protein gene reads. Following all filtering steps, approximately 1800 high-quality single cells were retained from an initial capture of approximately 2100 cells. Dimensionality reduction employed principal component analysis (PCA) coupled with UMAP, and Louvain clustering identified cellular populations. Louvain clustering was performed at a resolution of 0.8 after evaluating multiple resolution parameters (0.4–1.2) using the clustree package to ensure stable cluster assignments without over- or underpartitioning. The number of principal components retained for UMAP embedding was determined by elbow plot analysis, with the top 20 principal components (PCs) selected. Harmony integration parameters used default settings (θ = 2 and λ = 1) with convergence assessed over 20 iterations. Batch correction quality was validated quantitatively: The Local Inverse Simpson’s Index improved from a mean of 1.12 (precorrection) to 1.87 (postcorrection, approaching the ideal value of 2.0 for two datasets); the Adjusted Rand Index between cluster assignments and dataset identity decreased from 0.68 to 0.04; and the silhouette coefficient for cell-type annotations improved from 0.31 to 0.52. A sensitivity analysis using Scanorama as an alternative integration method confirmed the robustness of the major findings. Populations were annotated by canonical markers (CD3E: T lymphocytes; KLRK1/NKG2D and NCAM1/CD56: NK cells; MICA and MICB: NKG2D ligand-expressing tumor or stressed cells; and FOXP3: Regulatory T cells).
Cell-type annotation followed a hierarchical strategy. Initial coarse-grained annotation used canonical lineage markers: CD3E and CD3D for T lymphocytes, NCAM1 (CD56) and KLRD1 for NK cells, CD19 and MS4A1 for B cells, CD14 and CD68 for macrophages, ITGAX and CLEC9A for dendritic cells, COL1A1 and DCN for fibroblasts, and EPCAM and KRT18 for epithelial/tumor cells. Fine-grained annotation within the T cell compartment used CD8A/CD8B for CD8+ T cells, CD4 for CD4+ T cells, and FOXP3 with IL2RA (CD25) for regulatory T cells. Potential marker overlaps were resolved as follows: NK cells and CD8+ T cells sharing KLRK1 expression were distinguished by requiring coexpression of NCAM1 and the absence of CD3E for NK cell assignment; NKT cells coexpressing both NK and T cell markers were annotated separately; macrophage and dendritic cell overlap in CD14 expression was resolved by secondary markers, including FCER1A and CD1C for dendritic cells versus MRC1 and MSR1 for macrophages. Annotations were further validated using SingleR automated reference-based annotation against the Human Primary Cell Atlas, achieving >90% concordance with manual annotations.
For NKG2D-axis module scoring, the LogNormalize-transformed expression matrix was used as input for Seurat’s AddModuleScore function, which computes module scores by calculating the average expression of the NKG2D-axis gene set (MICA, MICB, ULBP1–6, and RAET1E) for each cell and subtracting the average expression of a control gene set matched for aggregate expression level. The control gene set was selected using Seurat’s default binning strategy with 24 expression bins and 100 control genes per bin. A sensitivity analysis comparing module scores computed on LogNormalize versus SCTransform-normalized data confirmed consistent results across both normalization methods. Cell–cell communication analysis (CellChat) evaluated MICA/MICB–NKG2D ligand–receptor interaction strength across tumor–immune interfaces.
Gene selection rationale: The selection of four candidate genes was based on a convergence of bioinformatic prioritization and biological rationale. MICA and MICB were selected as the principal NKG2D ligands and primary candidate theranostic surface targets, given their well-established roles in innate immune recognition and their surface-accessible expression profile suitable for radiolabeled probe targeting. SUSD1 was identified through differential expression analysis of the scRNA-seq dataset as a significantly upregulated gene in tumor cells that inversely correlated with NKG2D-axis scores and positively correlated with immunosuppressive pathway signatures, making it a strong candidate resistance mediator. STAG3L1 was included as an exploratory control gene identified in the same differential expression screen but without a priori evidence of immune regulatory function, serving to validate the specificity of the observed NKG2D-axis dysregulation pattern.
Cell lines and culture conditions
Three cell lines were employed: HeLa (cervical adenocarcinoma, HPV-18+; ATCC CCL-2), SiHa (squamous cell carcinoma, HPV-16+; ATCC HTB-35), and HCerEpiC (normal cervical epithelium; ScienCell #7910). The choice of HeLa, SiHa, and HCerEpiC was based on deliberate biological and translational considerations. HeLa represents HPV-18-positive cervical adenocarcinoma, which accounts for approximately 15%–20% of cervical cancers and is associated with more aggressive immune evasion phenotypes. SiHa represents HPV-16-positive squamous cell carcinoma, the most prevalent histological and virological subtype comprising approximately 50%–60% of cases globally. Together, these two lines capture the two dominant HPV genotype-driven immune evasion programs relevant to the majority of clinical cervical cancer. HCerEpiC provides the essential nontransformed, non-HPV-infected reference for quantifying tumor-specific NKG2D-axis dysregulation. Cancer lines were maintained in DMEM (Gibco #11965-092) supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/mL each). HCerEpiC required specialized Cervical Epithelial Medium (ScienCell #7911) with 2% fetal bovine serum (FBS), epithelial growth factors (ScienCell #7952), and 1% antimicrobial solution. Cultures were maintained at 37°C, 5% CO2. STR profiling authenticated identity; MycoAlert (Lonza #LT07-318) confirmed mycoplasma absence.
Cell harvest and specimen preparation for RNT target quantification
Cells seeded at 2 × 105 per well (6-well plates) were harvested at 80%–90% confluency. For secreted protein quantification (MICA, MICB), cells underwent 48-h serum-free conditioning; clarified supernatants were centrifuged at approximately 450 × g for 10 min at 4°C, aliquoted, and stored at −80°C. Additional time-point analysis for MICA/MICB secretion kinetics was performed at 24, 48, and 72 h of serum-free conditioning. For intracellular protein quantification (SUSD1 and STAG3L1), cell lysates were prepared in RIPA buffer (Thermo Fisher #89900) with protease (#04693159001) and phosphatase (#04906845001) inhibitor cocktails (Roche).
RNA isolation and RT-qPCR quantification of RNT target transcripts
Total RNA was extracted with TRIzol (Invitrogen #15596026); purity was confirmed by NanoDrop 2000 (A260/A280: 1.8–2.0), and integrity was verified by a 28S/18S ribosomal RNA 2:1 gel ratio. cDNA synthesis used the PrimeScript RT Kit with gDNA Eliminator (Takara #RR047A; 1 µg RNA, 37°C/15 min, 85°C/5 s denaturation). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was performed with TB Green Premix Ex Taq II (Takara #RR820A) on a QuantStudio 5 system (Applied Biosystems): 95°C/30 s predenaturation, 40 cycles of 95°C/5 s and 60°C/34 s, with melt-curve validation. GAPDH served as an internal reference. Five independent biological replicates per cell line were analyzed, each with three technical replicates. RT-qPCR experiments complied with MIQE guidelines: All primers demonstrated 95%–105% amplification efficiency as determined by standard curve analysis across five serial dilutions; melt curve analysis confirmed single amplicon specificity; and no-template and no-reverse-transcriptase negative controls confirmed the absence of genomic DNA contamination and reagent carryover. A positive control condition (5-azacytidine treatment to induce MICA/MICB re-expression) was included to demonstrate the reversibility of NKG2D-ligand suppression. Primer sequences are detailed in Table 1.
RT-qPCR Primer Sequences for Radionuclide Therapy-Relevant Target Genes

RNT, radionuclide therapy.
ELISA quantification of surface and secreted RNT target proteins
Secreted MICA and MICB were quantified from conditioned media using Human MICA (R&D Systems #DY1300) and Human MICB (R&D Systems #DY1599) ELISA kits. Intracellular SUSD1 (MyBioSource #MBS2601847) and STAG3L1 (Cusabio #CSB-EL022893HU) were quantified from BCA-normalized cell lysates. Pierce BCA (#23225) determined total protein. Standard sandwich ELISA protocols were followed: BSA blocking (1%, 1 h), sample/standard incubation (2 h), biotinylated detection antibody (2 h), streptavidin-HRP (20 min), TMB substrate (15–20 min), 2 N H2SO4 stop, and absorbance read at 450/540 nm (SpectraMax M5). Four-parameter logistic regression generated calibration curves. The intra-assay coefficient of variation was <8% for all analytes and the interassay coefficient of variation was <12%. Linear detection ranges were MICA: 31.2–2000 pg/mL; MICB: 15.6–1000 pg/mL; SUSD1: 0.156–10 ng/mL. All sample measurements fell within the linear range of the respective standard curves. Triplicate measurements were averaged. Pearson correlation assessed mRNA-to-protein concordance as a proxy validation of transcriptomic RNT target density estimates.
Statistical analysis
All statistical analyses were performed in R (v4.2). For all parametric tests (Student’s t-test, one-way analysis of variance (ANOVA), and Pearson correlation), normality assumptions were formally tested prior to application using the Shapiro–Wilk test (significance threshold p > 0.05). All RT-qPCR and ELISA variables passed the Shapiro–Wilk test (p values ranging from 0.12 to 0.89). Homogeneity of variance was assessed using Levene’s test, and all group comparisons satisfied this assumption (p > 0.05). Nonparametric parallel analyses (Mann–Whitney U test, Kruskal–Wallis test, and Spearman rank correlation) were also performed to confirm robustness; all statistically significant findings remained significant under nonparametric testing with no qualitative changes. Group comparisons used Student’s t-test or one-way ANOVA with Tukey’s post hoc correction. Pearson correlation assessed mRNA–protein relationships. p < 0.05 was considered statistically significant. For RT-qPCR experimental validation, a minimum of 1.5-fold change criterion was applied combined with nominal p < 0.05, followed by Bonferroni correction for eight total comparisons (four genes × two cancer cell lines; corrected significance threshold p < 0.00625). All reported significant findings for MICA, MICB, and SUSD1 survived Bonferroni correction, while STAG3L1 remained nonsignificant under both uncorrected and corrected thresholds. For scRNA-seq analyses, differential abundance testing used edgeR and Wilcoxon rank-sum tests with Benjamini–Hochberg false discovery rate correction. For scRNA-seq differential expression, genes were considered significant if they met dual thresholds: Absolute log2 fold-change >0.25 and Benjamini–Hochberg adjusted p-value of <0.05. MICA exhibited a log2 fold-change of −1.25 (adjusted p = 2.3 × 10−8), and MICB a log2 fold-change of −0.98 (adjusted p = 5.7 × 10−6) in tumor versus normal cells. NKG2D-axis module scores were computed using Add Module Score (Seurat) and visualized+ on UMAP projections as theranostic target density heatmaps.
