
Abstract
Background
Anti-amyloid-β (Aβ) immunotherapies are emerging as treatments for Alzheimer's disease (AD).
Objective
This review examines the structure-activity relationships of anti-Aβ therapeutics tested in phase 3 trials.
Methods
We analyzed crystallographic data and molecular models to elucidate the Aβ binding mechanisms of donanemab, lecanemab, aducanumab, bapineuzumab, gantenerumab, solanezumab, and crenezumab.
Results
Lecanemab recognizes minimally degraded Aβ missing 1–2 residues, avoiding common Aβ in circulation and further degraded material sequestered in plaques. Bapineuzumab buries the N-terminus of Aβ requiring Asp1 and is reactive with benign, common Aβ. Donanemab buries the truncated N-Glu3 terminus with strong contacts engaging the cyclized pyro-Glu3 modification. Gantenerumab shows lecanemab-like properties but also binds common Aβ. Aducanumab likely needs mAb-mAb cooperation to scavenge a spectrum of Aβ oligomers explaining higher doses. Solanezumab and crenezumab target a pre-amyloid epitope resulting in off-target engagement, including monomers and likely excluding Aβ-ApoE complexes.
Conclusions
Preventing primary Aβ nucleation failed due to limitations imposed by the blood-brain barrier, intracellular aggregation routes, and the natural abundance of Aβ. Anti-Aβ monoclonal antibody therapies in clinical use capture Aβ at various stages of decay where post translational modifications have been used effectively as proxies for time spent in vivo. By targeting a relatively labile epitope of aging Aβ, lecanemab selects more biologically active species of Aβ avoiding both benign monomers and old fortified species. This focal point may account for the significant cognitive effects of lecanemab. The structure of aducanumab suggests a broadly neutralizing role has evolved for natural immunity to AD.
Keywords
Introduction
Alzheimer's disease (AD) is characterized by the accumulation of amyloid-β (Aβ) peptides, derived from the amyloid-β protein precursor (AβPP). While Aβ is normally rapidly turned over, it can form toxic β-sheet-rich oligomers, protofibrils, and fibrils.1–3 Astrocytes readily engulf Aβ oligomers but cannot fully degrade the material which is stored and ejected in vesicles. 4 Microglia have been shown to phagocytose or compact fibrillar material suggesting these cells also work to contain persistent Aβ in the extracellular space. Early work found cored plaques are comprised mainly of truncated Aβ (∼88% N-Phe4, N-Ser8 and N-Gly9). 5 Surface Aβ deposits may catalytically propagate circulating Aβ oligomers, suggesting that clearance of these deposits might help disrupt soluble Aβ oligomer production. 6
Passive immunotherapies targeting Aβ have been prioritized above active approaches for better control of antibody titer, therapeutic window, and immune signaling. Seven monoclonal antibodies (mAbs) have reached phase 3 trials to date, with varying degrees of success in Aβ clearance and cognitive outcomes.
Aducanumab, a human IgG1 mAb derived from healthy elderly subjects, demonstrated Aβ clearance and effects on phosphorylated tau biomarkers.7,8 Despite broad reactivity for Aβ species, the therapy required high doses to show effect. Its unique kinetic profile and immune signature warrant further investigation.1,9
Lecanemab, a humanized IgG1 antibody, significantly lowers Aβ in prodromal and early AD, likely through microglial phagocytosis. 10 Its selectivity for protofibrils and particularly significant impact on cognitive decline requires detailed understanding of the antibody's mechanism of action to enhance this clinical benefit. 11
Donanemab, an IgG1 mAb targeting pyroglutamate-modified Aβ (pE3-Aβ), most effectively lowers Aβ measured by positron emission tomography (PET).12,13 Understanding its selectivity mechanism is critical, especially as novel vaccine candidates emerge based on pE3-Aβ epitopes. 14
Previous studies have revealed structural insights into bapineuzumab, solanezumab, and crenezumab binding to Aβ.15–17 Recent clinical trials, including the A4 trial of solanezumab, have yielded complex results including accelerated decline at very high doses highlighting the complexities of antibody selectivity, Fc composition and cognitive effect. 18
Amyloid-related imaging abnormalities (ARIA) remain a concern, particularly for higher doses and in APOE4 carriers.4,19,20 The interaction between Aβ and APOE, especially in APOE ε4 carriers, adds another layer of complexity to therapeutic approaches.21,22 Anti-Aβ therapies may enhance clearance of pathogenic APOE forms or expose them.
