
Abstract
Alzheimer’s disease (AD) is characterized by progressive impairment of learning, memory, and cognitive deficits. Glutamate is the major excitatory neurotransmitter in the central nervous system and plays an important role in learning, memory, and cognition. The homeostasis and reutilization of glutamate are dependent on astrocytic uptake by glutamate transporter-1 (GLT-1) and the subsequent glutamate-glutamine cycle. Increasing evidence showed impairments in GLT-1 expression and uptake activity and glutamate-glutamine cycle in AD. Ceftriaxone (Cef) has been reported to upregulate the expression and uptake of GLT-1. Therefore, the present study was undertaken to explore whether Cef can improve cognitive deficits of APP/PS1 mice in early stage of AD by upregulating GLT-1 expression, and then promoting the glutamate-glutamine cycle. It was shown that Cef treatment significantly alleviated the cognitive deficits measured by Morris water maze test and upregulated GLT-1 protein expression in the hippocampus of APP/PS1 mice. Particularly, the activity of glutamine synthetase (GS) and the protein expression of system N glutamine transporter 1 (SN1), which are the key factors involved in the glutamate-glutamine cycle, were significantly upregulated as well after the Cef treatment. Furthermore, inhibition of GLT-1 uptake activity by dihydrokainic acid, an inhibitor of GLT-1, blocked the Cef-induced improvement on the cognitive deficits, GS activity, and SN1 expression. The above results suggested that Cef could improve cognitive deficits of APP/PS1 mice in early stage of AD by upregulating the GLT-1 expression, GS activity, and SN1 expression, which would lead to stimulating the glutamate-glutamine cycle.
INTRODUCTION
Alzheimer’s disease (AD) is a typical neurodegenerative disease characterized by progressive impairments in learning, memory, and cognitive function [1, 2]. Glutamate is the major excitatory neurotransmitter in the central nervous system, and plays an important role in the course of learning, memory, and cognition by regulating synaptic plasticity and neural circuit function [3, 4]. The homeostasis and reutilization of glutamate as neurotransmitter in the central nervous system are associated with many links, of which the glutamate uptake by astrocytic glutamate transporter-1 (GLT-1) and the subsequent glutamate-glutamine cycle play an important role [5, 6].
In recent years, dysregulations in GLT-1 and glutamate-glutamine cycle in AD patients or animals have been paid attention [7–10]. For example, the expression and uptake activity of GLT-1 were significantly downregulated in AD patients [11–13] and AD model mice [14, 15]. Knockdown of GLT-1 accelerated the development of dementia in APP/PS1 transgenic AD mice [16]. Furthermore, glutamine synthetase (GS), a key enzyme in glutamate-glutamine cycle, was reported to be downregulated [9, 15] or oxidized injury [17] in transgenic AD mice. These dysfunctions in GLT-1 and GS would impede the glutamate uptake and subsequent glutamate-glutamine cycle, decrease the reutilization efficacy of glutamate as neurotransmitter, impair synaptic plasticity and neural circuit function, and then contribute to the cognitive disorders in AD. Therefore, modulating the expression and/or uptake activity of GLT-1 and stimulating the glutamate-glutamine cycle would facilitate synaptic transmission and plasticity and then might be beneficial to improve learning and memory deficits in AD.
Rothstein et al. reported that Ceftriaxone (Cef), a kind of β-lactam antibiotics, could significantly and selectively upregulate the expression of GLT-1 in vitro and in vivo studies, and this upregulation was shown to be protective for cerebral neurons in animal models of stroke, oxygen glucose deprivation and for motor neuron degeneration [18–20]. Our previous studies also indicated that Cef could protect pyramidal neurons in the CA1 hippocampus against ischemic insult by upregulating GLT-1 expression and its uptake activity for glutamate [21, 22]. Therefore, the present study was undertaken to explore whether Cef could alleviate learning and memory deficits by upregulating GLT-1 expression and then promoting glutamate-glutamine cycle in early stage of APP/PS1 transgenic AD mice.
MATERIALS AND METHODS
Animals and grouping
The APPswe/PS1dE9 (APP/PS1) transgenic AD model mice obtained from Chinese Academy of Medical Science were used in this study. This type of mice is bred in a C57BL/6J genetic background and over-expresses human amyloid precursor protein (APP) with the Swedish (K594M/N595L) mutation and presenilin 1 (PS1) deleted in exon 9. These mice show learning and memory deficits in the age of 3–5 months and plentiful senile plaques in 12 months, and manifest similar pathological characteristics with AD patients [23]. The housing conditions were controlled in temperature about 25°C, light from 07:00 to 19:00; humidity 50–60%, and sterilized food and water was freely available. The animal care and experimental procedures were conducted according to the ‘ARRIVE’ guidelines [24] and approved by the Committee of Ethics on Animal Experiments of Hebei Medical University. All efforts were made to minimize the suffering and numbers of the animals.
The experiment was primarily designed to three groups consisted of wild type, APP/PS1 and Cef groups using 6- and 7-month-old mice. In the Cef group, the APP/PS1 mice were administrated with Cef by intraperitoneal injection once a day for continuous 14 days which started at the age of 5.5 months for 6-month-old mice and at 6.5 months for 7-month-old mice. According to the dose of Cef administrated, the Cef group was further divided into 100 mg/Kg, 200 mg/Kg, and 300 mg/Kg subgroups. The doses were determined according to previous reports [18, 26]. The age-matched wild type mice and APP/PS1 mice received normal saline injection by the same protocols with those of the Cef group.
After completion of the administration, the mice in all groups received the Morris water maze test to evaluate their functions in spatial learning and reference memory. To avoid systemic errors in behavioral tests, the behavioral tests were repeated in 6- and 7-month-old mice. During the behavioral tests, the mice in each group were continuously administrated with Cef in the same protocols mentioned above. After the behavioral tests, the 6-month-old mice were sacrificed by decapitation under deep anesthesia and the cerebral hemispheres including the hippocampus were collected for assaying the expression of GLT-1 in protein level and changes of glutamate-glutamine cycle-related proteins including the expression and catalyzing activity of GS and the expression of system N glutamine transporter 1(SN1).
In addition, to determine whether the mice had manifested cognitive deficits when they began to receive Cef administration, we further designed a control group using 5.5-month APP/PS1 mice to test their functions in spatial learning and reference memory.