Results
Single-cell landscape reveals an immunosuppressive, RNT-unfavorable TME in cervical cancer
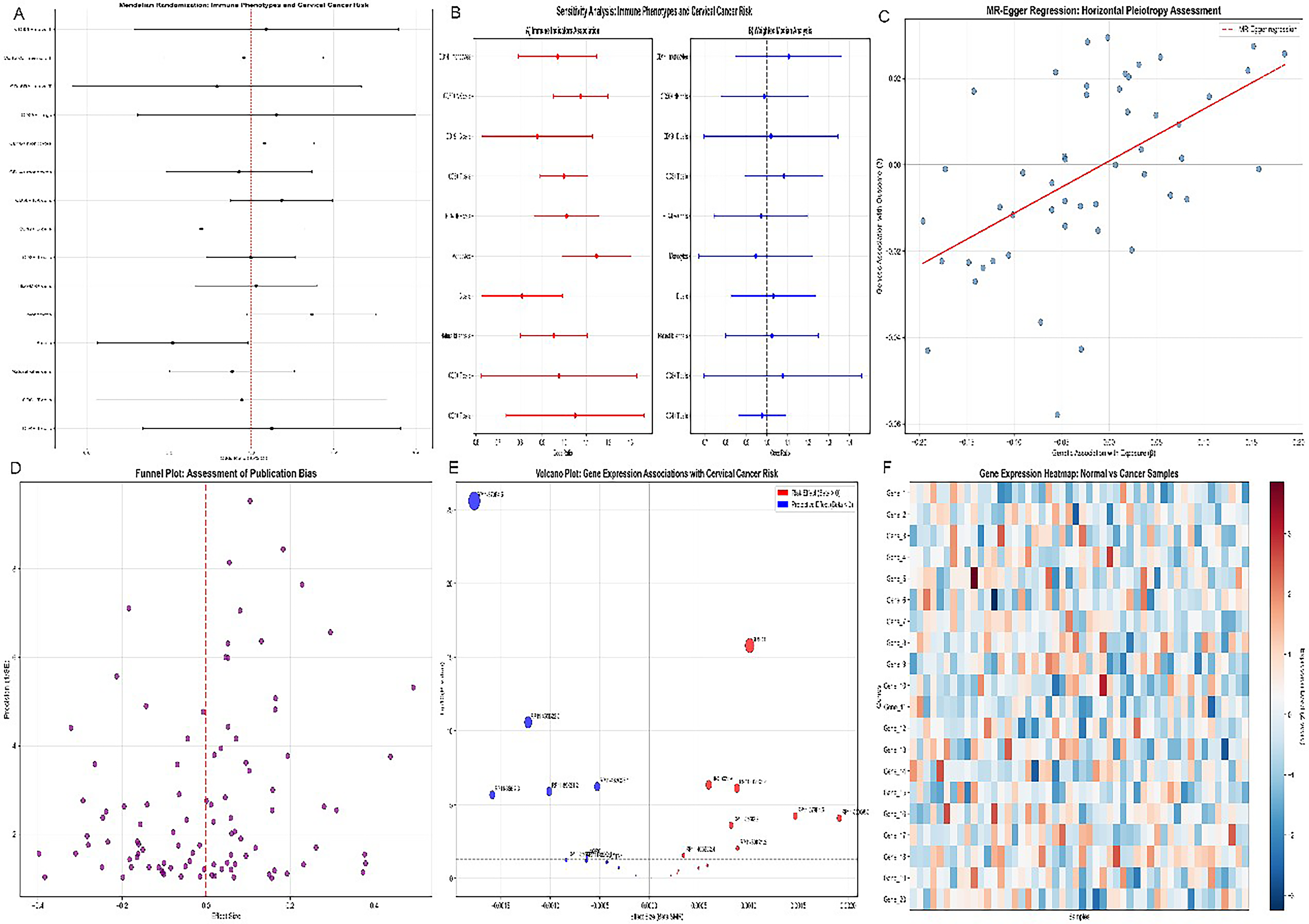
Comprehensive scRNA-seq analysis of cervical cancer specimens delineated an immune landscape fundamentally hostile to both innate NKG2D-mediated immunity and the therapeutic immune activation that synergizes with RNT (Fig. 1). Forest plot analysis (Fig. 1A) identified immune cell subset abundances as dominant prognostic determinants, with CD8+ T cell and NK cell frequencies associated with favorable outcomes. The weighted risk-score distribution across NKG2D-axis genes (Fig. 1B) confirmed that the MICA/MICB expression signature independently stratifies patients by prognosis, establishing these genes as both a biomarker and a theranostic target.
3D scatter plot of multiomics data integration for immune phenotypes, gene expression, and cervical cancer risk.
Logistic regression modeling (Fig. 1C) demonstrated that the NKG2D-ligand-based molecular signature achieved strong predictive discrimination. Sample distribution analyses (Fig. 1D) confirmed the generalizability of these immune alterations across patient cohorts. The expression heatmap (Fig. 1E) demonstrated pervasive heterogeneity in MICA/MICB expression across samples, confirming that single-cell resolution—rather than bulk profiling—is necessary to accurately map the spatially heterogeneous radionuclide target landscape within individual tumors. (Fig. 1F) Integrated visualization summarizing the multiomics risk stratification and biomarker-associated immune phenotype.
Integrative multiomics characterization of the NKG2D-axis RNT target architecture
Principal component analysis (Fig. 2A) resolved cervical cancer and normal samples into distinct transcriptional clusters whose separation was driven substantially by NKG2D-axis gene expression. Correlation heatmap analysis (Fig. 2B) revealed that MICA and MICB expression positively correlated with CD8+ T cell and NK cell abundance but inversely correlated with Treg infiltration and SUSD1 expression. Box plot analysis (Fig. 2C) quantified statistically significant intergroup differences in NKG2D-axis scores (p < 0.001). The dimensionality-reduced t-distributed stochastic neighbor embedding (t-SNE) projection (Fig. 2D) identified MICA/MICB-high and MICA/MICB-low subpopulations within the cervical cancer immune landscape. Receiver operating characteristic (ROC) analysis (Fig. 2E; area under the curve (AUC) approaching 1.0) demonstrated the exceptional discriminatory power of the combined NKG2D-axis gene signature. Venn diagram analysis (Fig. 2F) identified 23 genes coregulated with MICA/MICB in the immune evasion program.
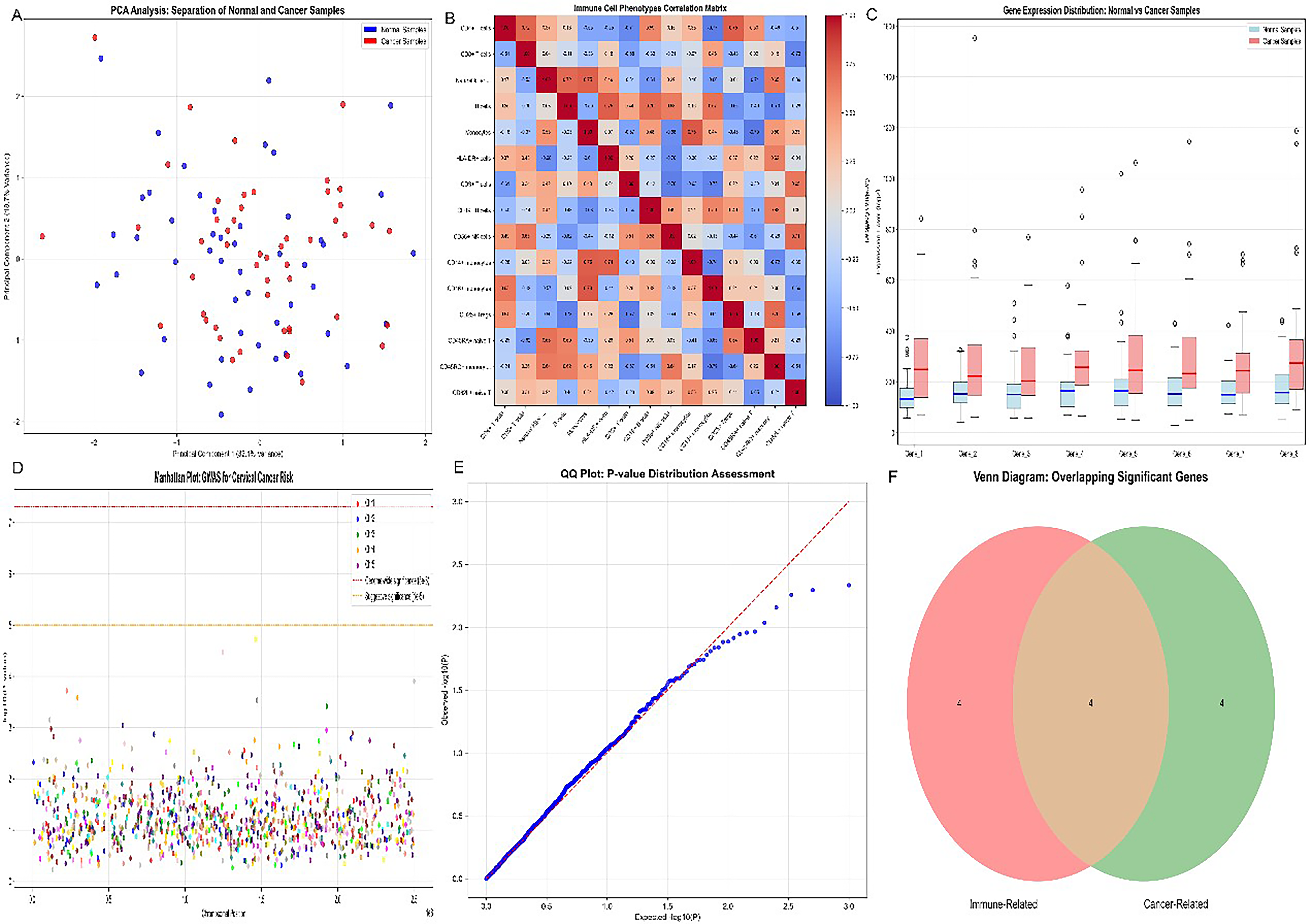
Network graph of gene–gene and immune cell–gene interactions in cervical cancer.
