
Abstract
Background
Individuals with Alzheimer's disease (AD) have a heightened risk of epilepsy. However, the underlying mechanisms are not well-understood.
Objective
We aimed to elucidate the role of the glutamate-glutamine cycle in this mechanism and test the effect of ceftriaxone, a glutamate transporter-1 (GLT-1) enhancer, on seizure susceptibility in the Tg2576 mouse model of AD.
Methods
First, we assessed expression levels of key proteins in the glutamate-glutamine cycle in Tg2576 (n = 7) and wild-type littermates (n = 7), and subsequently in the kindling model of epilepsy (n = 6) and sham (n = 6). Then, kindling susceptibility was assessed in three groups: 200 mg/kg ceftriaxone-treated Tg2576 (Tg-Ceft, n = 9); saline-treated Tg2576 (Tg-Sal, n = 9); and saline-treated wild-type (WT-Sal, n = 15). Mice were treated for seven days before kindling, and seizure susceptibility compared between groups.
Results
Protein levels of GLT-1 (p = 0.0093) and glutamine synthetase (p = 0.0016) were reduced in cortex of Tg2576 mice, compared to WT. Kindling increased GLT-1 (cortex: p < 0.0001, hippocampus: p = 0.0075), and glutaminase (cortex: p = 0.0044) protein levels, compared to sham. Both Tg-Ceft and WT-Sal displayed Class IV seizures in response to the first stimulation (p > 0.99), while Tg-Sal displayed Class V seizure (p = 0.0212 versus WT-Sal). Seizure susceptibility of Tg-Ceft was not different from Tg-Sal (p > 0.05), and kindling rates did not differ between groups.
Conclusions
Disruptions to key components of the glutamate-glutamine cycle are observed in models of AD and epilepsy. However, increasing GLT-1 through ceftriaxone treatment did not influence seizure susceptibility in Tg2576 mice, suggesting this is not an effective strategy to lower seizure susceptibility in AD, or a higher dosage is needed.
Keywords
Introduction
Alzheimer's disease (AD) is the most prevalent type of dementia and is defined by progressive deterioration in two or more cognitive domains, such as episodic memory and executive function, which result in the impairment of daily task performance. 1 The hallmark pathologies for AD include amyloid-β (Aβ) plaques and neurofibrillary tau tangles, although the exact mechanism that causes neurodegeneration remains undetermined. 2
Epilepsy is a debilitating neurological disorder that affects individuals across all age ranges, although incidence peaks in the young, and in older age. The disease is characterized by the presence of unprovoked and recurrent seizures caused by the brain neuronal network hyperexcitability. Epileptogenesis, the development of epilepsy, refers to the process by which a healthy brain is transformed into one which exhibits such hyperexcitable networks capable of generating seizure activity. In addition to seizures, epilepsy patients also commonly suffer from neurological and psychiatric comorbidities. 3
There is evidence to show that AD patients have up to 10-fold higher risk of developing epilepsy compared to healthy age-matched controls,4,5 rates which are even higher in familial AD.6,7 Seizures in this population are often non-convulsive events,8,9 which are difficult to identify and diagnose. Seizures in dementia patients are important clinical factors: these patients appear to experience accelerated cognitive decline in multiple domains, such as short and long-term visual and verbal memory, executive function and attention, compared to those without. In addition, increased mortality rate is observed in dementia patients with seizures,10,11 and these patients also appear to present a different treatment responsivity profile.12–15 Interestingly, dementia and epilepsy appear to exhibit bidirectional relationships. For example, temporal lobe epilepsy and AD affect similar regions of the brain, and evidence of the pathological hallmarks of AD have been identified in surgical specimens from epilepsy patients.16–18 In addition, seizures often occur prior to AD diagnosis, and accelerate disease onset, 19 and many patients with epilepsy experience cognitive decline. 20 Therefore, identifying the common mechanisms between these neurological disorders and developing treatments for seizures in AD will be beneficial for both epilepsy and AD patients.
Despite the evidence that AD is a risk factor for epilepsy, 21 the mechanism that links AD pathologies to epileptogenesis and higher seizure susceptibility is still poorly understood. Recently, it was found that a number of rodent models of AD, such as Tg2576,22–24 are more prone to seizures than non-transgenic controls. In addition to susceptibility to induced seizures, Tg2576 mice also show spontaneous epileptiform activities, including spikes early in disease development,25–28 as well as rare spontaneous seizures in later life. 26 Since the Tg2576 mice exhibit cognitive impairments at 6 months old, 29 gliosis at 10 months old, 30 and Aβ plaques at 11 months old, 31 it could be utilized to elucidate the mechanism that links AD pathologies to epilepsy.
With the treatment of epilepsy in AD, there is evidence to show that some of the currently available antiseizure medications (ASMs) can cause cognitive impairment as an adverse effect in patients, and this can potentially aggravate the symptoms of AD.15,32 Therefore, development of ASMs with different mechanisms of action than the existing compounds would be highly valuable for this patient population. One such potential candidate mechanism is abnormal glutamate homeostasis. A recent randomized clinical trial demonstrated that levetiracetam, an ASM targeting the SV2A protein, and which can suppress the release of glutamate, 33 improves cognitive outcome of AD patients who experience seizures. 14 Further, interruption to the glutamate-glutamine cycle has been implicated in the pathogenesis of both AD34,35 and epilepsy. 36 There is also evidence to suggest that altering the expression levels of key proteins in this cycle, such as reducing the levels of glutamate transporter-1 (GLT-1) and glutamine synthetase (GS) proteins, may result in lethal seizures 37 and cognitive impairment 38 in rodent models. This may suggest that the disruption in glutamate-glutamine cycle has a role in the pathogenesis of both epilepsy and AD, representing a shared mechanism between these two neurological disorders. 39 If so, enhancing GLT-1 expression might be a viable strategy in preventing or delaying epileptogenesis in AD.
One GLT-1 protein level enhancer under active investigation is ceftriaxone, a third-generation cephalosporin antibiotic. 40 Treating rodent models of AD with 200 mg/kg ceftriaxone for 7 consecutive days significantly increased the level of GLT-1 expression in the hippocampus41,42 via transcription associated with the nuclear factor kappa B (NF-κB) pathway, and this was accompanied by improved cognitive function.41–43 However, the effect of ceftriaxone treatment on seizure susceptibility in rodent AD model has not been investigated.