To determine whether the alterations in cognitive function tested by Morris water maze and the expression of proteins involved in glutamate-glutamine cycle after Cef treatment are mediated by GLT-1, we further designed a DHK+Cef group using 6-month-old APP/PS1 mice. In this group, the mice were administered DHK (dihydrokainic acid, 10 mg/Kg), a selective GLT-1 inhibitor, by intraperitoneal injection once daily 30 min before Cef injection. This dose and delivery route of DHK has been reported to effectively inhibit the uptake activity of GLT-1 in the brain [27, 28]. The Cef administration was in the same protocols mentioned above and only the dose of 200 mg/Kg was selected. Other treatments after the completion of the administrations were the same as above.
Morris water maze test
A plastic tank with a diameter of 120 cm containing a white submerged escape platform was used. The escape platform was 9 cm in diameter and 29 cm high centered in the first quadrant, and 1.0 cm below the water surface. An overhead video camera coupled to a computer was used to track animal movements. The test was performed in a dimly light, sound proof test room with various visual cues. The temperature of the water was kept 20±1°C throughout the experiment.
The Morris water maze test was composed of space navigation test and retention test. The space navigation test consisted of 4 consecutive days of testing, with five trials per day. If the mouse failed to find the escape platform within the maximum time of 60 s, it was gently guided to the platform. The mouse was allowed to remain on the platform for 15 s after escaping to it. The time reached to the platform was measured as escape latency if the swimming speed >8 cm/s, and the average escape latency of five trials was determined. The starting position of the navigation varied randomly among four constant locations at the pool rim. Mice were placed in the water with their nose pointing toward the wall at one of the starting points. The retention test was conducted by probe trial. On the 5th day, the platform was removed, and the mice were allowed to swim for 60 s to determine their search bias. The time spent in the platform quadrant and the frequency crossed the platform area were measured according to the search bias.
The data for the behavioral tests were collected and analyzed using the JLBehv behavioral analysis software (JiLiang Science and Technology Company Limited, Shanghai, China), and the experimenters for the behavioral tests were blinded to the experimental design.
Chemicals and antibodies
Ceftriaxone (Cef) (Rocephin, Roche) was obtained from Roche. Dihydrokainic acid (DHK) was purchased from Sigma Chemical Co. Cef and DHK were both dissolved with normal saline and were administered by i.p. injection. Primary antibodies for immunohistochemistry and western blotting were as follows: GLT-1, guinea-pig polyclonal anti-GLT-1 (Lot: 2673084, Millipore); GS, rabbit polyclonal anti-GS (Lot: GR220419-1, Abcam); SN1, rabbit polyclonal anti-SN1 (Lot: 14315-1-AP, Proteintech). The secondary antibodies were biotin labeled-goat anti-guinea pig IgG for GLT-1 (Lot: XH051, KPL) and horseradish peroxidase (HRP) labeled-goat anti-rabbit IgG for GS and SN1 (Lot: 140012, KPL). The assay kit for GS activity was obtained from GENMED (Lot: GMS50375.2, Shanghai, China).
Immunohistochemistry
This method was used to assay the expressions of GLT-1 and SN1 in the hippocampus of 6-month-old mice. The cerebral hemisphere of one side was dissected and embedded in paraffin after fixation in paraformaldehyde. For each sample, serial sections in thickness of 5μm were cut in the largest coronal plane of the hippocampus. After deparaffinization by xylene, hydration in descending alcohol and incubation with 3% (v/v) H2O2 for 30 min to eliminate endogenous peroxidase and with 10% (w/v) goat serum to block nonspecific antigen, the sections were incubated with the primary antibody against GLT-1 (1:500) and SN1 (1:500) overnight at 4°C. In the next day, the sections were incubated with the secondary antibody (1:1000) at 37°C for 30 min. The immune reaction was visualized using diaminobenzidine (ZSGB, Beijing, China). Sections in different groups being compared were treated in the same immunostaining run. Because the diffuse distribution of GLT-1 [29] and SN1 [30] in the hippocampus, we selected the area in the peak of the arch of the CA1 subfield in 400×magnification for the quantitative analysis of the immunoreactive density (the area indicated by the box in the inset A in Figs. 3 and 5). The mean optical density of the selected area was measured by Image-Pro Plus (IPP) software (version 6.0, Media Cybernetics, Silver Spring, MD) to quantify the density of immunoreactivity, and the average of the mean optical density was used for statistical analysis. Three sections were randomly selected per mouse for the analysis.
Western blotting
The expressions of GLT-1, GS, and SN1 in the hippocampus of 6-month-old mice were assayed using western blotting analysis. The hippocampus was dissected and stored at –80°C. The hippocampus was homogenized using tissue lysis buffer supplemented with 1 mM PMSF and protease inhibitors (Roche). Protein concentrations in the supernatants were determined by bicinchoninic acid and assayed by Synergy-HT microplate reader (BioTek, Gene company Limited, USA). Fifty-microgram protein were loaded on SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) and then probed with the primary antibodies of GLT-1 (1:1000), GS (1:1000), and SN1 (1:1000) overnight at 4°C. After washing with TPBS (Tween-20 and PBS mixed solution), the membranes were incubated with the secondary antibody (1:2000) at room temperature for 1 h. β-actin or GAPDH was used as a loading control. The immunoblot bands were visualized using enhanced chemiluminescent (ECL) substrate and quantitated by measurement of integral optical density (densitometry) using Amersham Imager 600 (GE Healthcare UK Limited, UK) and analyzed with an image analyzing software (Alpha Imager, USA). The ratios of integral optical densities between aimed proteins and controls from the same homogenate were used to present the relative expression of the aimed proteins.
The assay of GS activity
The catalyzing activity of GS was determined using the assay kit mentioned above. The kit was composed of continuous reaction enzyme system including GS, pyruvate kinase, and lactic dehydrogenase. Firstly, the hippocampus was homogenized using lysis buffer provided by the kit, and then the homogenate was subsequently reacted with the components included in the kit according to the manufacturer’s instruction. In the final reaction, NADH was oxidized to NAD+. According to the content of oxidized NADH assayed by spectrophotometry, the activity of GS was calculated and expressed with μmol NADH/min/mg protein.
Statistics
All statistical analyses were performed using SPSS for Windows 16.0 software (SPSS Inc, Chicago, IL, USA). All data were presented with Means±SEM and analyzed using one-way ANOVA and LSD-t for comparison between groups, except that the escape latency in Morris water maze test was analyzed using repeated measures ANOVA. Statistical significance is reported as p < 0.05.