While all anti-Aβ mAbs show promise in murine models, only aducanumab, lecanemab, and donanemab have demonstrated cognitive benefits in humans.10,23 As other protein targets also show potential in AD, 24 a thorough analysis of anti-Aβ therapeutics is essential to guide future drug development.
This review examines publicly available structural data to clarify the mechanisms of target engagement by anti-Aβ passive immunotherapies. We re-analyze crystallographic data, propose models for antibodies lacking public structural information, and compare binding motifs across various mAbs. Our analysis suggests that clinical effects may be significantly mediated by engagement of modified Aβ species near and distant from cored amyloid plaques. Low-affinity-high avidity as used by pattern recognition receptors such as CD33 and CLEC7a has proven to be an effective means to distinguish between endogenous protein aggregates and benign abundant forms.25,26 We discuss how efficient engagement of these species, moderated Fc potency, optimized dosing, and early detection may improve the safety and efficacy of immunotherapies for AD.
By elucidating the structural basis of antibody-Aβ interactions, this review aims to inform the development of safer and more effective AD therapeutics, potentially impacting future clinical strategies and patient outcomes.
Methods
Data sources and sequence analysis
Crystallographic structures were downloaded from the Protein Data Bank (PDB). Sequences for donanemab and lecanemab were sourced from the International Non-proprietary Names for Pharmaceutical Substances (INN) Lists 120 and 84, respectively. Murine versions mE8 (donanemab) and m158 (lecanemab) sequences were retrieved via BLAST search of published sequences (Genebank accession numbers AJL00377.1, AJL00378.1, AEP60320.1, AEP60319.1).
Homology modeling
We used the Swiss-Model Server for homology modeling, focusing on variable regions and core epitopes to avoid humanization bias. For m158, PDB 5my4 (Fab C#17) was the top-ranked template (GMQE: 0.85, QSQE: 0.85, 84.2% sequence identity with C#17). For mE8, PDB 5myx (Fab C#24) was selected (GMQE: 0.89, QSQE: 0.97, 86.8% sequence identity with C#24).
Model refinement
Initial models were built against crystal structure templates in PyMol. For m158, which has a two-residue (Gly-Gly) insertion in CDR3(H), we first modeled without these residues, then refined the full-length sequence. CDR1(H) and CDR2(H) of m158 were modeled on PDB entry 1mf22. pE3 Aβ ligands were incorporated into preliminary models, with clashes resolved manually. For m158, pE3 was mutated to Glu3 and modeled with local energy minimization, followed by manual addition of Ala2 to investigate alternative ligand binding modes.
Final optimization
Final models and template crystal structures underwent energy minimization using Swiss-Model Server in User-Template mode. Model statistics and interface parameters for energy-minimized m158, mE8, and templates C#17 and C#24 in complex with Aβ are provided in Supplemental Table 1.
Supplemental material
Sequence alignments are presented in Supplemental Figures 1–3. Supplemental Figure 1 shows light chain variable regions of murine N-terminal homologues including mAb C#17 and m158 VL homologues. Supplemental Figure 2 displays m158 variable heavy region alignments with PDB homologues [7r9d, 1mf2, 4kvc, 1igc] and pairwise alignment with PDB 5my4. Supplemental Figure 3 presents pairwise alignments of light and heavy chain variable regions for mE8, mAb C#24, and humanized mE8 (donanemab). Software used for each analysis step are detailed in the Supplemental Methods.