In this study, we aimed to generate insights to reveal potential pharmacological targets for reducing seizure susceptibility in AD patients by studying the Tg2576 mouse model of AD. The Tg2576 model is a well-characterized model of mutant human amyloid precursor protein (APP) overexpression. 31 We hypothesized that the disruption of glutamate-glutamine cycle in the Tg2576 mouse model of AD causes an increase in seizure susceptibility and counteracting this change through GLT-1 enhancement would normalize seizure susceptibility.
Methods
Study design
This study consisted of a series of experiments which aimed to characterize the role of glutamate-glutamine cycle in the pathogeneses of epilepsy and AD using model systems, and the potential of this cycle as a pharmacological target for the treatment of epilepsy in AD. In a first experiment, expression levels of key components of the glutamate-glutamine cycle were investigated in the Tg2576 mouse model of AD, compared to WT. In a second experiment, the effects of electrically-induced seizures on the levels of these same components were investigated in WT mice, compared to sham. The aim of the third experiment was to investigate GLT-1 as a pharmacological target for reducing seizure susceptibility of Tg2576 mice. For this, first a study was performed to validate the effect of ceftriaxone on GLT-1 expression in Tg2576 mice. Then in the treatment study, additional mice were separated into 3 groups: 1) WT mice treated with saline; 2) Tg2576 mice treated with saline; and 3) Tg2576 mice treated with ceftriaxone. After 7 days of treatment, amygdala kindling epileptogenesis—an assessment of seizure/epilepsy susceptibility— was initiated on all mice and compared between groups. Following this, the mice were euthanized and the level of key proteins associated with glutamate-glutamine cycle were measured. The overall design of experiment three is shown in Supplemental Figure 1.
Animals
Tg2576 and WT littermates (6 months old, n = 7/group), bred on a c57xSJL background, were used for the first experiment to determine early changes in glutamate-glutamine cycle associated with the APP overexpression pathology in Tg2576 mice. For the second experiment, WT mice (aged, 12 to 14 months old) were employed to determine changes in glutamate-glutamine cycle associated with kindling-induced seizures (n = 6/group) in the later stage of life. As epilepsy and dementia tend to be comorbid in later stages of life, 32 observing changes associated with kindling-induced in aged mice would be relevant. In the third experiment, n = 34 aged Tg2576, and n = 21 WT littermates mice were used. Again, aged WT mice were used to study changes in glutamate-glutamine cycle associated with epileptic seizures in the presence of mutant human APP overexpression in later stage of life. Additionally, the effect of ceftriaxone on the seizure susceptibility of Tg2576 mice during the late pathological stage of AD was also investigated. Aged mice were used with the same rationale as the second experiment.
All mice were obtained from our colonies at the Alfred Research Alliance (Melbourne, Australia). Sample size calculation for t-test with continuous variables was performed to estimate the minimum sample size needed in each experiment. Expected effect size and standard deviation were acquired from previous study 44 and our initial study (Supplemental Figure 2). All experiments were conducted in the Department of Neuroscience (Alfred Hospital), Monash University with the approval from the Alfred Research Alliance Animal Ethics Committee (AEC approved project number: E/1955/2019/M).
The genotype of each mouse was determined by extracting the DNA from a tail snip with Wizard® Genomic DNA Purification Kit (Promega, NSW, Australia), then the DNA for mutant human APP overexpression was amplified with primers (ThermoFisher, VIC, Australia) through polymerase chain reaction. Finally, gel electrophoresis was performed, and DNA for the mutant human APP overexpression was identified at 466 bp, while the DNA for actin, the loading control, was identified at 300 bp.
Drug treatment
To validate the effect of ceftriaxone on GLT-1 levels, mixed-sex Tg2576 mice were alternated into 2 groups (n = 4M, 4F/group) to receive daily ip injections of either 200 mg/kg ceftriaxone (Cayman Chemical, Ann Arbor, MI, USA) or vehicle (saline) for 7 days. For the drug treatment study, mice were randomly assigned into groups: 1) wild-type mice treated with saline (WT-Sal, 15 mice - 4M, 11F); 2) Tg2576 mice treated with saline (Tg-Sal, 9 mice – 3M, 6F); and 3) Tg2576 mice treated with ceftriaxone (Tg-Ceft, 9 mice – 5F, 4M). The investigators were blinded to the genotype and treatment of the mice. After 7 consecutive days of treatment via ip injection, kindling commenced—during kindling, all mice continued to receive treatment each day ∼1 h before stimulations.
Kindling procedure
Electrode implantation surgery was performed in experiments two and three to allow kindling and electroencephalography (EEG) recording as described previously, with modifications. 45 Briefly, a bipolar depth electrode (Science Products, Hofheim, Germany) was implanted into the amygdala (anteroposterior [AP]: −0.8; mediolateral [ML]: 3.1 relative to bregma; dorsoventral [DV]: −5.0 relative to dura) and a monopolar depth electrode was implanted into the hippocampus (AP: −2.0; ML: −2.5; DV: −2.0). After recovery from surgery, the after-discharge thresholds (ADT) of all mice, except the sham, were tested by stimulating the bipolar electrode once every minute using battery-operated ISO-Flex stimulus isolator (Microprobes for Life Science, Gaithersburg, MD, USA) until the stimulation triggered a seizure. The intensity of these stimulations began at 200 μA and was increased with 20 μA increments after each stimulation until the current reached 400 μA. For Experiment 3, mice were treated according to their groups for 7 days prior to determination of ADT. To qualify as a seizure, the abnormal activity on the EEG must be: 1) ≥6 s long; 2) ≥2× the amplitude of the baseline; 3) synchronized; 4) evolving with increasing spike frequency per minute; and 5) biological, not mechanical (movements). An example of a seizure trace is shown in Supplemental Figure 3. To quantify behavioral severity of each seizure, a modified Racine scale 46 was used, as described in our previous work. 47 Briefly, seizure severity was classified as follows: class I = mouth and facial automatism; class II = head nodding and jerks; class III: class II plus forelimb clonus; class IV: class III plus rearing or Straub tail, and class V = class IV plus falling over and/or jumping/running. EEG data were recorded with the ML870 PowerLab data acquisition device (ADInstruments, NSW, Australia) attached to the ML136 Animal Bio Amp signal amplifier (ADInstruments). LabChart 7 software (ADInstruments) was used for controlling the stimulation and visualizing the EEG data. Once the ADTs were established, each mouse was subjected to a once-per-day stimulation at their respective ADT current. Mice were then stimulated until they had 5 Class V seizures or 15 total induced seizures. Seizure severity displayed by each animal was measured as the primary outcome, while the secondary outcome measured the number of stimulations needed for the animal to develop its first “Class V” seizure.