RESULTS
Cef treatment improved cognitive deficits in early stage of APP/PS1 AD mice
Compared with age-matched wild type mice, the APP/PS1 mice showed an increased escape latency (Fig. 1A) with a decreased time spent in target quadrant (Fig. 1B, C) and decreased crossings in the platform site (Fig. 1D). There was no difference in the speed of swimming between the two groups of mice (Fig. 1E). These results indicated that the APP/PS1 mice at 5.5 months old had already manifested cognitive impairments when they began to be administrated with Cef.
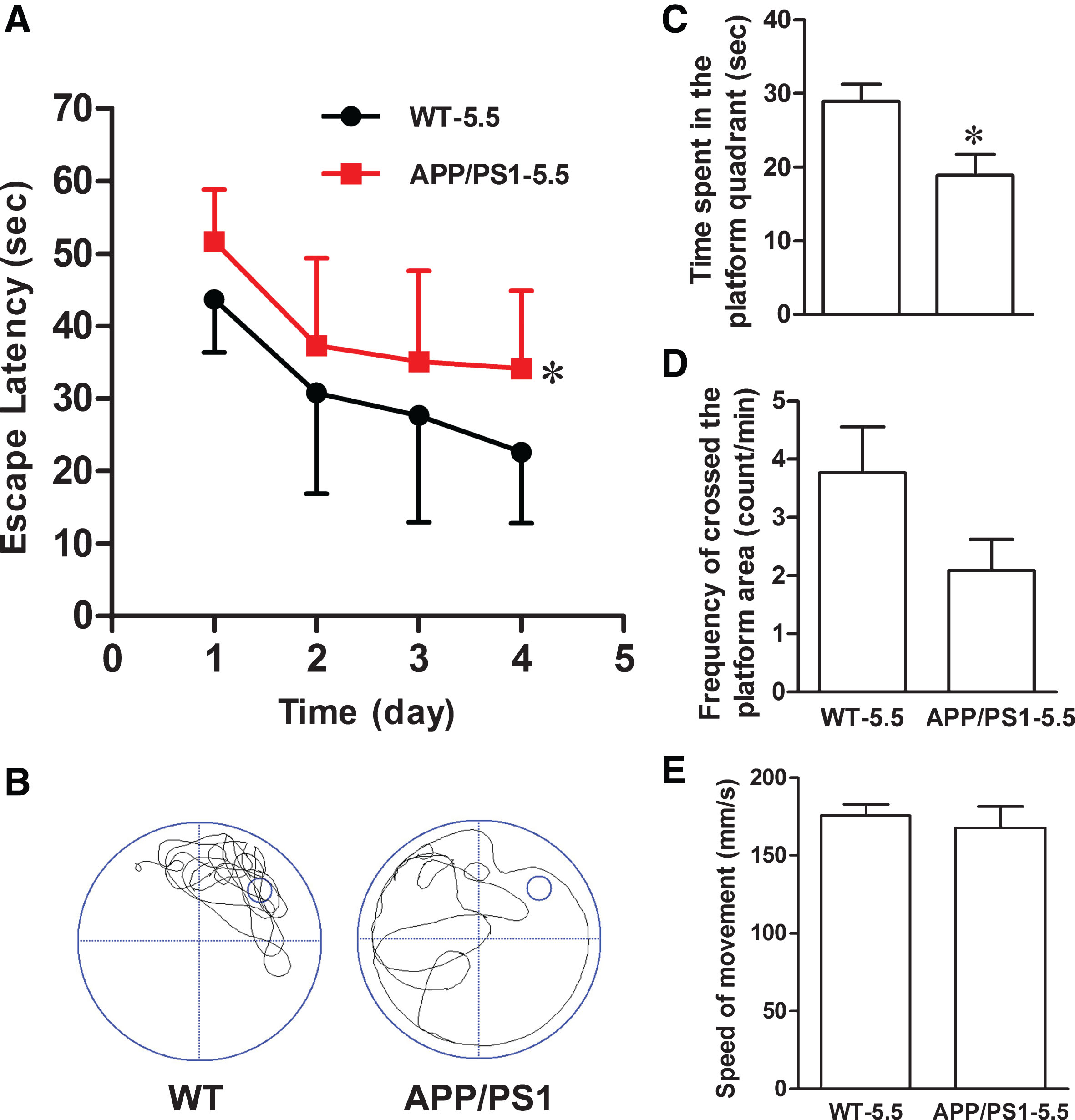
APP1/PS1 mice show cognitive deficits at 5.5 months. The cognitive functions in 5.5-month-old APP/PS1 transgenic and the age- and sex-matched wildtype control mice were measured by Morris water maze test. APP/PS1 mice show spatial learning and memory deficits represented with the increased escape latency during the training trial for 4 days (A), and the decreased time spent in target quadrant (B, C) and the decreased crossing times in the platform site (D) measured at day 5 by removed the platform (n = 11 each group). There was no difference in the speed of swimming between the two groups (E). Data were presented as mean±s.e.m; *p < 0.05 versus wild type (repeated measures ANOVA, Bonferroni’s post hoc test).
Then, we observed the effect of Cef on the cognitive deficits of the APP/PS1 mice. In the 6-month-old APP/PS1 group, the escape latency was prolonged in the navigation test compared with wild type group (Fig. 2A). The time spent in the platform quadrant (Fig. 2B, C) and the frequency crossed the platform area were decreased (Fig. 2D), which further confirmed the cognitive deficits of the APP/PS1 mice. Compared with the APP/PS1 group, Cef treatment at 200 mg/Kg and 300 mg/Kg significantly decreased the escape latency (Fig. 2A), and increased the time spent in the platform quadrant and frequency crossed the platform area (only Cef 300 mg/Kg group) of the APP/PS1 mice in the Cef treatment group (Fig. 2C, D). No obvious change was observed in the speed of swimming in each group (Fig. 2E). The search bias delineated in the retention test showed that wild type mice swam around the platform and crossed it repeatedly when it “missed” the platform, while the APP/PS1 mice swam around the rim of the pool and had no idea to find out the platform (Fig. 2B). Cef treatment increased the exploratory ability of the APP/PS1 mice. The effect of Cef on the 7-month-old APP/PS1 mice in the Morris water maze test (Fig. 2F-J) was similar with that on the 6-month-old group. The results indicated that Cef alleviated the spatial learning and reference memory deficits in APP/PS1 mice in early stage of AD.