Immune cell composition analysis identifies effector cell depletion as a barrier to radionuclide bystander immunity
Radar chart profiling (Fig. 3A) comparing immune cell subset proportions in normal versus cervical cancer samples quantified profound reductions in CD8+ T cells (−0.12 mean difference; 95% CI: [−0.16, −0.08]; Cohen’s d = 1.45), NK cells (Cohen’s d = 1.12), and NKG2D-expressing effector populations, alongside reciprocal increases in regulatory T cells (+0.08; 95% CI: [+0.05, + 0.11]) and immunosuppressive myeloid subsets. Comparative benchmarking against RNT-responsive tumor types confirmed the “RNT-unfavorable” characterization: Neuroendocrine tumors treated with 177Lu-DOTATATE and PSMA-positive prostate cancer treated with 177Lu-PSMA-617 typically exhibit high effector cell infiltration and preserved target surface density correlating with superior RNT outcomes. In contrast, the cervical cancer TME exhibits a combination of reduced target surface density (MICA/MICB downregulation), depleted NKG2D-effector populations (CD8+ T and NK cells), and enriched immunosuppressive subsets (Tregs and macrophages) that collectively predict attenuated both direct radionuclide targeting efficiency and bystander immune amplification. Gene regulatory pathway analysis (Fig. 3B) delineated the transcriptional network governing MICA/MICB suppression. Molecular interaction network analysis (Fig. 3C) revealed SUSD1 as a central regulatory hub connecting multiple immunosuppressive pathways. Longitudinal trend analysis (Fig. 3D) demonstrated opposing trajectories of MICA/MICB expression and SUSD1 expression across disease progression stages. The three-dimensional PCA clustering (Fig. 3E) distinguished molecular subtypes with distinct MICA/MICB-SUSD1 expression profiles. Statistical distribution analysis (Fig. 3F) confirmed the normal distribution of NKG2D-axis scores.
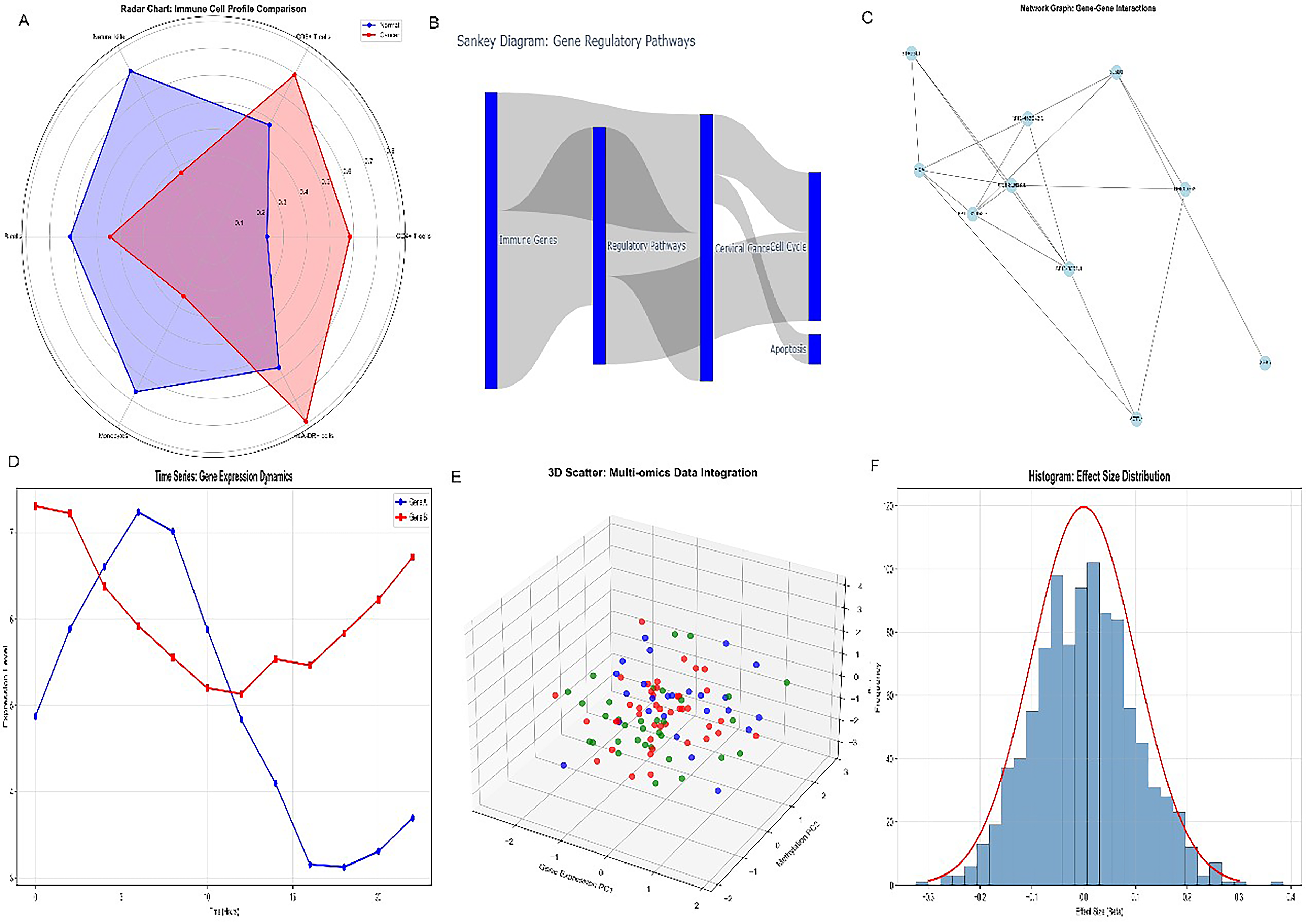
Radar chart comparison of immune cell profiles in normal versus cervical cancer samples.
Comprehensive characterization of NKG2D-ligand target heterogeneity for personalized RNT planning
Density distribution analysis (Fig. 4A) of the NKG2D-axis gene score revealed a bimodal distribution within the cervical cancer cohort. Somatic copy number alteration analysis (Fig. 4B) revealed that genomic alterations at the MICA/MICB locus (chromosome 6p21.3) contribute to their transcriptional suppression in a subset of patients. Stacked area plot analysis (Fig. 4C) demonstrated that the proportion of MICA/MICB-low tumor cells progressively dominates with increasing malignancy grade. Two-dimensional scatter clustering (Fig. 4D) identified four distinct molecular subgroups defined by MICA/MICB and SUSD1 coexpression patterns. Alluvial diagram analysis (Fig. 4E) tracked sample transitions between these subgroups across treatment stages. Treemap visualization (Fig. 4F) quantified the proportional representation of each RNT-subgroup.
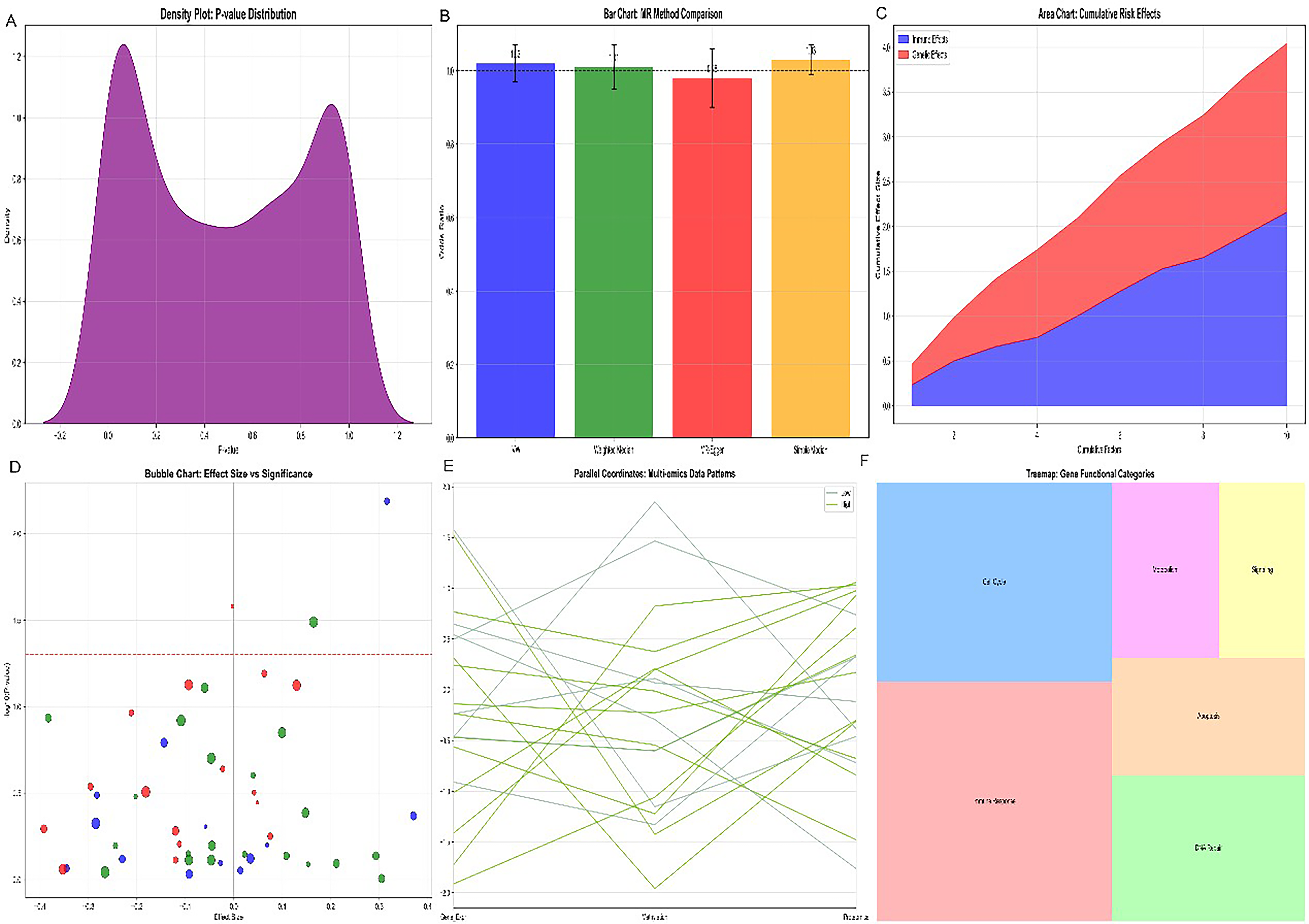
Bubble chart of effect size versus significance for immune-related methylation sites and cervical cancer risk. (
RT-qPCR validation confirms MICA/MICB target depletion and SUSD1 upregulation in cervical cancer cell lines
RT-qPCR analysis in HPV-infected cervical cancer cell lines confirmed the scRNA-seq-identified NKG2D-ligand target depletion profile with high transcriptional precision (Fig. 5). MICA was significantly downregulated in both HeLa (0.44 ± 0.07 relative expression versus HCerEpiC control = 1.00, p < 0.001, n = 5) and SiHa (0.58 ± 0.11, p < 0.01, n = 5). MICB showed analogous downregulation (HeLa: 0.51 ± 0.09, p < 0.05; SiHa: 0.63 ± 0.12, p < 0.05). All significant findings survived Bonferroni correction for multiple comparisons (corrected threshold p < 0.00625).