Protein analysis
The levels of key proteins in the glutamate-glutamine cycle (GLT-1, GS, GLS (glutaminase), and GFAP (glial fibrillary acidic protein)) were measured in homogenized cortical and hippocampal samples. Briefly, all mice were euthanized via lethal injection with pentobarbitone (80 mg/kg) (Provet, VIC, Australia), then both the left and right cortex and hippocampus were surgically extracted.
Two methods of western blotting were utilized, the robot-assisted western blot (WES system, protein Simple, CA, USA) and traditional western blot. The traditional western blot was utilized in first experiment, while the WES system was utilized for the second and third experiments. Sample preparation for WES was performed according to the manufacturer's instructions. The 12–230 kDa separation modules (cat#: SM-W004) was used with anti-mouse (cat#: DM-002) and/or anti-rabbit (cat#: DM-001) detection modules. GLT-1 (cat#: sc-365634, Santa Cruz, Tullamarine, VIC, Australia), GS (cat#: ab64613, Abcam), GLS (cat#: ab93434, Abcam) and GFAP (cat#: Z0334, Agilent Technologies) antibodies were used. GAPDH (cat#: 2118, Cell Signalling) was the housekeeping protein. All consumables used in WES were supplied by ProteinSimple (San Jose, CA, USA). Area under the curve for each band was normalized against the housekeeping protein, then the average expression compared between groups.
With the traditional western blot, GLT-1 (cat#: 701988, ThermoFisher) and GS (cat#: ab64613, Abcam, Melbourne, VIC, Australia) antibodies were used. GAPDH (cat#: 2118, Cell Signalling, Karrinyup, WA, Australia) and β-actin (cat#: ab8224, Abcam) were used as housekeeping proteins. For the secondary antibodies, the swine anti-rabbit antibody (cat#: P0217, Agilent Technologies, Melbourne, VIC, Australia) and the goat anti-mouse immunoglobulin HRP (cat#: P0447, Agilent Technologies) were used. The intensity of each band was normalized with the housekeeping protein, then the average was compared between groups.
Gene expression analysis
RNA was extracted from cortical samples acquired from the first experiment with the RNeasy Plus Mini Kit (cat#: 74034, Qiagen, Chadstone, Melbourne, Australia). Then, cDNA conversion was done via Omniscript RT Kit (Qiagen). Samples were then mixed with TaqMan Fast Advanced Master Mix (cat#: 4444556, ThermoFisher) and TaqMan Gene (ThermoFisher) according to the manufacturer's protocol. The GLT-1 (assay ID: Mm00441457_m1) and GS (assay ID: Mm00725701_s1) genes, reference genes were TBP (assay ID: Mm01277042_m1), YWHAZ (assay ID: Mm03950126_s1), GAPDH (assay ID: Mm99999915_g1) and β-actin (assay ID: Mm02619580_g1). QUANTStudio 7 system (ThermoFisher) was used to perform quantitative PCR (qPCR). The cycle threshold values for GLT-1 and GS genes from Tg2576 and WT littermates were used to calculate the relative gene expression values compared to the geometric mean of the reference genes, using the ΔΔCt method.
Data analysis
Statistical analyses were conducted using Prism software (GraphPad, La Jolla, CA, USA). First, data normality was tested using Kolmogorov-Smirnov test. Normally distributed data are presented as mean ± standard error of the mean (SEM), while nonparametric data are presented as median ± interquartile range (IQR). Statistical significance was assessed with ANOVA followed by Tukey's multiple comparison test (>2 groups comparison) or unpaired t-tests for normally distributed data, whereas ANOVA with Kruskal-Wallis test followed by Dunn's test for multiple comparison (>2 groups comparison) or Mann-Whitney U test for nonparametric data. We used Kaplan-Meier survival analysis with Chi square to assess the proportion of animals experiencing Class V seizures over time. Data collected from qPCR were analyzed according to the method described by Livak and Schmittgen. 48 The significance thresholds were set at p < 0.05.
Results
AD modeling and kindling disrupt protein levels of key targets in glutamate-glutamine cycle
We identified significant reductions in the levels of key proteins in the glutamate-glutamine cycle in Tg2576, compared to WT mice. GLT-1 (p = 0.0093, unpaired t-test, Figure 1A and B) and GS (p = 0.0016, unpaired t-test, Figure 1C and D) proteins were significantly decreased by 65% and 50%, respectively, in Tg2576 cortex, compared to WT. However, these differences were not observed in hippocampus (GLT-1, p = 0.767 & GS, p = 0.0589, unpaired t-test). GLS (Supplemental Figure 4) and GFAP (Supplemental Figure 5) proteins were not different between Tg2576 and WT mice. The results from experiment one are summarized in Table 1. Despite striking changes in protein levels, mRNA expression of these proteins in AD mice did not differ from WT mice (GLT-1: p = 0.205; GS: p = 0.525, unpaired t-test, Supplemental Figure 6).