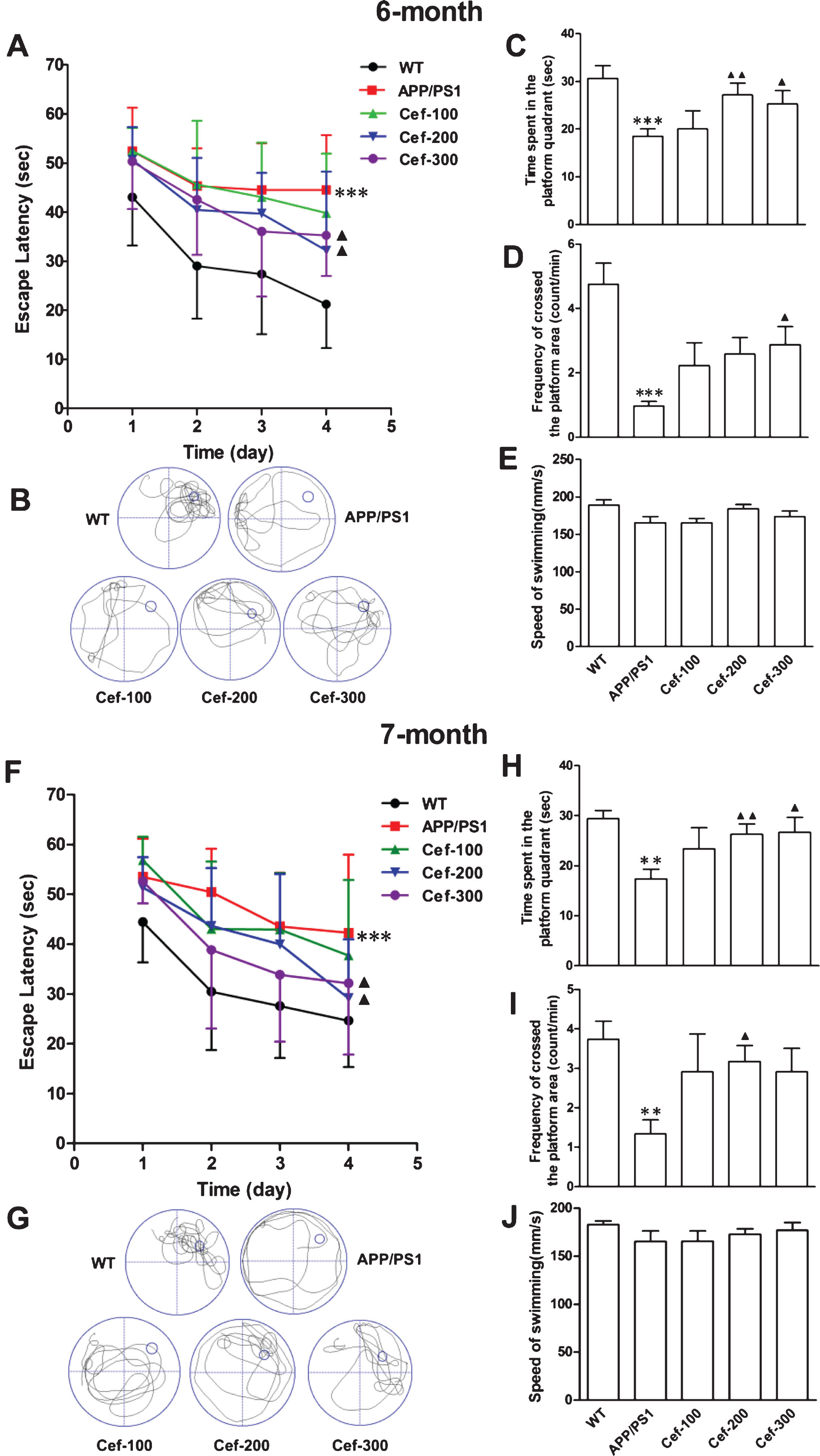
Cef treatment improves cognitive deficits in 6- and 7-month-old APP/PS1 mice. In the Morris water maze test, the untreated APP/PS1 mice in the 6-month-old group showed increased escape latency (A), and decreased time spent in the target quadrant and decreased crossing times in the platform site (B-D) (n = 17). Cef treatment in doses of 200 mg/Kg (n = 11) and 300 mg/Kg (n = 14) decreased the escape latency (A), increased the time spent in the target quadrant, and increased the crossing times in the platform site (B-D). There was no difference in the speed of swimming in each group of mice (E). In 7-month-old APP/PS1 mice (F-J), Cef treatment shows similar improvement on the spatial learn and memory function with that in 6-month-old APP/PS1 mice. **p < 0.01, ***p < 0.001 versus Wild type group. ▴p < 0.05, ▴▴p < 0.01 versus APP/PS1 group.
Cef upregulated GLT-1 expression, GS activity, and SN1 expression in the hippocampus of APP/PS1 AD mice
To investigate the role of GLT-1 and glutamate-glutamine cycle in the Cef-induced cognitive improvement in APP/PS1 mice, we measured the expressions of GLT-1 and glutamate-glutamine cycle-related proteins including GS and SN1 by western blotting and immunohistochemistry, and the GS catalyzing activity in the hippocampus of 6-month-old APP/PS1 mice.
For GLT-1 expression, immunohistochemical staining showed that brown fine GLT-1 immunoparticles diffusely distributed in the whole hippocampus including the CA1, CA3 subfields, and dental gyrus in the wild type mice (Fig. 3A). In high magnification (400×), the immunoparticles were found in layers of pyramidal neurons, polymorphous cells, and stratum radiatum (Fig. 3B). This staining pattern is consistent with other reports [21, 29]. Compared with wild type mice, APP/PS1 mice showed decreased immunoreactivity of GLT-1 in CA1 subfield of the hippocampus (Fig. 3C). The densitometry showed significant decreases in the mean optical density (Fig. 3G). Cef treatment in all the three doses of 100 mg/Kg, 200 mg/Kg, and 300 mg/Kg induced significant increase in GLT-1 immunoreactivity (Fig. 3D-F) and mean optical density (Fig. 3G) compared with the untreated APP/PS1 group.