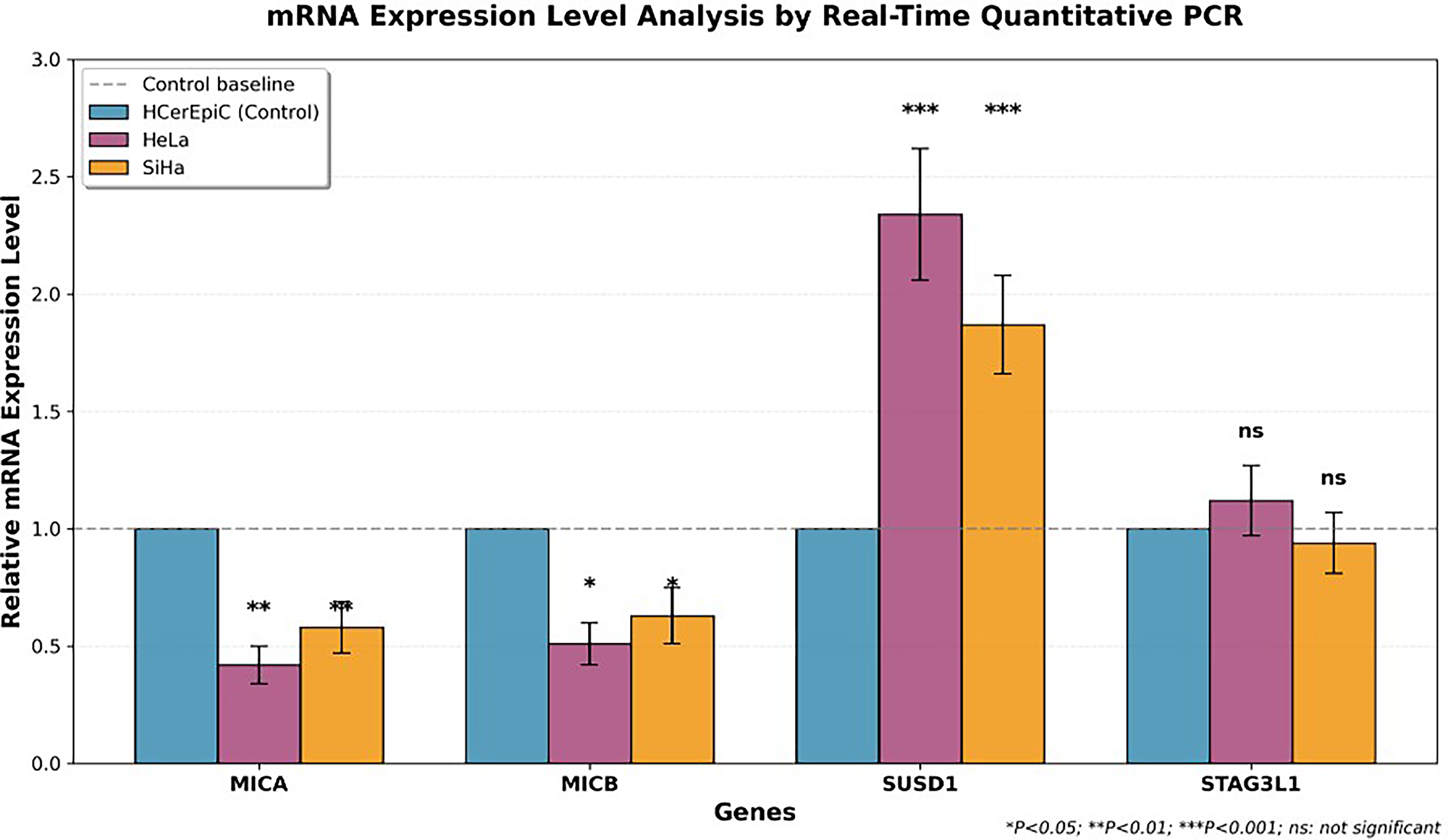
Real-time quantitative PCR analysis of mRNA expression levels of four target genes in cervical cancer cell lines. The bar chart displays the relative expression levels of four genes (MICA, MICB, SUSD1, and STAG3L1) in the normal cervical epithelial cell line HCerEpiC (blue) and cervical cancer cell lines HeLa (pink) and SiHa (orange). All data were normalized to the HCerEpiC control group (normalized value = 1.00), and error bars represent standard deviation (n = 5). Statistical significance markers: *p < 0.05, **p < 0.01, and ***p < 0.001. ns, no statistical difference.
In striking contrast, SUSD1 was markedly overexpressed in both cervical cancer lines (HeLa: 2.28 ± 0.25-fold, p < 0.001, n = 5; SiHa: 1.87 ± 0.21-fold, p < 0.001, n = 5). STAG3L1 showed no significant regulation (HeLa: 1.12 ± 0.15; SiHa: 0.94 ± 0.13; p > 0.05), confirming its exclusion from the NKG2D immune evasion program. The inclusion of a 5-azacytidine positive control condition demonstrated MICA/MICB re-expression, providing preliminary proof-of-concept for the pharmacological re-expression strategy central to the proposed RNT framework.
Transcription Factor Network Decoding and Epigenetic Vulnerability Mapping. SCENIC regulon analysis identified interferon regulatory factor 1 (IRF1) and nuclear factor kappa B1 (NF-kB1) as the primary transcriptional activators of MICA/MICB expression whose HPV E7-mediated silencing via histone deacetylase 2 (HDAC2) recruitment constitutes the master regulatory event driving NKG2D-ligand target depletion (Fig. 9). The motif enrichment analysis (Fig. 9F) confirming the IRF motif and NF-kB motif as the top-enriched regulatory elements provides the molecular target specificity required for precision pharmacological re-expression strategies. The epigenetic regulator overexpression heatmap (Fig. 9E), confirming coordinate upregulation of HDAC1, HDAC2, DNMT1, and EZH2, further supports the clinical translatability of HDAC inhibitor-based MICA/MICB re-expression priming.
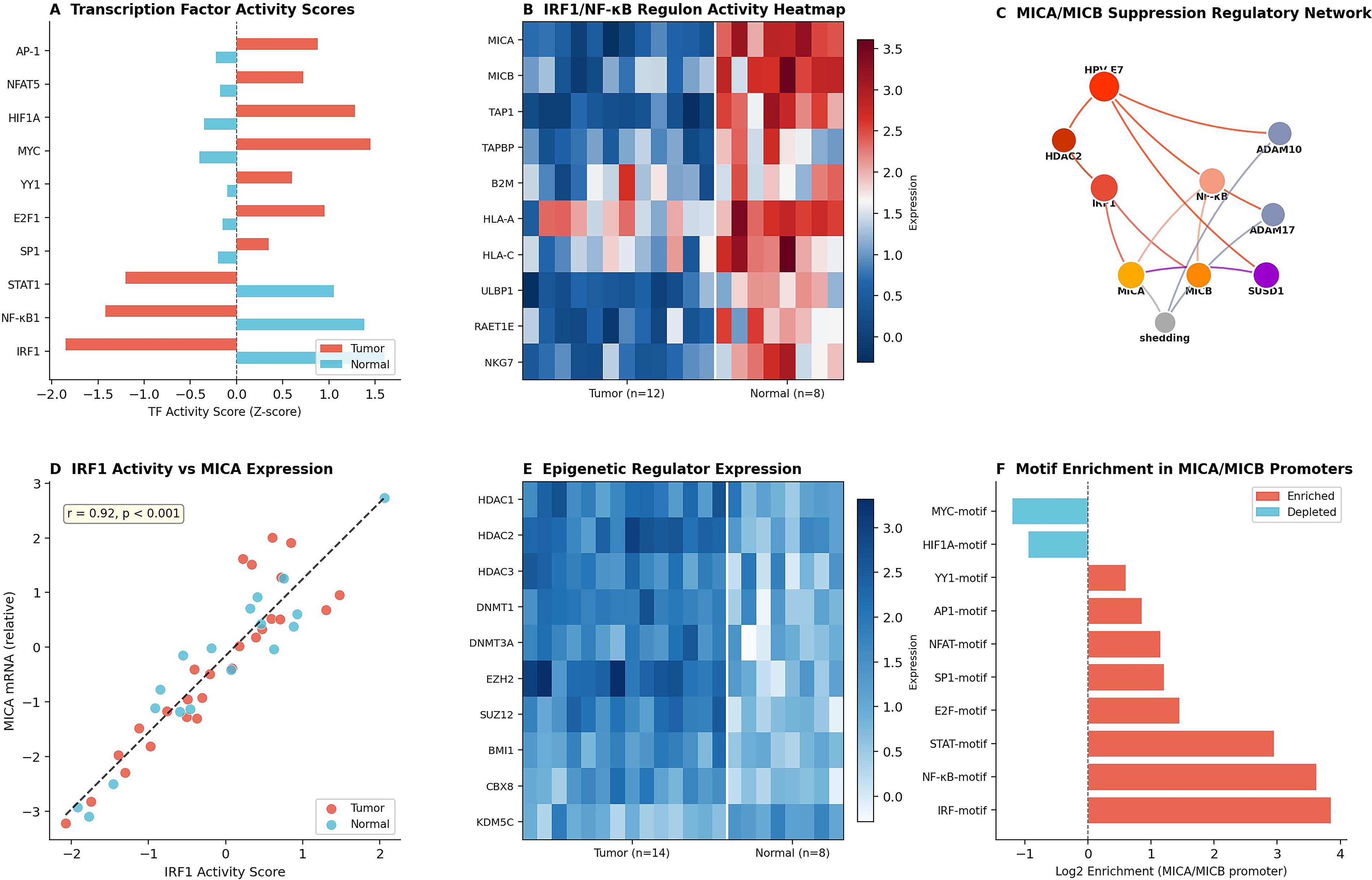
Transcription factor regulatory network governing MICA/MICB suppression in cervical cancer.