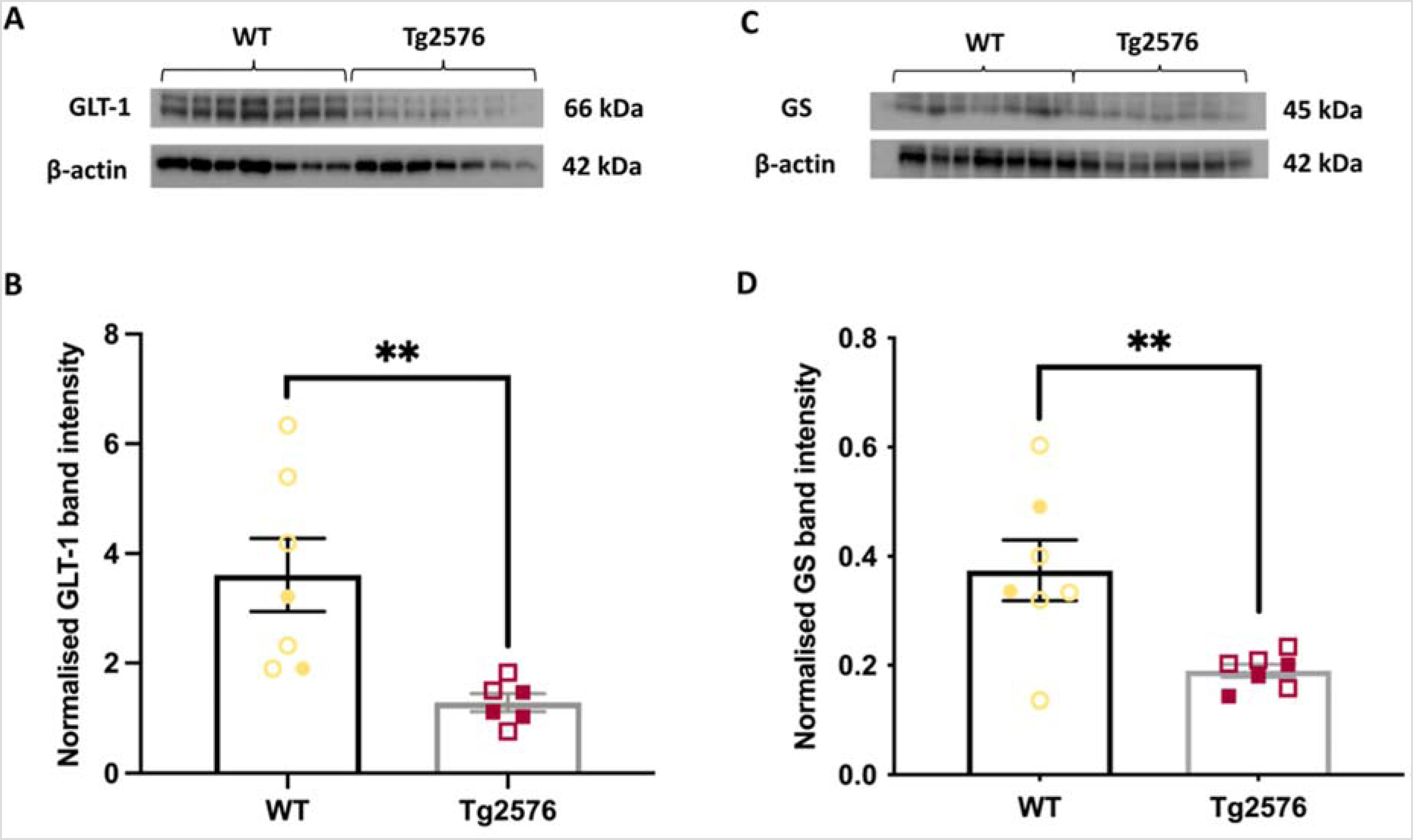
Naïve 6-month Tg2576 mice show altered levels of key proteins in the glutamate-glutamine cycle in cortex. (A) Representative western blots of cortical samples from WT (5M, 2F) and Tg2576 mice (4M, 3F) showing immunoreactivity of GLT-1. (B) The GLT-1 band intensity was significantly decreased in the cortex of Tg2576 mice, compared to WT. (C) Representative blots of GS protein. (D) The level of GS protein was significantly decreased in the cortex of Tg2576, compared to WT. **p < 0.01, data represent mean ± SEM. Open symbols represent male samples, closed symbols represent female samples. No significant effects of sex were noted (p > 0.05).
Summary of changes in the levels of key proteins in the glutamate-glutamine cycle. Differences in the protein levels were compared between Tg2576 (AD model) mice and WT littermates in the second and the third column from the left. The levels of GLT-1 and GS proteins were significantly decreased in the cortical samples of Tg2576 mice. In the fourth and fifth columns, changes in the levels of key proteins in the glutamate-glutamine cycle were compared between kindled (epilepsy model) WT mice and sham WT mice. Kindling significantly increased the levels of GLT-1 and GLS proteins in the cortex and GLT-1 in the hippocampus. The differences in the level of GFAP protein were not significant in any comparison.

↓↓ = p < 0.01, ↑↑ = p < 0.01, ↑↑↑ = p < 0.001, n.s. = not significant (p > 0.05).
GLT-1: glutamate transporter-1; GS: glutamine synthetase; GLS: glutaminase; GFAP: glial fibrillary acidic protein.
Kindling significantly altered expression of some key protein targets in the glutamate-glutamine cycle. Compared to sham mice, GLT-1 was significantly increased by 134% in the cortex (p < 0.0001, unpaired t-test, Figure 2A and B) and 20% in the hippocampus (p < 0.01, unpaired t-test, Figure 2C and D) of kindled WT mice. GLS also significantly increased by 35% in the cortex (p = 0.0044, unpaired t-test, Figure 2E and F), but not the hippocampus (p = 0.0767, unpaired t-test, Figure 2G and H). Kindling did not alter the GS (Supplemental Figure 7) or GFAP (Supplemental Figure 8). The results from aim two are shown in Table 1.