Cef treatment increases GLT-1 protein level in the hippocampus of 6-month-old APP/PS1 mice. APP/PS1 mice showed decreased immunoreactivity of GLT-1 in hippocampal CA1 subfield, and administration with Cef increased the immunoreactivity of GLT-1 measured by immunohistochemistry (A-G, A: scale bar = 200μm; B: scale bar = 20μm) and western blotting analysis (H) (n = 5 per group). *p < 0.05, **p < 0.01 versus Wild type group. ▴p < 0.05, ▴▴p < 0.01, ▴▴▴p < 0.001 versus APP/PS1 group (One-way ANOVA).
Western blotting analysis showed that there was basal expression of GLT-1 in the hippocampus of wild type mice. The GLT-1 expression was downregulated in APP/PS1 mice compared with wild type mice. Cef treatment in the doses of 200 mg/Kg and 300 mg/Kg significantly upregulated the GLT-1 expression, with no evident change in the expression of GLT-1 in Cef 100 mg/Kg group compared with the untreated APP/PS1 group (Fig. 3H).
These results together indicated that Cef treatment upregulated the expression of GLT-1 in APP/PS1 mice.
For GS expression and activity, western blotting analysis showed that there was obvious basal GS expression in the hippocampus of either wild type or APP/PS1 mice, and the Cef treatment in all three doses had no effect on the GS expression (Fig. 4A, B). However, the activity of GS was significantly declined in the APP/PS1 mice compared with wild type mice. After Cef treatment, the activity of GS in APP/PS1 mice was significantly upregulated in all Cef treated groups compared with untreated APP/PS1 group (Fig. 4C). These data suggested that GS activity was inhibited in APP/PS1 mice although the total protein level was not significantly changed, while Cef treatment attenuated the inhibition of GS activity.
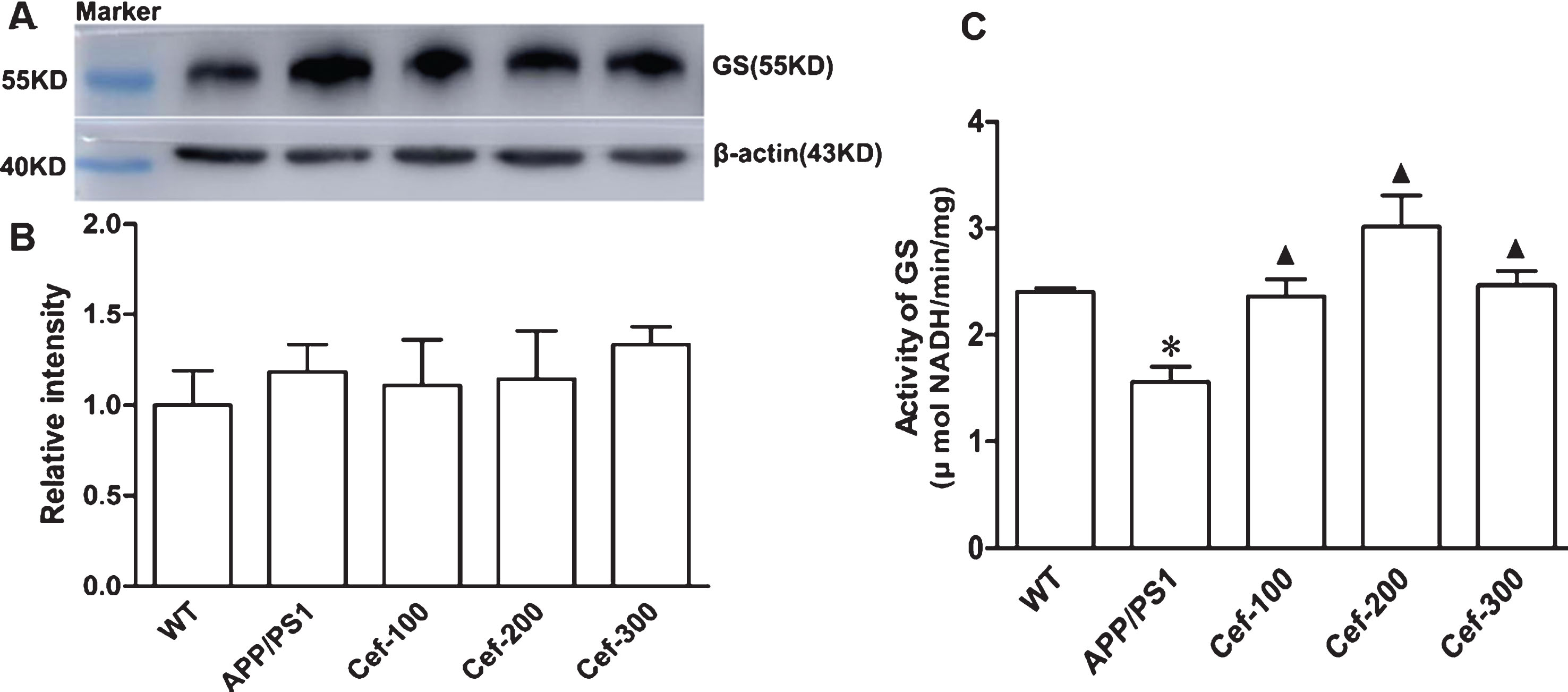
Cef treatment increases glutamine synthetase (GS) activity without changing GS protein level in the hippocampus of 6-month-old APP/PS1 mice. By western blotting, no change of GS immunoreactivity normalized to β-actin was detected in all groups (n = 5 per group) (A, B). However, APP/PS1 mice showed decreased catalyzing activity of GS and Cef treatment increased the activity of GS (C) (n = 4 per group). *p < 0.05 versus Wild type group. ▴p < 0.05 versus APP/PS1 group.
For SN1 expression, immunohistochemistry assay showed that brown SN1 immunoparticles were diffusely distributed in the hippocampus (Fig. 5A). In high magnification (400×), the SN1 immunoparticles were found in layers of polymorphous cells, stratum radiatum, and pyramidal neurons. Some brown fiber-like stripes in different length were observed in the stratum radiatum in wild type mice (Fig. 5B). This staining pattern is consistent with others reports [7, 31]. Compared with wild type mice, APP/PS1 mice showed an obvious decrease in the SN1 immunoreactive particles (Fig. 5C). Compared with the APP/PS1 mice, Cef treatment induced a prominently increase in the immunoparticles of SN1 in Cef treatment group, especially in the large dose of 300 mg/Kg (Fig. 5D-F). The densitometry showed corresponding changes with the immunostaining (Fig. 5G). Western blotting analysis showed that there was basal SN1 expression in the hippocampus of wild type mice. Compared with wild type mice, SN1 expression was decreased in APP/PS1 mice. Cef treatment in doses of 200 mg/Kg and 300 mg/Kg significantly upregulated SN1 expression in Cef treatment group compared with APP/PS1 group (Fig. 5H). These data together suggested that SN1 was decreased in APP/PS1 mice, while Cef treatment partially restored the level of SN1.