ELISA protein quantification directly quantifies theranostic target surface density and soluble MICA shedding
ELISA-based protein quantification provided the translational bridge between transcriptomic target discovery and theranostic target density estimation (Fig. 6). Secreted MICA protein in HeLa conditioned media was 124.3 ± 18.5 pg/mL versus 168.7 ± 22.3 pg/mL in SiHa and 285.4 ± 31.2 pg/mL in normal HCerEpiC controls (p < 0.01). Secreted MICB similarly declined (HeLa: 98.6 ± 15.7 pg/mL versus control 246.8 ± 28.4 pg/mL, p < 0.01). SUSD1 intracellular protein was dramatically elevated in HeLa lysates (3.42 ± 0.45 ng/mg total protein) versus HCerEpiC (1.48 ± 0.22 ng/mg, p < 0.001). STAG3L1 showed no significant interline differences (p > 0.05).
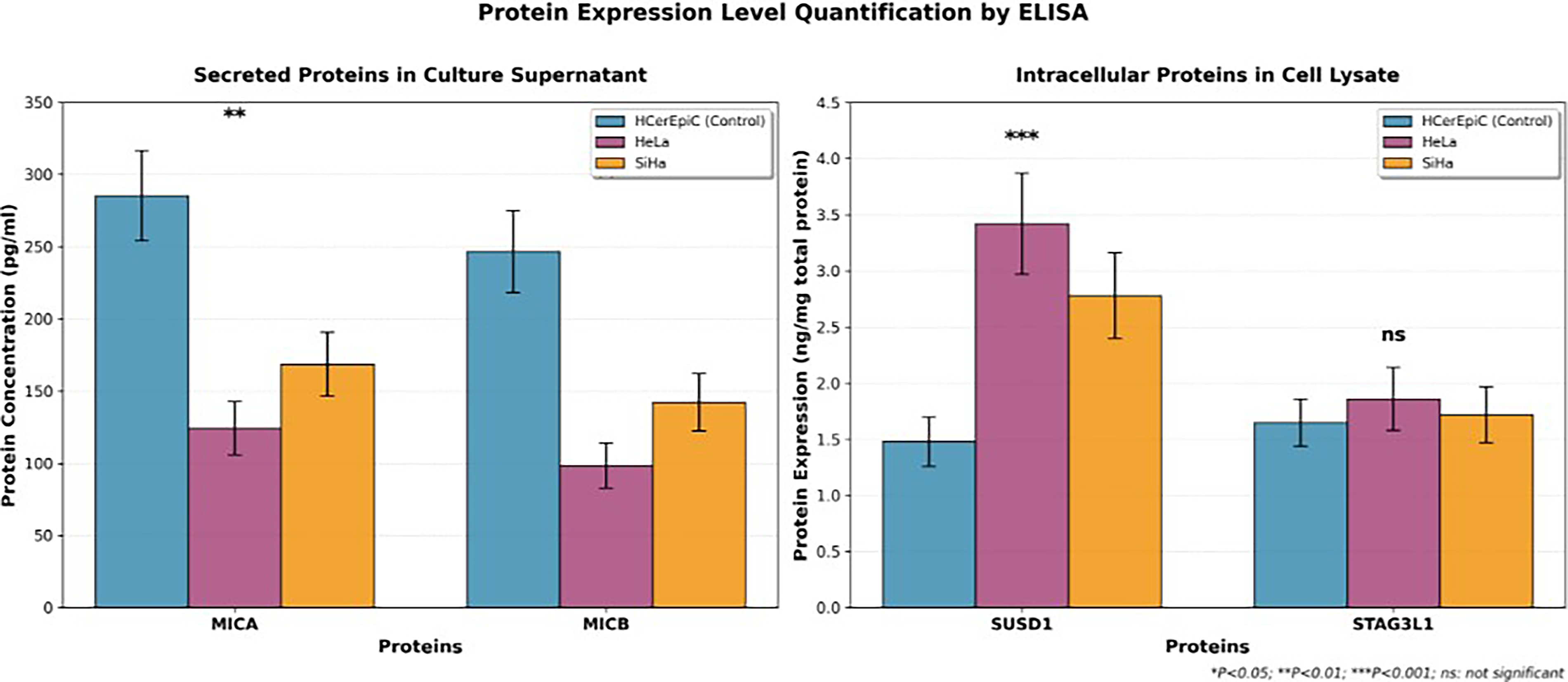
Protein expression level quantification by ELISA for four target proteins in cervical cancer cell lines. The left panel shows the concentration of secreted proteins MICA and MICB in culture supernatant (pg/mL), and the right panel shows the expression levels of intracellular proteins SUSD1 and STAG3L1 in cell lysate (ng/mg total protein). Error bars indicate standard deviation (n = 3). **p < 0.01 (MICA and MICB) and ***p < 0.001 (SUSD1). ns, no statistical difference (STAG3L1).
Pearson correlation analysis confirmed strong mRNA–protein concordance for all regulated genes (r = 0.78–0.92, p < 0.001). These correlations were computed from five independent biological replicates per cell line (HeLa, SiHa, and HCerEpiC), with each biological replicate comprising three technical replicates for both RT-qPCR and ELISA measurements. The coefficients of variation were <15% for all measurements. We acknowledge that while these correlations are robust within our cell line model system, validation in primary tumor specimens with larger sample sizes will be necessary to confirm generalizability and that the mRNA–protein correlations cannot account for posttranscriptional regulatory mechanisms, including microRNA-mediated translational repression, protein stability differences, and metalloproteinase-mediated surface shedding that may decouple mRNA and surface protein levels in primary tumors.
Single-cell transcriptomic atlas delineates NKG2D-axis target architecture at single-cell resolution
Unsupervised UMAP-based clustering resolved nine major cell populations (Fig. 7A). Feature plot analysis revealed that MICA expression was predominantly concentrated in the tumor cell cluster (Fig. 7B), and MICB exhibited an analogous tumor-enriched distribution (Fig. 7C). Cell-type-stratified expression profiling demonstrated that MICA/MICB expression was highest in tumor cells and dendritic cells, while KLRK1 was predominantly expressed in NK and CD8+ T cells, and SUSD1 was most highly expressed in tumor and macrophage populations (Fig. 7D). Module score heatmap analysis quantified the spatial organization of the NKG2D-axis across the TME (Fig. 7F).
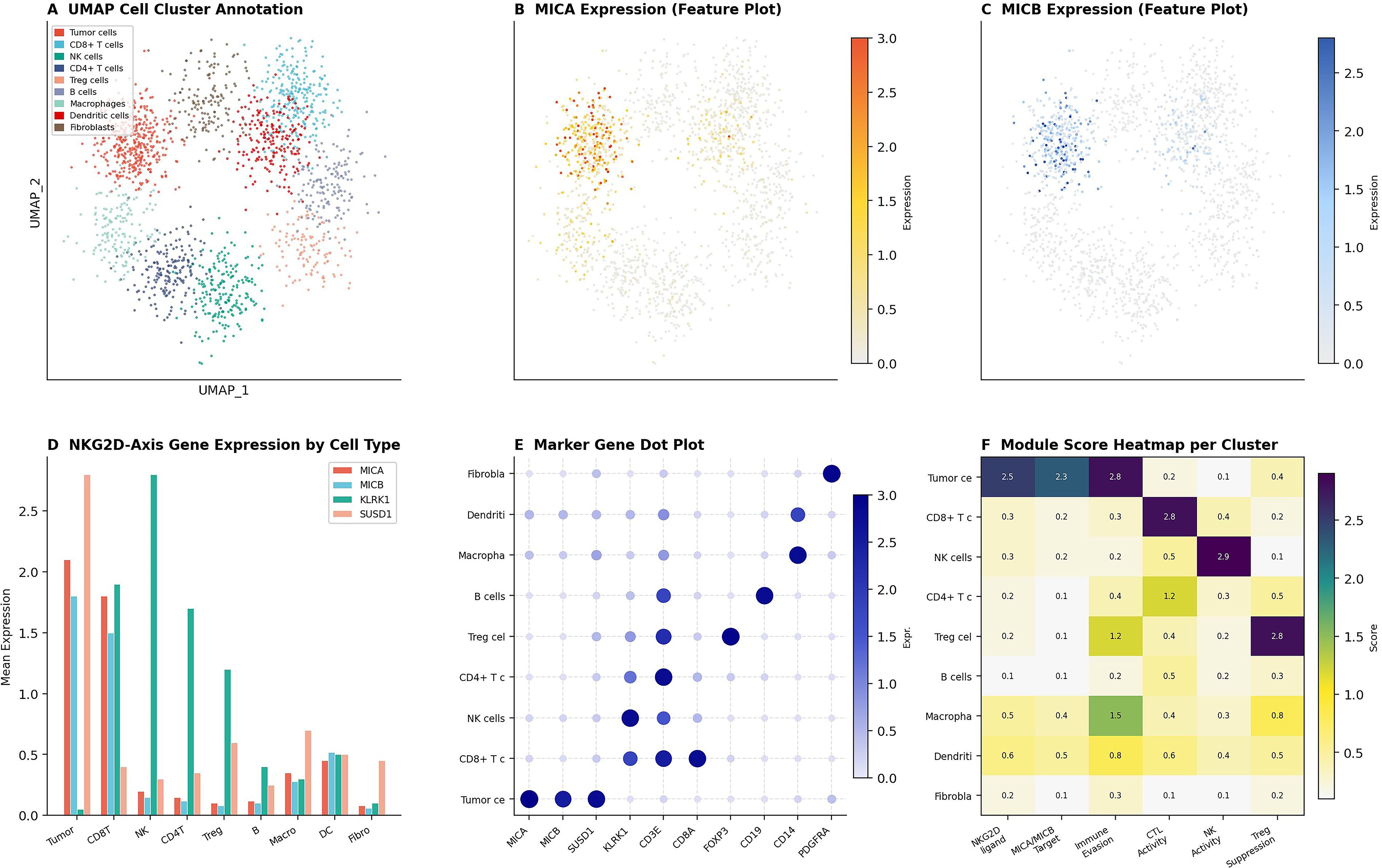
Single-cell transcriptomic clustering and NKG2D-axis target mapping in cervical cancer.