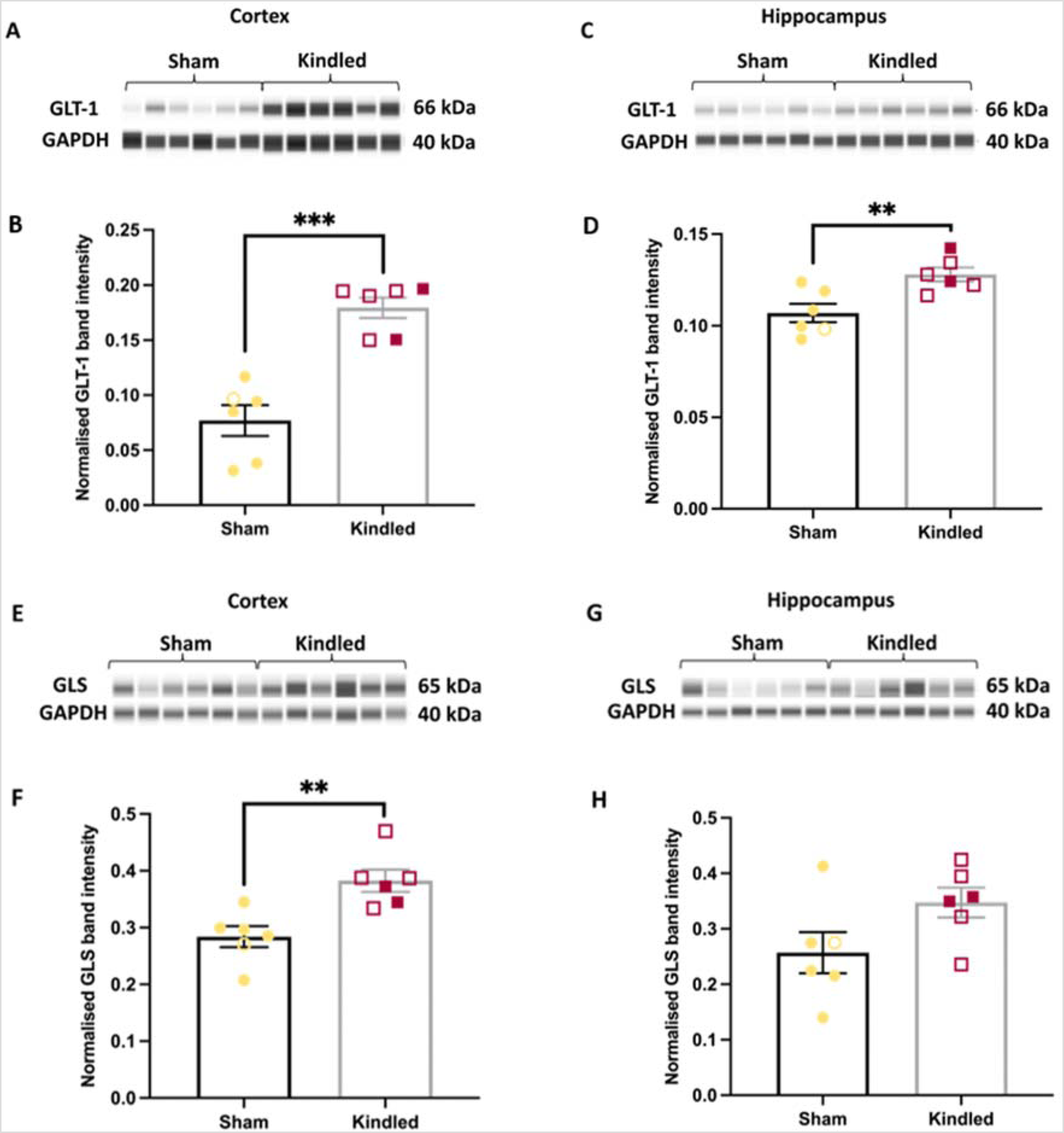
Kindling-induced seizures alter the expression of key proteins in the glutamate-glutamine cycle in aged wt mice. (A) Representative western blots of GLT-1 protein band from cortical samples of sham (1M, 5F) and kindled WT mice (4M, 2F). (B) GLT-1 immunoreactivity was significantly elevated in the cortex of kindled mice, compared to WT mice. (C) Representative blots of GLT-1 protein from hippocampal samples. (D) GLT-1 protein levels are significantly upregulated in the hippocampus of kindled mice, compared to sham. (E) Immunoreactivity of GLS protein in cortex. (F) GLS protein levels in the cortex of kindled mice was significantly upregulated compared to sham. (G) Western blot of GLS protein in the hippocampus. (H) No difference in GLS expression was observed in the hippocampus between kindled and sham mice. ***p < 0.001, ** p < 0.01, data represent mean ± SEM. Open symbols represent male samples, closed symbols represent female samples. No significant effects of sex were noted (p > 0.05).
Ceftriaxone did not impact seizure susceptibility or kindling in TG2576 mice
In agreement with literature, Tg2576 mice were more susceptible to kindling-induced seizures than WT littermates (p = 0.01, U = 3.5, Mann-Whitney, Supplemental Figure 9A). We therefore commenced an intervention study designed to reduce this susceptibility using ceftriaxone (Ceft).
We found that the behavioral severity of first seizure did not significantly differ between Tg-Ceft and Tg-Sal groups (adjusted p = 0.206, Kruskal-Wallis with Dunn's post-hoc, Figure 3A), nor between Tg-Ceft and WT-Sal groups (p > 0.99). However, we again identified a significant difference between Tg-Sal and WT-Sal groups (adjusted p = 0.0212, Kruskal-Wallis with Dunn's post-hoc, Figure 3A). We also examined the average after-discharge durations of this first seizure, and a similar trend emerged. Tg-Sal mice exhibited longer seizures than WT-Sal mice (p = 0.0005), but this measure was not different between Tg-Sal and Tg-Ceft-treated mice (p = 0.09; Figure 3B). When comparing the number of stimulations required to experience the first Class V seizure, mice from Tg-Sal group required significantly fewer stimulations than WT-Sal mice (adjusted-p = 0.0393, Kruskal-Wallis with Dunn's post-hoc, Figure 3C), but there was no difference between Tg-Ceft mice and Tg-Sal mice (p > 0.99). Likewise, there were no differences between the groups when comparing the seizure classes elicited over the entire kindling process (Figure 3D). Finally, a survival analysis assessing when mice experienced their first Class V seizure showed that this happened significantly earlier in Tg-Sal compared to WT-Sal mice (p = 0.0002), but no difference was found between Tg-Sal and Tg-Ceft mice (p = 0.33; Figure 3E). These findings suggest that ceftriaxone treatment does not influence seizure susceptibility or kindling in Tg2576 mice.