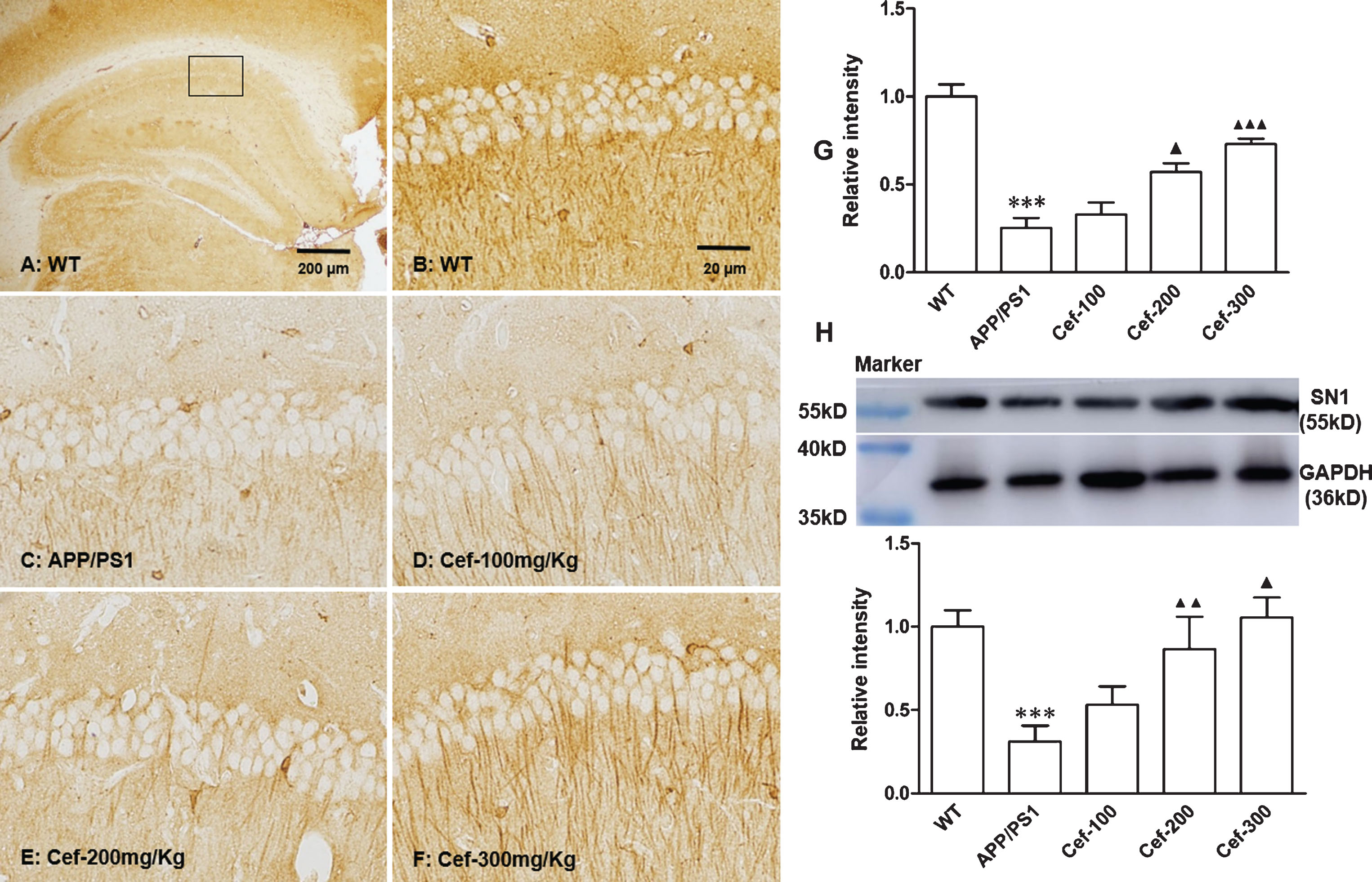
Cef treatment upregulates the expression of SN1 in the hippocampus of 6-month-old APP/PS1 mice. Immunohistochemistry (A-G, A: scale bar = 200μm; B: scale bar = 20μm) and western blotting analysis (H) showed that SN1 immunoreactivity in hippocampal CA1 subfield was decreased, while administration with Cef increased the SN1 immunoreactivity (n = 5 per group). ***p < 0.001 versus Wild type group. ▴p < 0.05, ▴▴p < 0.01, ▴▴▴p < 0.001 versus APP/PS1 group.
DHK reversed the Cef-induced improvements on cognitive function, GS activity, and SN1 expression in APP/PS1 AD mice
To determine whether Cef-induced cognitive improvement and the upregulation of GS activity and SN1 expression were mediated by GLT-1, we further measured the effect of DHK, a selective inhibitor of GLT-1, on the GS activity and SN1 expression after Cef treatment in 6-month-old APP/PS1 mice.
In the Morris water maze test, the escape latency was significantly prolonged, the time spent in the platform quadrant and the frequency crossed the platform area were declined after the administration of DHK in DHK+Cef group compared with Cef group (Fig. 6C-E). The upregulation in GS activity and SN1 expression induced by Cef treatment was also reversed by the administration of DHK in DHK+Cef group compared with Cef group (Fig. 6F, G). These results indicated that Cef-induced improvements in the recognition memory deficits, spatial learning and reference memory deficits and GS activity and SN1 expression were inhibited by administration DHK.