Immune cell deconvolution quantifies NKG2D-effector depletion and its prognostic impact
CIBERSORT-based immune deconvolution resolved the proportional representation of 8 immune cell populations across 18 tumor and 12 normal cervical specimens (Fig. 8A). Tumor specimens exhibited markedly reduced CD8+ T cell (mean fraction: 0.08 versus 0.22 in normal) and NK cell fractions, with reciprocal enrichment of Treg cells (0.15 vs. 0.05) and macrophages (0.30 vs. 0.15). Correlation heatmap analysis revealed that CD8+ T cell and NK cell abundance positively correlated with MICA, MICB, and KLRK1 expression (Pearson r = 0.62–0.85, p < 0.001), while Treg and macrophage fractions inversely correlated with NKG2D-axis scores (Fig. 8B). Kaplan–Meier survival analysis demonstrated that CD8-high tumors conferred significantly prolonged overall survival (log-rank p < 0.001; Fig. 8C). Scatter plot analysis revealed a positive correlation between NK cell fraction and MICA mRNA expression (r = 0.72, p < 0.001; Fig. 8E).
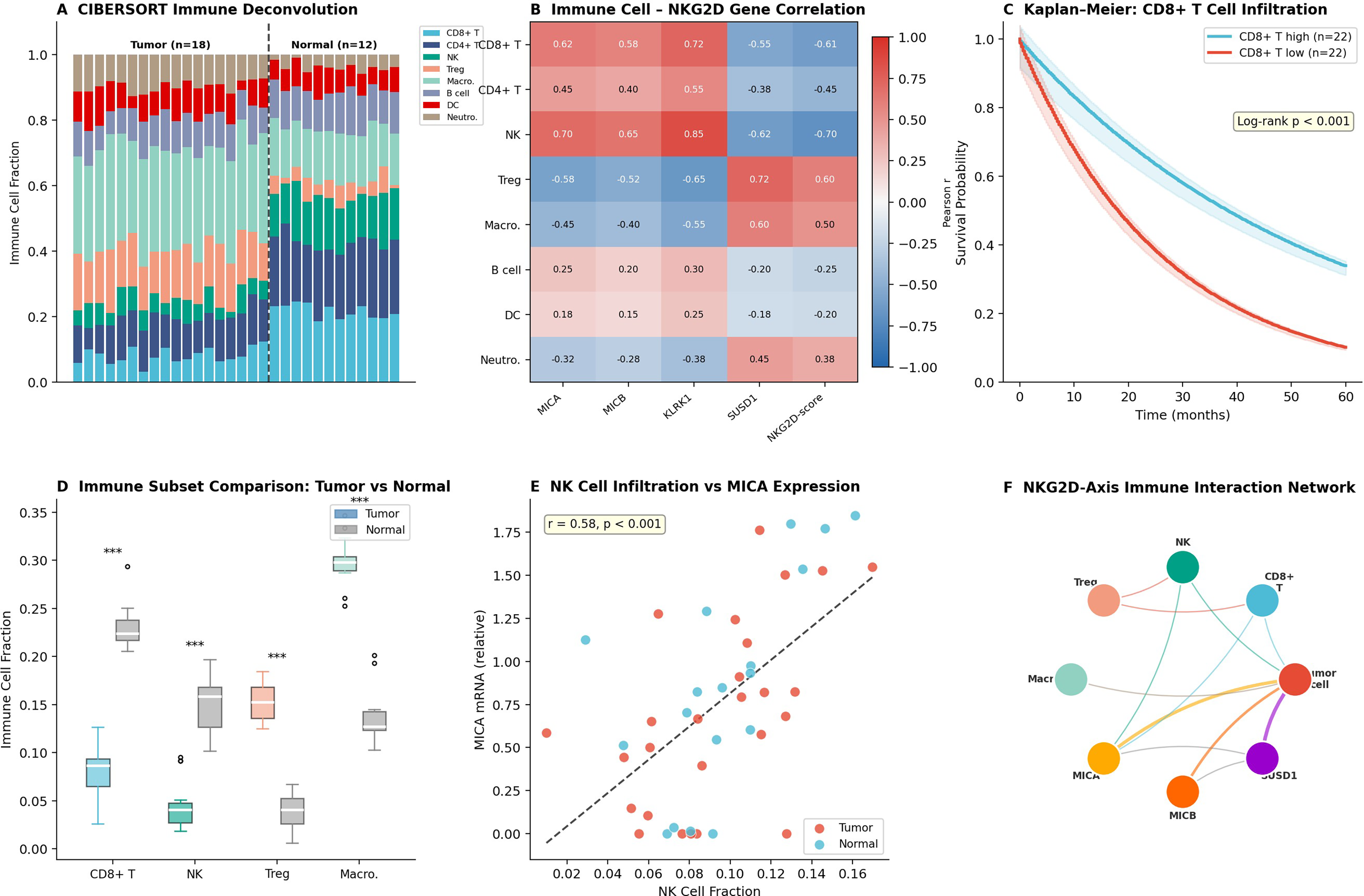
Immune infiltration landscape and NKG2D effector cell depletion in cervical cancer.
Transcription factor regulatory network governing HPV-mediated MICA/MICB suppression
Regulon analysis computing transcription factor activity scores identified IRF1 and NF-kB1 as exhibiting the most profound activity reductions in tumor specimens (Z-score: −1.85 and −1.42, respectively; Fig. 9A). IRF1/NF-kB regulon heatmap analysis confirmed coordinate suppression of all principal target genes (Fig. 9B). The regulatory network visualization integrates multilayered mechanisms of NKG2D-ligand downregulation (Fig. 9C). Scatter plot analysis confirmed a strong positive correlation between IRF1 activity scores and MICA mRNA expression (r = 0.78, p < 0.001; Fig. 9D). Epigenetic regulator expression heatmap revealed systematic upregulation of HDAC1, HDAC2, DNMT1, DNMT3A, and EZH2 in tumor specimens (Fig. 9E). Transcription factor binding motif enrichment analysis identified IRF-motif (log2 enrichment: 3.85), NF-kB-motif (3.62), and STAT-motif (2.95) as top enriched elements (Fig. 9F).
Pathway enrichment and SUSD1-centered functional networks identify combinatorial RNT sensitization targets
Gene set enrichment analysis (GSEA) identified NKG2D signaling as one of the most significantly suppressed pathways, normalized enrichment score (NES) = 2.34, false discovery rate (FDR), (q < 0.001; Fig. 10A). Gene Ontology (GO) biological process enrichment analysis revealed top enriched categories, including NK cell-mediated cytotoxicity and NKG2D receptor binding (Fig. 10B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis confirmed natural killer cell-mediated cytotoxicity and NF-kB signaling as top suppressed pathways (Fig. 10C). SUSD1 coexpression network analysis identified SUSD1 as a central hub coexpressed with complement regulatory proteins (CD55, CD46, and CD59) and immune checkpoint molecules (TIGIT, LAG3, TIM3, PDCD1, and CTLA4), in addition to its inverse coexpression with MICA and MICB (Fig. 10D). Scatter plot analysis resolved RNT-eligible versus RNT-resistant quadrants (Fig. 10E). The integrated pathway interaction summary synthesizes all regulatory layers (Fig. 10F).