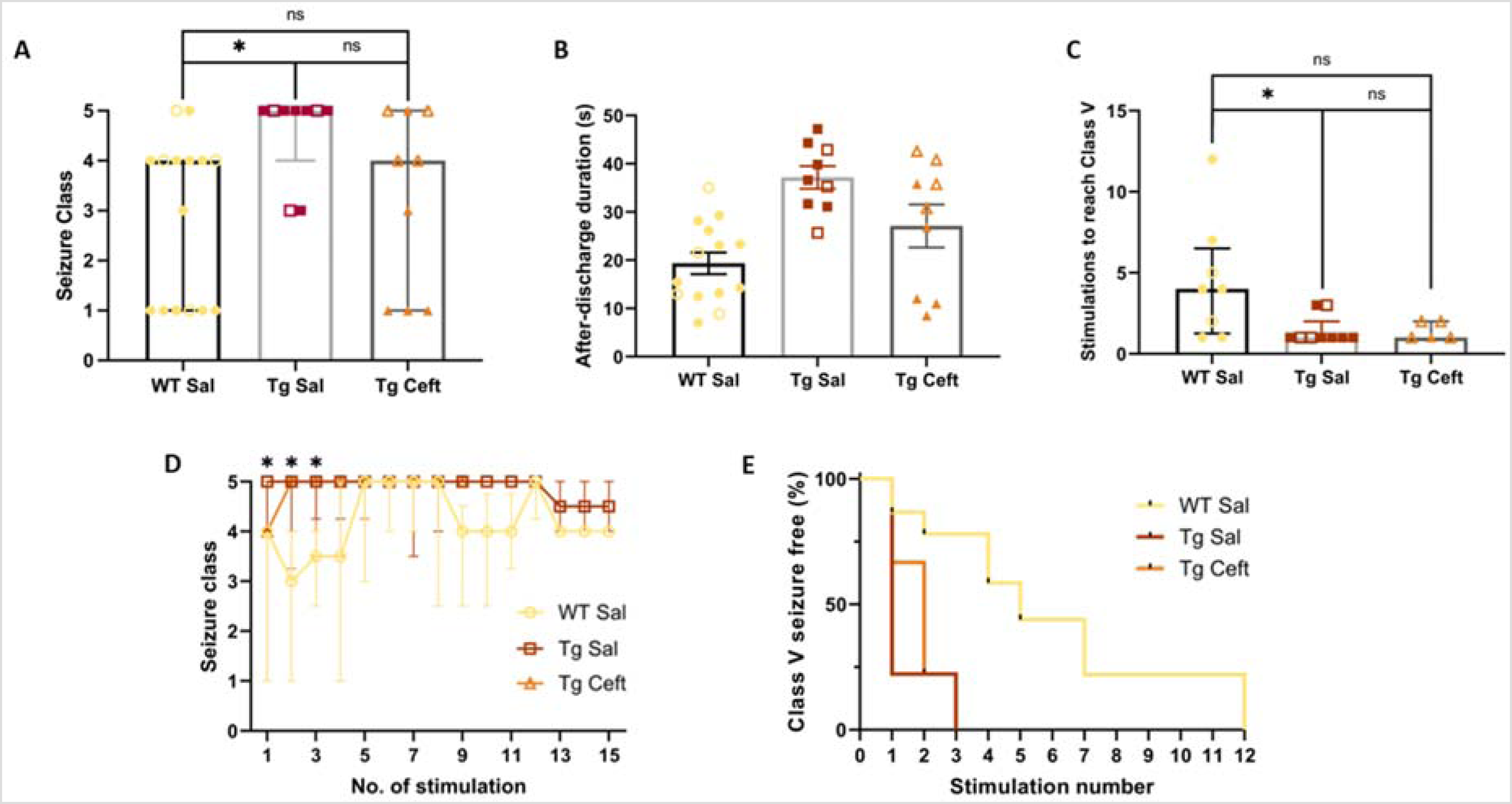
Ceftriaxone does not impact seizure susceptibility or kindling rates in Tg2576 mice. (A) Severity of the first seizure in the Tg-Ceft group was not significantly different from either Tg-Sal and WT-Sal groups. (B) Ceftriaxone treatment did not significantly influence the average duration of seizures in Tg2576 mice, compared to Saline treatment. (C) Ceftriaxone did not alter the number of stimulations needed to trigger a Class V seizure in Tg2576 mice. (D) Seizure class evolutions during kindling from each group. The kindling susceptibility of Tg-Ceft group was not significantly different from both Tg-Sal and WT-Sal groups. (E) Kaplan-Meier survival curve demonstrates that WT mice kindle slower than Tg-Sal mice, but that Ceftriaxone treatment did not influence this outcome. *p < 0.05 (WT-Sal versus Tg-Sal), data = medians ± IQR. Sample sizes: WT Saline (4M, 11F), Tg Saline (3M, 6F); Tg Ceftriaxone (4M, 5F). No significant effects of sex were noted (p > 0.05).
Kindling diminished the effects of ceftriaxone on GLT-1 and GLS levels in TG2576 mice
We then conducted biochemistry studies to assess the effects of ceftriaxone on GLT-1 and GS levels, both in naïve and kindled mice. We found that ceftriaxone augmented GLT-1 (p = 0.0177, unpaired t-test, Figure 4A) and GLS (p = 0.0105, unpaired t-test, Figure 4C) protein expression in the hippocampus of naïve Tg2576 mice. However, in kindled mice, we did not find any effects of drug treatment GLT-1 (p = 0.938, unpaired t-test, Figure 4B) or GLS (p = 1, unpaired t-test, Figure 4D) in Tg2576 mice. This suggests that the kindling itself counteracts the effects of drug on the expression levels of these proteins. The drug itself was well tolerated, with no overt changes in weight, compared to saline treatment (p = 0.52, ANOVA, Supplemental Figure 3).
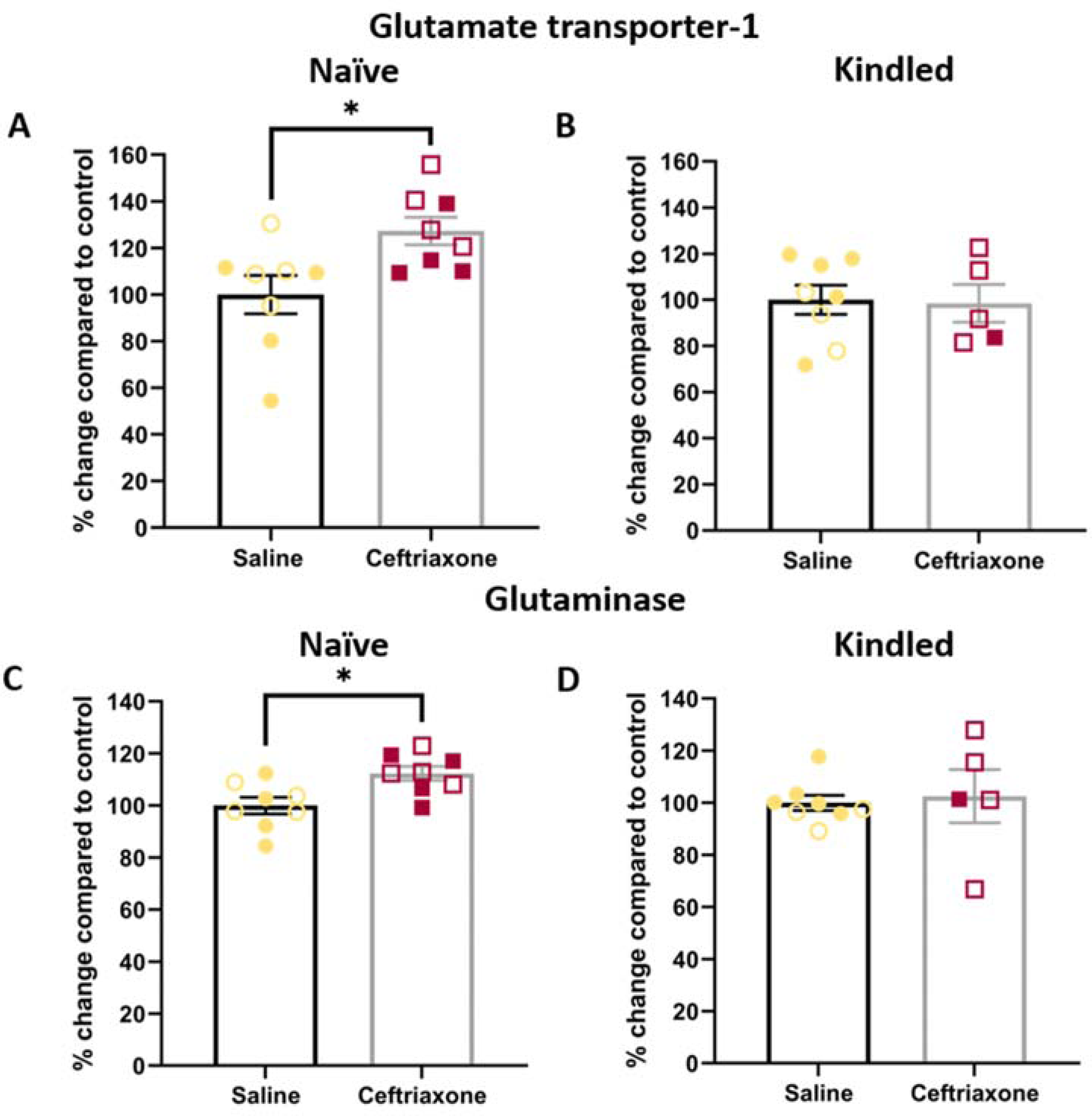
Kindling diminished the effects of ceftriaxone treatment on the glutamate-glutamine cycle in Tg2576 mice. Ceftriaxone treatment significantly increased GLT-1 protein level in the hippocampus of (A) naïve, but not (B) kindled Tg2576 mice, compared to saline (vehicle) treatment. Ceftriaxone also significantly increased the level of GLS protein in the cortex of (C) naïve, but not (D) kindled Tg2576 mice. *p < 0.05, data represent mean ± SEM. Sample sizes: Naïve Saline (4M, 4F), Naïve Ceftraixone (4M, 4F); Kindled Saline (3M, 5F), Kindled Ceftriaxone (4M, 1F). Open symbols represent male samples, closed symbols represent female samples. No significant effects of sex were noted (p > 0.05).
Discussion
In agreement with previous studies, 22 our results demonstrate that Tg2576 mice are more susceptible to kindling than WT mice. Furthermore, disruptions to the glutamate-glutamine cycle are observed in both AD and epilepsy models. Ceftriaxone treatment significantly heightened the expression levels of GLT-1 protein in the brain but did not impact seizure susceptibility of Tg2576 mice.
Glutamate-glutamine cycle is disrupted in the Tg2576 AD model