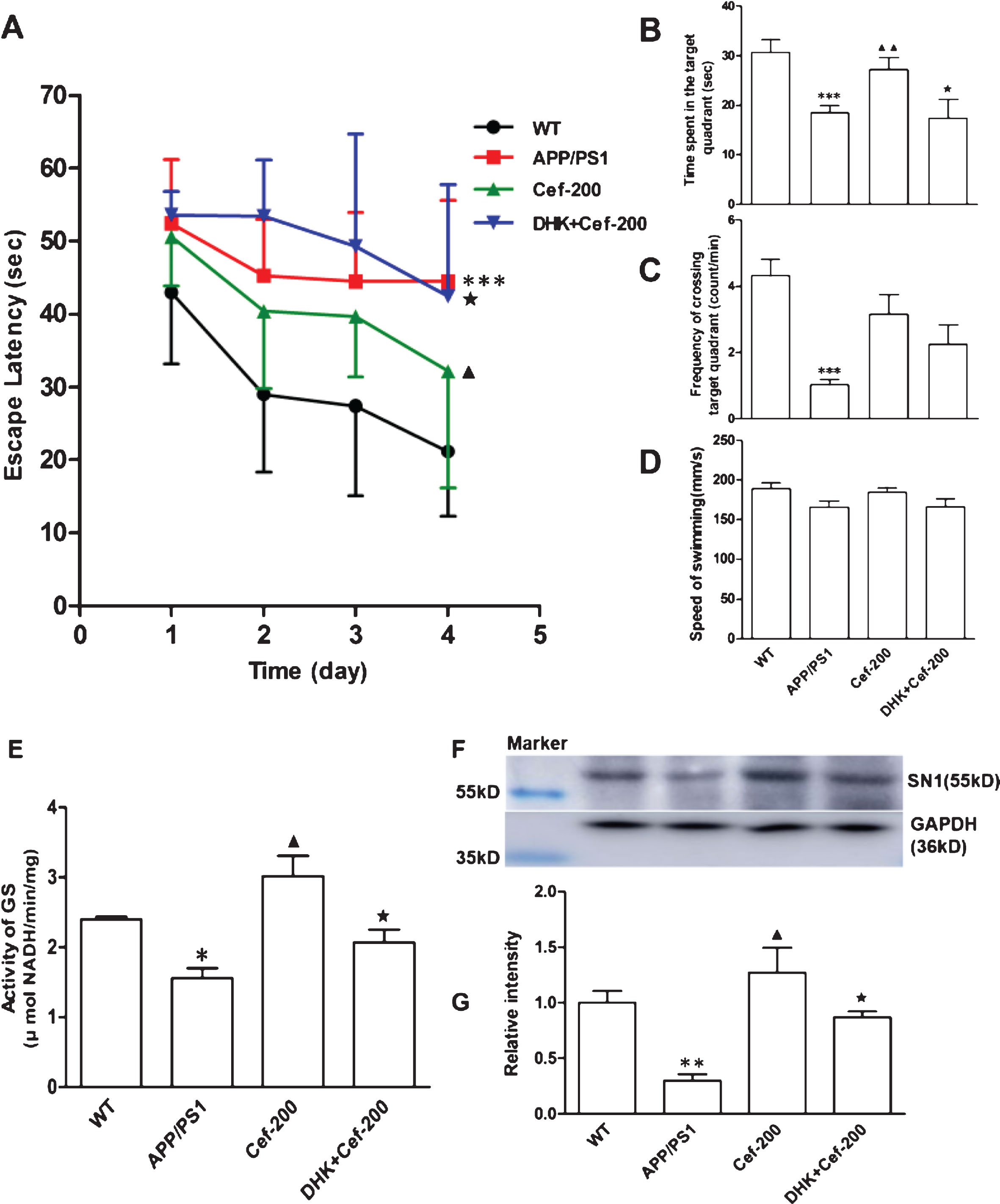
Simultaneous inhibition of GLT-1 by DHK abolishes Cef-induced improvement on behavior and metabolism in the hippocampus of 6-month-old APP/PS1 mice. Simultaneous inhibition of GLT-1 function by DHK abolished the Cef-induced cognitive improvement evidenced by the prolonged escape latency (A) and decreased time spent in the target quadrant (B) and the decreased crossing times in the platform site (C) in the Morris water maze test (n = 10). The administration of DHK inhibited the increase of GS activity (E) (n = 4) and prevented the upregulation of SN1 expression induced by Cef (F, G) (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001 versus Wild type group. ▴p < 0.05, ▴▴p < 0.01 versus APP/PS1 group. ★p < 0.05 versus Cef-200 group.
DISCUSSION
Glutamate is the most important excitatory neurotransmitter in the mammalian central nervous system and plays an important role in the course of learning, memory and cognition by participating in synaptic plasticity regulation and keeping neural circuit function [3, 4]. The glutamate uptake activity of excitatory amino transporters, especially GLT-1, plays a crucial role in regulating the homeostasis and reutilization of glutamate. A variety of evidence has shown that GLT-1 expression and/or uptake activity were downregulated during the occurrence and development of learning and memory deficits in AD patients or AD transgenic animal model [13–15, 32]. The present study further confirmed the dysregulation of GLT-1 expression in the APP/PS1 AD mice. The downregulated expression of GLT-1 would lessen the amount of glutamate uptake from the synaptic cleft and then reduce the substrate for glutamate-glutamine cycle in astrocytes. As a result, the efficient reutilization of glutamate as neurotransmitter was impeded, which would induce disturbance in synaptic transmission and neural circuit function, and sequentially lead to impairments of learning, memory, and cognition [33, 34].
Cef is a typical β-lactam antibiotic and reported to significantly upregulate GLT-1 expression and its uptake activity [18]. This property of Cef has been shown benefits to some pathological processes such as stroke, oxygen glucose deprivation, cerebral ischemia, and amyotrophic lateral sclerosis [19, 35]. Our previous studies using a rat global brain ischemic model have shown that Cef can effectively upregulate GLT-1 expression and its uptake activity, reduce the excessive accumulation of glutamate and its excitotoxicity and then protect hippocampal neurons against global brain ischemia [21, 22]. The present study tried to explore the improving effect of Cef on the learning and memory deficits of APP/PS1 AD mice according to the important role of glutamate in learning and memory and the disturbance of glutamate-glutamine cycle in AD. It was found that Cef could alleviate the learning and memory deficits either in 6- or 7-month-old APP/PS1 mice as indicated by the decreased escape latency, increased time spent in the platform quadrant and frequency crossed platform area in the Morris water maze test. The same tests indicated that the mice in age of 5.5 months when they began to receive Cef administration have manifested learning and memory deficits. This characteristic is consistent to the report that the mice showed learning and memory deficits in the age of 3–5 months [23]. Meanwhile, Cef treatment in the present study significantly upregulated the GLT-1 expression in APP/PS1 mice as shown in immunohistochemistry and western blotting analysis. Particularly, the Cef-induced improvement in the cognitive deficits of APP/PS1 AD mice was prevented by prior administration of DHK, the selective inhibitor of GLT-1. These results suggested the role of the GLT-1 in the Cef-induced improvement in cognitive deficits of the APP/PS1 AD mice. Zumkehr et al. reported similar results in which Cef administration in dose of 200 mg/Kg to 10-month-old AD mice for 2 months ameliorated cognitive decline and upregulated GLT-1 expression of the mice [26]. These results support each other and reinforced the suggestion that Cef-induced upregulation of GLT-1 contributed to the improvement of the cognitive deficits in AD mice.