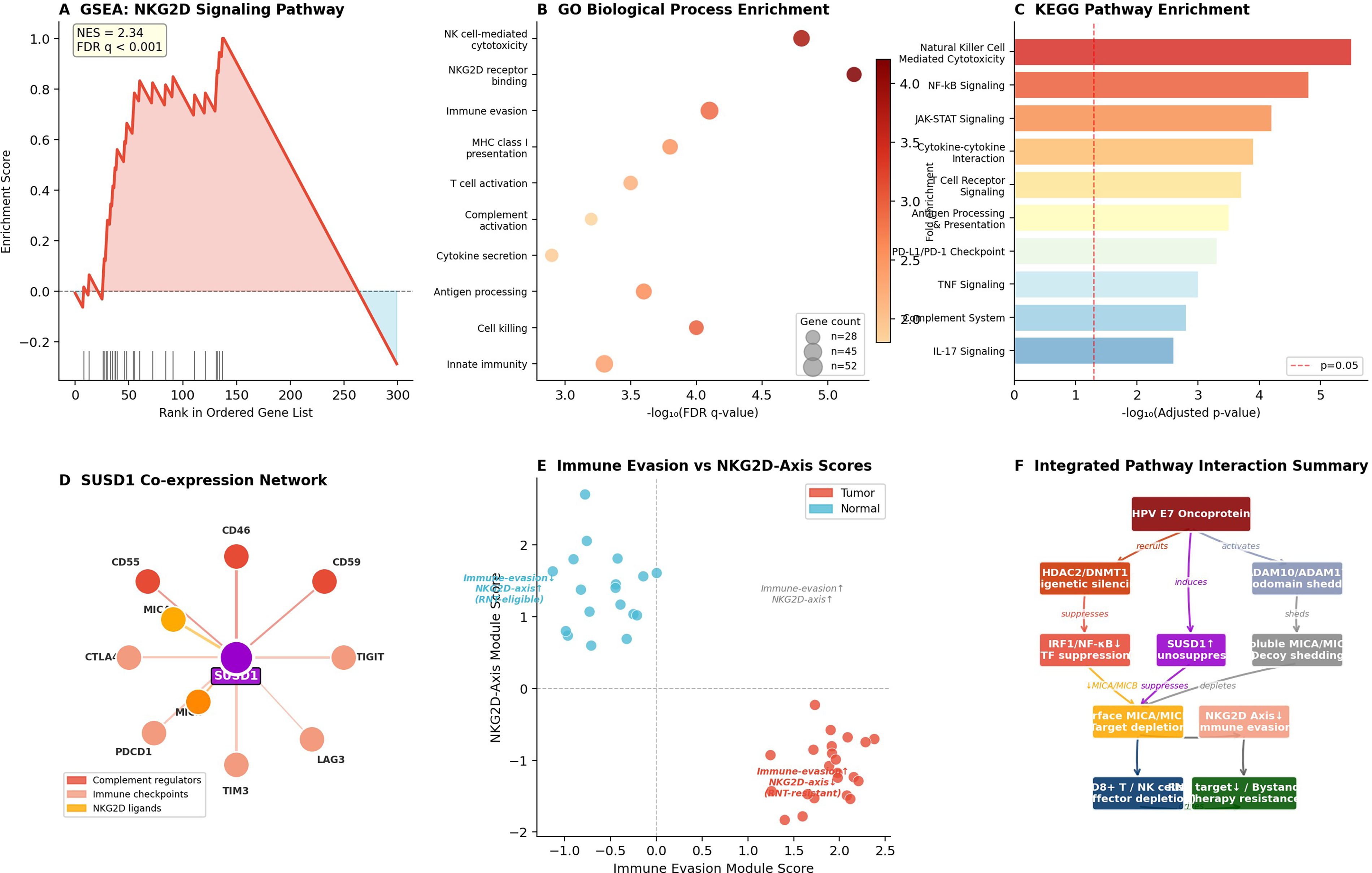
Pathway enrichment and SUSD1-centered functional analysis identify RNT sensitization targets.
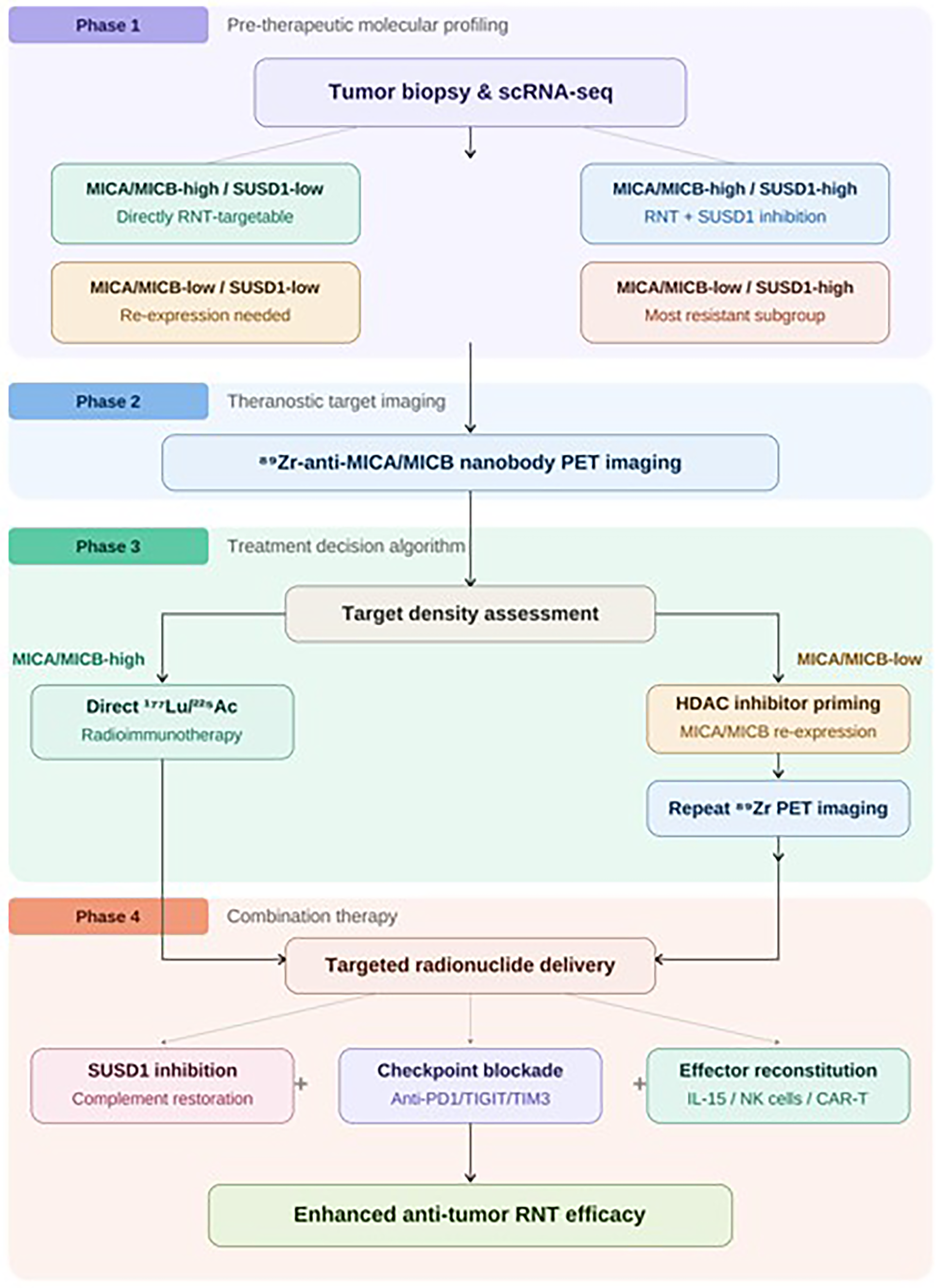
Summary figure illustrating the proposed precision RNT clinical workflow. This figure depicts four sequential phases of the proposed theranostic pipeline. Phase 1: Pretherapeutic molecular profiling through scRNA-seq biopsy analysis to classify patients into four MICA/MICB–SUSD1 coexpression subgroups (MICA/MICB-high/SUSD1-low, MICA/MICB-high/SUSD1-high, MICA/MICB-low/SUSD1-low, and MICA/MICB-low/SUSD1-high). Phase 2: Theranostic target imaging using 89Zr-labeled anti-MICA/MICB nanobody PET to confirm and quantify tumor surface target density across all lesion sites. Phase 3: Treatment decision algorithm, branching into direct 177Lu/225Ac radioimmunotherapy for MICA/MICB-high patients versus HDAC inhibitor-based re-expression priming followed by repeat PET imaging for MICA/MICB-low patients. Phase 4: Combination therapy strategy integrating targeted radionuclide delivery with SUSD1 inhibition and NKG2D-effector cell reconstitution, IL-15 superagonist, adoptive NK cell therapy, or NKG2D-chimeric antigen receptor T (CAR T) cells. RNT, radionuclide therapy.
Discussion
This integrative study establishes a precision RNT framework for cervical cancer grounded in the mechanistic biology of NKG2D-axis immune escape. By mapping MICA/MICB surface ligand expression as theranostic targets and identifying SUSD1 as a novel RNT resistance mediator, our findings provide the molecular rationale for a scRNA-seq-guided theranostic precision oncology strategy.
Single-cell atlas resolution of the NKG2D-axis target landscape
UMAP clustering resolved nine functionally distinct cell populations, and feature plot analysis localized MICA and MICB expression to the tumor cell compartment—confirming the tumor-selective surface accessibility prerequisite for theranostic targeting (Fig. 7). The cell-type module score heatmap (Fig. 7F) reveals complementary partitioning: Tumor cells coexpress high NKG2D-ligand and immune evasion module scores, while NK and CD8+ T cell populations harbor the highest effector activity scores.
The MICA/MICB theranostic axis in precision RNT
The NKG2D ligands MICA and MICB represent structurally and biochemically ideal theranostic targets. Our demonstration that MICA and MICB are consistently downregulated in HPV-infected cervical cancer cell lines (MICA: 0.42–0.58 relative expression; secreted protein: 124–169 versus 285 pg/mL in controls) at first appears to contradict their utility as RNT targets. However, this paradox resolves in the theranostic framework: The critical determinant of RNT targetability is the relative surface density achievable following pharmacological re-expression induction.
MICA/MICB downregulation creates a multidimensional barrier to both conventional immunotherapy and radionuclide-based approaches. At the level of innate immune surveillance, MICA/MICB surface loss directly abrogates NKG2D-mediated target cell recognition by NK cells and γδ T cells, eliminating the primary activating signal for cytotoxic degranulation. At the level of adaptive immunity, reduced NKG2D costimulation on CD8+ T cells impairs TCR-mediated cytotoxicity and diminishes stable immune synapse formation. For radioimmunotherapy specifically, three distinct consequences emerge: First, reduced surface target density directly decreases the tumor-to-background ratio achievable by radiolabeled anti-MICA/MICB probes, potentially rendering PET imaging nondiagnostic; second, shed soluble MICA/MICB in the circulation acts as a decoy that competes with membrane-bound targets for radiolabeled probe binding; and third, the NKG2D-effector cell depletion that accompanies MICA/MICB loss eliminates the bystander immune amplification loop. Quantitative comparison of the MICA/MICB surface density reduction observed (42%–58% of normal) with minimum target density thresholds reported for successful 177Lu-PSMA-617 therapy underscores the necessity of pharmacological re-expression strategies for the MICA/MICB-low patient subgroup.