We hypothesized that early disturbances in the glutamate-glutamine cycle in Tg2576 mice is one of the causes of enhanced seizure susceptibility and indeed, dramatic reductions in the expression levels of key proteins such as GLT-1 and GS were observed in this study. It is well-documented that astrocytes undergo morphological and physiological changes in the presence of AD pathologies, therefore, processes which rely on the functionality of astrocytes may be disturbed in AD. 39 There is evidence to suggest that the glutamate-glutamine cycle is one of the cycles affected in AD, which may disturb the balance of brain glutamate and glutamine levels.39,49 Several studies, through techniques such as nuclear magnetic resonance spectroscopy, have identified that changes in brain glutamate and glutamine levels in AD,50,51 supporting this concept. Using the glutamate amine exchange saturation transfer (GluCEST) protocol in magnetic resonance imaging (MRI) another study demonstrated that the overall level of glutamate in the brain was reduced by approximately 30% in aged APP/PS1 mice (a model of AD), compared to WT. 52
The reductions in the expression levels of key proteins in the glutamate-glutamine cycle may reflect the interaction between Aβ42 oligomers and astrocytes. There is evidence to suggest that Aβ42 oligomers cause astrocytes to undergo structural, functional and morphological changes (astrogliosis). 39 However, the expression level of GFAP in mature Tg2576 mice was not altered in our experiment, which suggests that astrogliosis might not be significant at this age. The disruption to the glutamate-glutamine cycle observed in Tg2576 mice may therefore be caused by the direct interaction between key proteins and mutant human APP along with its downstream products, such as the soluble Aβ species. Interestingly, reductions in GLT-1 and GS protein levels were only observed in the cortex at 6 months, not hippocampus, in Tg2576 mice. In human, it has been shown that the earliest detectable Aβ deposits are found in the neocortex, before being detectable in other regions as the disease progresses. 53 Therefore, it is possible that the effects of mutant human APP overexpression pathology on the glutamate-glutamine cycle may spread from cortex to other regions as the pathology progresses. One limitation of our study is that we did not examine protein expression levels in older mice, so we could not confirm this proposed expansion over time. Interestingly, we did not observe changes in GLT-1 and GS mRNA expression in the cortical samples of Tg2576 mice, indicating that the upregulation of GLT-1 and GS protein is driven by post-translational mechanisms. The signaling pathways associated with the NF-κB transcriptional factor governs the expression of GLT-1 proteins, 54 and dysregulation in NF-κB has been implicated in the pathogenesis of AD. 55 Since the pathways involved in the translation of GLT-1 protein have not been elucidated, targeting the transcription process is an alternate way to pharmacologically augment GLT-1 expression in the brain.
Disrupted glutamate-glutamine cycle and the relationship to epileptogenesis
Kindling-induced seizures heightened the expression levels of GLT-1 and GLS proteins in the brain. Since enhancing GLT-1 expression may grant neuroprotective effects against neuronal excitotoxicity, these changes might be one of the brain's mechanisms designed to increase the rate of glutamate uptake from the synaptic cleft to prevent overexcitation. 40 Supporting this, it has been shown that enhancing GLT-1 expression with ceftriaxone limited pentylenetetrazole-induced seizures in rodent models.56,57 Therefore, it is likely that the change in the expression level of GLT-1 in kindled mice imparts a compensatory effect designed to counter the increase in the brain excitability. One limitation of our investigation is that we did not explore the influence of seizures/kindling on GLT-1 levels in Tg2576 brain, and it is possible that the upregulation we observed in WT mice may be altered in pathological settings, such as in Tg2576 mice.