Concerning the specific mechanisms underlying the Cef-induced improvement by GLT-1 upregulation on the cognitive deficits of AD mice, Zumkehr et al. reported the effect of GLT-1 upregulation after Cef treatment on the tau and Aβ levels [26]. Unlike their report, we tried to focus on the glutamate-glutamine cycle to explore the mechanisms considering the important role of GLT-1 and the subsequent glutamate-glutamine cycle in the glutamate reutilization as neurotransmitter. GS is an astrocyte-specific key enzyme for the glutamate-glutamine cycle in the brain, and rapidly amidates glutamate to glutamine, a non-neuroactive amino acid [36, 37]. The dysregulation of GS such as downregulated expression [9, 15] and oxidized injury [17] was reported in transgenic AD mice. But in AD patients the GS expression levels in cerebrospinal fluid and serum were unchanged [38], and it was no different in retinal ganglion cells between APP/PS1 and wild type mice [39]. In the present study, it was found that the GS expression in protein level had no difference between APP/PS1 and wild type mice. However, the catalyzing activity of GS significantly declined in APP/PS1 mice compared with wild type mice. This decreased GS activity in APP/PS1 mice maybe associated with oxidative stress injury of Aβ [17, 40]. Furthermore, it is noticeable in the present study that Cef treatment remarkably reversed the declined of the catalyzing activity of GS in the APP/PS1 mice although the expression in protein level had no change after the Cef treatment. This reverse effect of Cef on the downregulated catalyzing activity of GS would elevate the amidation efficacy of glutamate to glutamine because GS is the key enzyme for the conversion [41]. Verma et al reported similar phenomenon that Cef upregulated the catalyzing activity but not the expression of GS in rat cerebral ischemia/reperfusion injury model, and attributed the mechanism of the phenomenon to a compensatory response to the increased substrate of glutamate resulted from the upregulated GLT-1 expression [42]. This report supports our finding above.
Another interesting finding in the present study is the upregulation of SN1 after Cef treatment. The transporter specifically expresses on astrocyte processes surrounding dendritic synapses, and serve as to transport glutamine efflux from astrocytes [43–45]. In our present study, immunohistochemistry assay showed that SN1 protein diffusedly expressed in the hippocampus, especially in the stratum radiatum. Relative quantitative analysis of the immunohistochemical staining and western blotting showed that the expression of SN1 was downregulated in APP/PS1 mice and Cef treatment significantly reversed the downregulated SN1 expression in APP/PS1 mice. This upregulation would improve the glutamine-glutamate cycle because of the important role of SN1 in glutamine efflux from astrocytes [43, 46]. Limited information exists regarding the SN1 expression in AD up to now. The present study indicated the change and the effect of Cef treatment on the expression of SN1 in APP/PS1 mice for the first time.
Some SN1-reactive fiber-like stripes in different length were observed in the stratum radiatum of the CA1 hippocampus in immunohistochemical staining (Fig. 5B). This staining pattern suggests neuronal distribution of SN1. Indeed, Zielińska et al. observed similar immunohistochemical staining pattern in chick cerebellar sections, and they attributed this staining to Bergmann glial cells [47]. Astrocyte is the main glial cell in the stratum radiatum of hippocampus [48]. Chaudhry et al had provided convincing evidence indicating the astrocytic, rather than neuronal, expression of SN1 in the hippocampus using in situ hybridization and double labeling staining [31]. Further study using double labeling of SN1 and neuron or astrocyte markers would provide direct evidence for its cell-type-specific distribution.
The upregulations in GS activity and SN1 expression after Cef treatment suggested that the Cef-induced improvement on the cognitive deficits of APP/PS1 AD mice at least partly is related to stimulating the glutamate-glutamine cycle, because GS and SN1 are key factor involved in the cycle. As known, GS is responsible for the conversion of glutamate to glutamine and SN1 transports glutamine efflux from astrocytes. The upregulation of the GS catalyzing activity and SN1 expression would make more glutamate convert to glutamine and promote more glutamine efflux from astrocytes, respectively, and thus stimulates the glutamate-glutamine cycle. Furthermore, we found that DHK prevented the Cef-induced improvement on GS activity and SN1 expression. This finding suggested that the upregulation of GLT-1 contributed to the stimulation of the glutamate-glutamine cycle. It has been reported that GLT-1 accounts for up to 70–90% of glutamate uptake [5, 6]. So, the upregulation GLT-1 induced by Cef would take more glutamate into astrocytes, afford more substrate for glutamate-glutamine cycle, and then stimulating the glutamate-glutamine cycle by substrate-induced enzyme activity mechanism at least partly. In other word, it could be concluded that Cef could stimulate the glutamate-glutamine cycle by upregulating GLT-1. Further studies direct focused on the transporting activity of SN1, concentrations of glutamate and glutamine in astrocytes would provide more convincing evidence to reinforce the above conclusion.
Cef is widely used in clinical therapy because it has wide antibacterial spectrum. Long-term administration would result in some side effects such as dysbacteriosis, bacterial resistance, etc., and rarely someone developed Cef-induced hemolytic anemia, which severely limit its long-term application. Therefore, the present study does not mean that Cef can be used as anti-AD medication. However, it could shed a new light on the study in the prevention and therapy of AD, if the side effects mentioned above could be avoided by some methods such as chemical structural transformation of the medication, or finding some other substitutes which have similar structure to Cef and could upregulate GLT-1 expression and the subsequent glutamate-glutamine cycle, but has no antibacterial activity. Additionally, it was somewhat unfortunate that the effect of Cef treatment in the present study showed no satisfactory dose-dependency between the doses of 200 mg/Kg and 300 mg/Kg. It may be related to the side effect of large doses of Cef and/or relatively small group size.