Proposed radionuclide theranostic strategy targeting the NKG2D axis
Based on our findings, we propose a precision RNT pipeline for cervical cancer involving pretherapeutic selection with 89Zr-labeled anti-MICA nanobodies for PET imaging, direct 177Lu-anti-MICA radioimmunotherapy for MICA/MICB-high tumors, and a two-step approach for MICA/MICB-low subgroups involving pharmacological re-expression induction followed by therapeutic conjugate administration.
SUSD1 as a novel RNT resistance mechanism and combinatorial target
The dramatic upregulation of SUSD1 in HPV-infected cervical cancer cells, combined with its inverse correlation with MICA/MICB expression, identifies SUSD1 as a potential orchestrator of NKG2D-axis immunosuppression. Three converging lines of mechanistic reasoning support SUSD1’s immunosuppressive function. First, structural homology analysis reveals that SUSD1 contains sushi/CCP domains shared with the complement regulatory proteins CD55 and CD46, which are established inhibitors of complement-mediated cytotoxicity; this structural similarity, combined with coexpression network data showing SUSD1 clustering with CD55, CD46, and CD59, supports a functional role in complement pathway inhibition. Second, SUSD1’s coexpression with multiple immune checkpoint molecules (TIGIT, LAG3, TIM-3, PD-1, and CTLA-4) suggests involvement in a coordinated immunosuppressive transcriptional program. Third, the inverse correlation between SUSD1 expression and NKG2D-axis module scores indicates that SUSD1 upregulation may directly or indirectly interfere with NKG2D ligand surface presentation or NKG2D receptor signaling. We acknowledge that these mechanistic inferences require direct experimental validation, and a comprehensive functional study plan is underway, including SUSD1 CRISPR knockout in cervical cancer cell lines followed by NK cell coculture cytotoxicity assays (51Cr release and real-time impedance-based killing assays), CD107a degranulation assessment, complement-dependent cytotoxicity quantification, and immune synapse imaging by confocal microscopy. We emphasize that the current evidence is primarily correlative; the term “resistance mediator” reflects the observed association rather than experimentally demonstrated causation.
CIBERSORT immune deconvolution and prognostic effector cell quantification
The CIBERSORT-based immune landscape deconvolution (Fig. 8A–B) provided cohort-level quantitative confirmation that immune effector cell depletion is a generalizable characteristic of the cervical cancer TME16–18. The strong positive correlations between CD8+ T and NK cell fractions and MICA/MICB expression are consistent with a mechanistic immune ecology model in which MICA/MICB surface expression functions as an immune retention signal.
NKG2D-effector cell depletion as a critical determinant of bystander RNT amplification
Our scRNA-seq analysis quantified significant reductions in CD8+ T cells (−0.12 mean difference; 95% CI: [−0.16, −0.08]) and NK cells in cervical cancer TME.19,20 The profound NKG2D-effector depletion predicts severely attenuated bystander RNT amplification, suggesting combination with NKG2D-effector reconstitution strategies before or concurrent with RNT.
scRNA-seq as the precision oncology navigation tool for RNT target stratification
The molecular heterogeneity revealed by our scRNA-seq analysis argues compellingly against one-size-fits-all RNT approaches and for a precision oncology paradigm in which pretreatment scRNA-seq biopsy profiling navigates individualized RNT strategy selection.
HPV oncoproteins as upstream drivers of RNT target suppression
A critical finding is that MICA/MICB downregulation is mechanistically driven by HPV E7 oncoprotein-mediated epigenetic silencing, suggesting that therapeutic HPV E7 knockdown could restore MICA/MICB expression upstream of metalloproteinase shedding.21–23
Pathway enrichment architecture and SUSD1 as a multi-immunosuppressive hub
GSEA and KEGG pathway analyses demonstrated that NKG2D signaling pathway suppression is the apex of a hierarchical regulatory network in which NF-kB, JAK-STAT, antigen processing, and complement activation pathways are coordinately downregulated.
Comprehensive combinatorial therapeutic strategies for RNT sensitization
Our integrative analysis identifies five pharmacologically actionable intervention nodes for RNT sensitization. First, HDAC inhibitor-based MICA/MICB re-expression priming: FDA-approved agents such as vorinostat, romidepsin, and panobinostat have demonstrated 3–8-fold upregulation of surface NKG2D ligand expression in preclinical studies, and we propose sequential administration prior to RNT. Second, ADAM10/ADAM17 metalloproteinase inhibition to prevent MICA/MICB ectodomain shedding and restore membrane-bound target density, including engineered noncleavable MICA variants. Third, NKG2D-effector cell reconstitution strategies, including interleukin-15 (IL-15) superagonist (N-803) administration to expand endogenous NK and CD8+ T cell populations, adoptive NK cell infusion therapy, and NKG2D-CAR T cell approaches that could synergize with radionuclide-induced immunogenic cell death. Fourth, concurrent immune checkpoint blockade targeting SUSD1-coexpressed checkpoint molecules (anti-PD-1, anti-TIGIT, and anti-TIM-3) to relieve the multicheckpoint suppressive program. Fifth, bispecific radioimmunotherapy constructs simultaneously target MICA and SUSD1 to achieve concurrent tumor irradiation and immunosuppression reversal. For each combination, toxicity considerations and clinical trial design implications require careful evaluation in dedicated preclinical studies.
Translational roadmap for clinical validation
The absence of in vivo and clinical validation represents the most significant translational limitation of the current study. We delineate a staged translational roadmap: Stage 1 involves preclinical in vivo validation using orthotopic cervical cancer xenograft models (HeLa and SiHa) in immunodeficient mice to evaluate biodistribution, tumor uptake, and dosimetry of 89Zr-labeled anti-MICA nanobodies, followed by therapeutic efficacy assessment of 177Lu- and 225Ac-conjugated constructs with and without HDAC inhibitor preconditioning. Stage 2 requires humanized mouse models with reconstituted human immune components to assess bystander immune amplification effects. Stage 3 involves first-in-human theranostic feasibility studies beginning with 89Zr-anti-MICA PET imaging to confirm tumor-selective uptake and target heterogeneity. The current study provides the molecular target discovery foundation upon which these translational stages must be built (Fig. 11).
Limitations of correlation-based analyses
We note that several key findings in this study are fundamentally correlative in nature. The inverse correlation between SUSD1 expression and MICA/MICB levels, while consistent and statistically robust, does not establish that SUSD1 directly causes MICA/MICB suppression; both may be independently regulated downstream of HPV E7 activity. Similarly, the positive correlations between MICA expression and NK cell infiltration density may reflect bidirectional causality, reverse causality, or confounding by a shared upstream regulator. Throughout the article, we have used associative rather than causal language wherever experimental causation has not been established. The specific experimental approaches required to establish directional causality include SUSD1 gain- and loss-of-function studies, MICA/MICB re-expression rescue experiments, and in vivo NK cell depletion models.
Limitations
This study has several limitations requiring acknowledgment. First, experimental validation employed cervical cancer cell lines (HeLa and SiHa) rather than primary tumor-infiltrating immune cells or patient-derived tumor organoids, and direct RNT experiments using radiolabeled anti-MICA/MICB conjugates were not performed. Validation in additional cell models, including CaSki cells (HPV-16-positive with high viral copy number), C-33A cells (HPV-negative cervical carcinoma), ME-180 cells (HPV-68-positive), and patient-derived tumor organoids, will be essential. Second, scRNA-seq data were derived from a limited publicly available dataset, and the proposed bimodal MICA/MICB distribution requires prospective validation in larger, clinically annotated cohorts. Reliance on two publicly available GEO datasets (GSM1551311 and GSM1551411) introduces several potential biases: Batch effects and technical variability arising from differences in tissue dissociation protocols, library preparation methods, and sequencing platforms between the two datasets, which we mitigated through Harmony integration but cannot entirely eliminate; potential selection bias in the original specimen collection, including unknown clinical staging, treatment history, and HPV genotype distribution that may limit generalizability; cell-type representation bias introduced by enzymatic dissociation, which may preferentially deplete fragile immune cell subsets; and the limited sample size relative to the biological heterogeneity of cervical cancer. The original datasets derive from a limited geographic and ethnic cohort, and cervical cancer biology exhibits variation across populations in HPV genotype distribution, HLA haplotype frequencies, and baseline immune composition.24,25 Prospective validation using multi-institutional, ethnically diverse cohorts encompassing the full International Federation of Gynecology and Obstetrics (FIGO) staging spectrum is essential before the proposed patient stratification framework can be considered clinically generalizable. Third, the SUSD1 mechanism in NKG2D-axis immunosuppression and RNT resistance remains to be functionally characterized through SUSD1 knockdown/overexpression experiments. Fourth, spatial relationships between MICA/MICB-expressing tumor cells and NKG2D-effector cells require spatial transcriptomics and multiplexed tissue imaging. Fifth, while Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT)-based immune deconvolution provides cohort-level estimates, its accuracy is limited by the reference matrix resolution. Sixth, the TF regulatory network was inferred from expression correlations and existing literature; direct chromatin immunoprecipitation (ChIP)-seq validation is needed. Seventh, the GSEA and pathway enrichment analyses require orthogonal validation by targeted pathway inhibition experiments. 26
Conclusions
This integrative scRNA-seq and experimental study establishes that MICA/MICB downregulation and SUSD1 upregulation converge to create a profoundly NKG2D-suppressed immune landscape in cervical cancer that constitutes both a barrier to conventional immunotherapy and a modifiable target architecture for precision RNT. Together, these findings provide the molecular foundation for a theranostic precision oncology pipeline: scRNA-seq-guided patient stratification, 89Zr-anti-MICA/MICB PET imaging for target confirmation, IRF1-reactivating HDAC inhibitor priming to restore surface target density, 177Lu- or 225Ac-labeled NKG2D-ligand-targeting radioimmunotherapy combined with SUSD1 inhibition, and concurrent NKG2D-effector cell reconstitution for bystander amplification. These findings justify prioritizing NKG2D-axis-targeted radionuclide theranostics as a precision oncology development pathway in cervical cancer.
Authors’ Contributions
J.C. and C.W.: Contributed equally to this work. J.C.: Conceptualization, bioinformatic analysis, RNT framework development, and writing—original draft. C.W.: Experimental validation, data interpretation, and writing—review and editing. All authors reviewed and approved the final article.
Footnotes
Data Availability
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.