Additionally, kindling-induced seizure also increased GLS expression level in the cortex. This may be because the presynaptic glutamate pools in the brain were exhausted due to kindling; thus, the pools would require replenishment via the enhancement of GLS expression level. 58 On the other hand, heightened GLS may be consequential to the increase in the rate of glutamate uptake as the result of enhanced GLT-1 expression. 59 Therefore, additional glutamate would be produced via GLS and released from the presynapse to compensate for the increase in glutamate uptake rate. The effect of kindling on the expression levels of key components in the glutamate-glutamine cycle should be investigated further both in WT and Tg2576 mice to study these intriguing possibilities.
Ceftriaxone treatment does not reduce kindling susceptibility in Tg2576 mice
Consistent with the literature, we found that ceftriaxone significantly increased the GLT-1 protein level in the hippocampus of Tg2576 mice.41,42 However, contrary to our hypothesis, ceftriaxone did not ameliorate the enhanced seizure susceptibility phenotype of the Tg2576 mice. Several technical and/or experimental possibilities exist which might explain this lack of drug effect. Firstly, this may be related to the observation that kindling itself can increase GLT-1 expression levels, at least in WT mice. This finding agrees with previous work which shows activation of the NF-κB pathway following kainate-induced seizures,60,61 and suggests kindling-induced seizures may also utilize the NF-κB pathway to elevate GLT-1 protein. Given that the targeted mechanism of ceftriaxone is to also increase GLT-1 levels, 54 the lack of drug effect might be explained by competing influences on this pathway. Additionally, as described above, we did not confirm sustained downregulation of the target protein in older Tg2576 mice. In humans, the reduction in GLT-1 protein is found in AD patients post-mortem, ie: at the end stage of disease, possibly due to APP overexpression. 62 Given that our mouse model of AD exhibits APP overexpression, this suggests that the early GLT-1 downregulation we observed would persist into the chronic phase of disease in Tg2576 mice. 63 But it is possible that the early changes we observe are not sustained, and so the lack of drug effect may be because the target is no longer relevant at this age. Future studies should explore the influence of ceftriaxone on epileptogenesis using 6-month mice to address this, and/or characterize GLT-1 expression levels in Tg2576 mice at 12–14 months to ensure relevance of the GLT-1 target at this age. Another potential explanation for the lack of drug effect relates to the possibility of insufficient dosage. We based our treatment regime (200 mg/kg ip daily for 7 days prior to initiating kindling) on published reports which demonstrate upregulation of GLT-1.41,42 Indeed, this dose was well-tolerated, and succeeded in upregulating GLT-1 in WT mice, validating this approach. However, ceftriaxone undergoes rapid metabolism in rodents (half-life ∼1–1.5 h), with almost complete removal from rat brain after 16 h following subcutaneous injection. 64 Given we used a once-daily routine, this would likely have resulted in substantial time when there was no drug present to influence brain neurochemistry. However, we used a chronic dosing regimen which one might expect to result in sustained increases in GLT-1, even with the peaks and troughs of circulating drug levels in blood. Moreover, in our experiments, we performed our interventions ∼1 h following drug injection, so we would anticipate that, at this time, drug effects were maximally present in the brain.
Our study was directed towards assessing the effects of ceftriaxone on susceptibility to kindled seizures, but it is possible that, despite the lack of success on this measure, other outcomes may be important to assess. Tg2576 mice exhibit interictal epileptiform spikes,25–28 as well as (rare) spontaneous seizures (e.g., Supplemental Figure 5), so it is possible that, although susceptibility to induced seizures was not impacted, ceftriaxone could improve these other epileptiform measures which are not necessarily mutually inclusive. It is also important to note that we did not explore measures of Aβ pathology in Tg2576 mice, and future studies should look to see whether Aβ plaques and APP expression are impacted by seizures, and by our intervention.
We chose to include both male and female mice in our studies, despite there being noted differences in the presentation of AD in both humans,65,66 and in animal models of AD,67,68 including Tg2576. 69 These consistently show that females exhibit greater risk, and accelerated decline in cognitive function. One relevant study demonstrated female-specific cognitive decline after repeated seizures in the PSEN2KO mouse model. 68 We enrolled both sexes in our study, but statistical analyses did not identify any sex-specific effects in any of the outcome measures here. However, we did not power our study sufficiently to assess sex as a primary variable, so this lack of effect should be noted with caution.
As is a cephalosporin antibiotic, it is possible that ceftriaxone may possess other qualities which can potentially be proconvulsant. Cephalosporin antibiotics, such as cefozopran, have demonstrated proconvulsive activities which are mediated by the inhibition of GABA(A) receptor function. 70 However, there is no evidence that ceftriaxone specifically exhibits any proconvulsive effect. On the contrary, there is evidence to demonstrate that 200 mg/kg and 400 mg/kg doses of ceftriaxone can reduce the severity of pentylenetetrazol-induced seizure in a dose-dependent manner. 71 Therefore, it is possible that higher doses of ceftriaxone may be necessary to produce beneficial effects against kindling induced seizure under the condition of this study. Furthermore, it may also be beneficial to investigate a more potent and specific GLT-1 expression enhancer 72 to avoid potential proconvulsive effect associated with cephalosporin antibiotic. Finally, the kindling model of seizure used in this study might be too severe, therefore it may leave little room for the anti-convulsant effects of ceftriaxone to be influential. Other models of seizure should be considered for future work.
Supplemental Material
sj-docx-1-alz-10.1177_13872877241289053 - Supplemental material for Effect of ceftriaxone on the glutamate-glutamine cycle and seizure susceptibility of Tg2576 mouse model of Alzheimer's disease
Supplemental material, sj-docx-1-alz-10.1177_13872877241289053 for Effect of ceftriaxone on the glutamate-glutamine cycle and seizure susceptibility of Tg2576 mouse model of Alzheimer's disease by Hattapark Dejakaisaya, Runxuan Lin, Anna Harutyunyan, Jianxiong Chan, Patrick Kwan and Nigel C Jones in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Author contributions
Hattapark Dejakaisaya (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Validation; Visualization; Writing – original draft; Writing – review & editing); Anna Harutyunyan (Conceptualization; Data curation); Runxuan Lin (Conceptualization; Data curation); Jianxiong Chan (Investigation); Patrick Kwan (Funding acquisition; Project administration; Supervision; Validation; Visualization; Writing – review & editing); Nigel C Jones (Funding acquisition; Project administration; Resources; Supervision; Validation; Visualization; Writing – review & editing).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available within the article and/or its supplemental material.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.