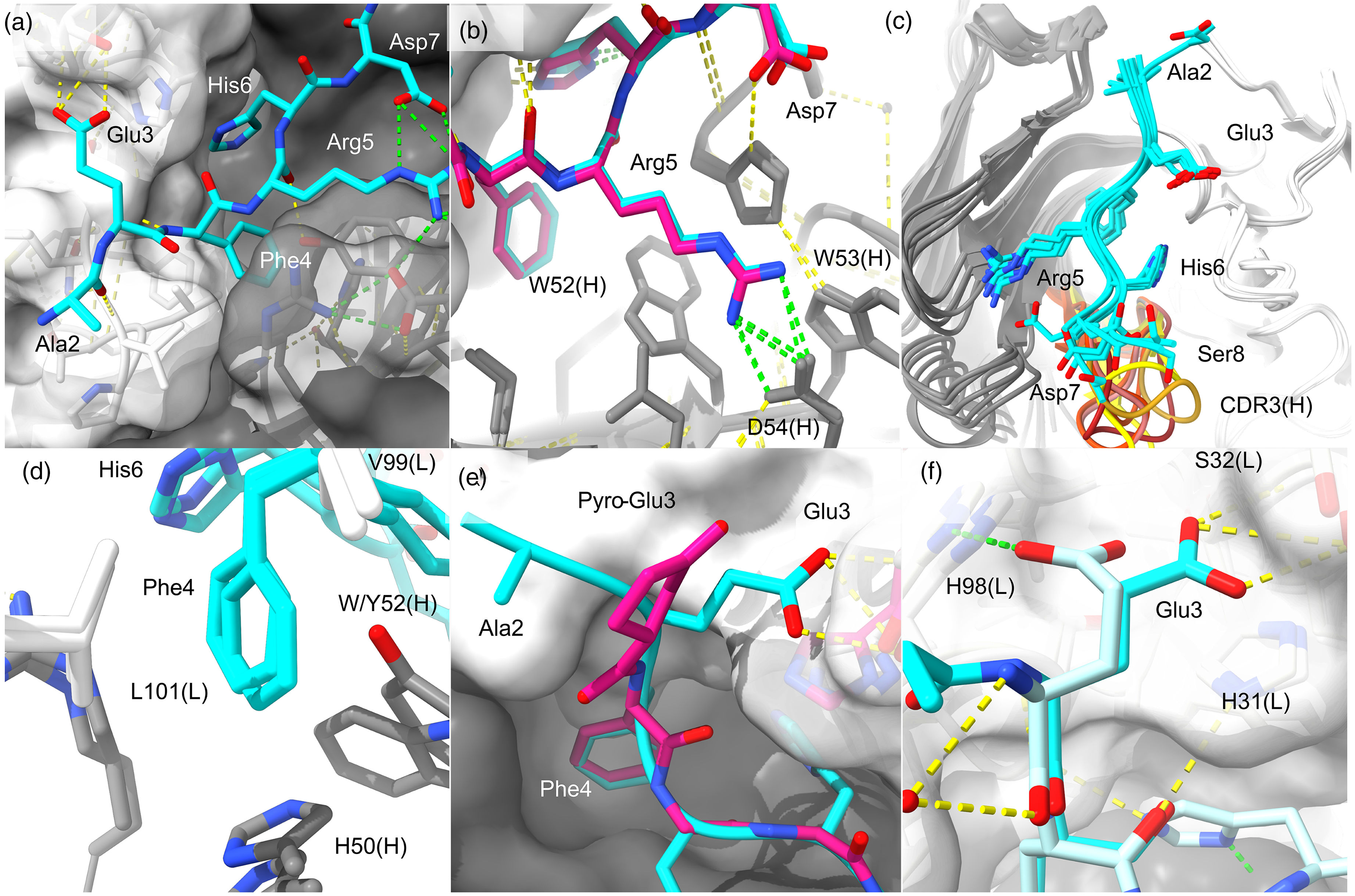
The immunodominant B-cell epitope in mice. Aβ-anti-Aβ structures reveal a dominant response to human sequence Aβ in mice. Antibody equivalent positions are labeled with chain identifier in parenthesis. (a) The 1.6 Å structure of W02 bound to Aβ1−28 [PDB 3bae].
29
The solvent accessible surface area of Aβ buried in W02 is ∼662 Å2. The positions of residues 2-AEFRHDS-8 are evident and well defined over residues 3-7. N-terminal residues contact VL (white surface), and C-terminal residues of the epitope contact VH (gray surface). Yellow dashes indicate H-bonds. Green dashes indicate salt bridges. (b) Superpositioned structures of PFA1 bound to Aβ1-8 (blue; PDB 2ipu) and pE3-8 Aβ (pink; PDB 3eys) show the same conformation overall, with Arg5 captured by a conserved 52-WWDDD-56 consensus motif.
32
(c) Six earliest anti-Aβ crystal structures are shown with CDR3(H) loops brightly colored, namely, with PDB codes, W02(3bae), PFA1(2ipu), PFA2(2r0w), 12A11(3ifl), 10D5(3ifo), and 12B4(3ifp). (d) Phe4 sits between VH-VL domains (W02 and PFA1) with L101(L) and VH aromatics forming a pocket to house the Phe4 sidechain. (e) PFA1 complexed to Aβ (blue) or pE3-8 Aβ (pink) shows that pE3 lacks contacts made by Glu3 with the VL domain. (f) PFA1 binds to Aβ (DA
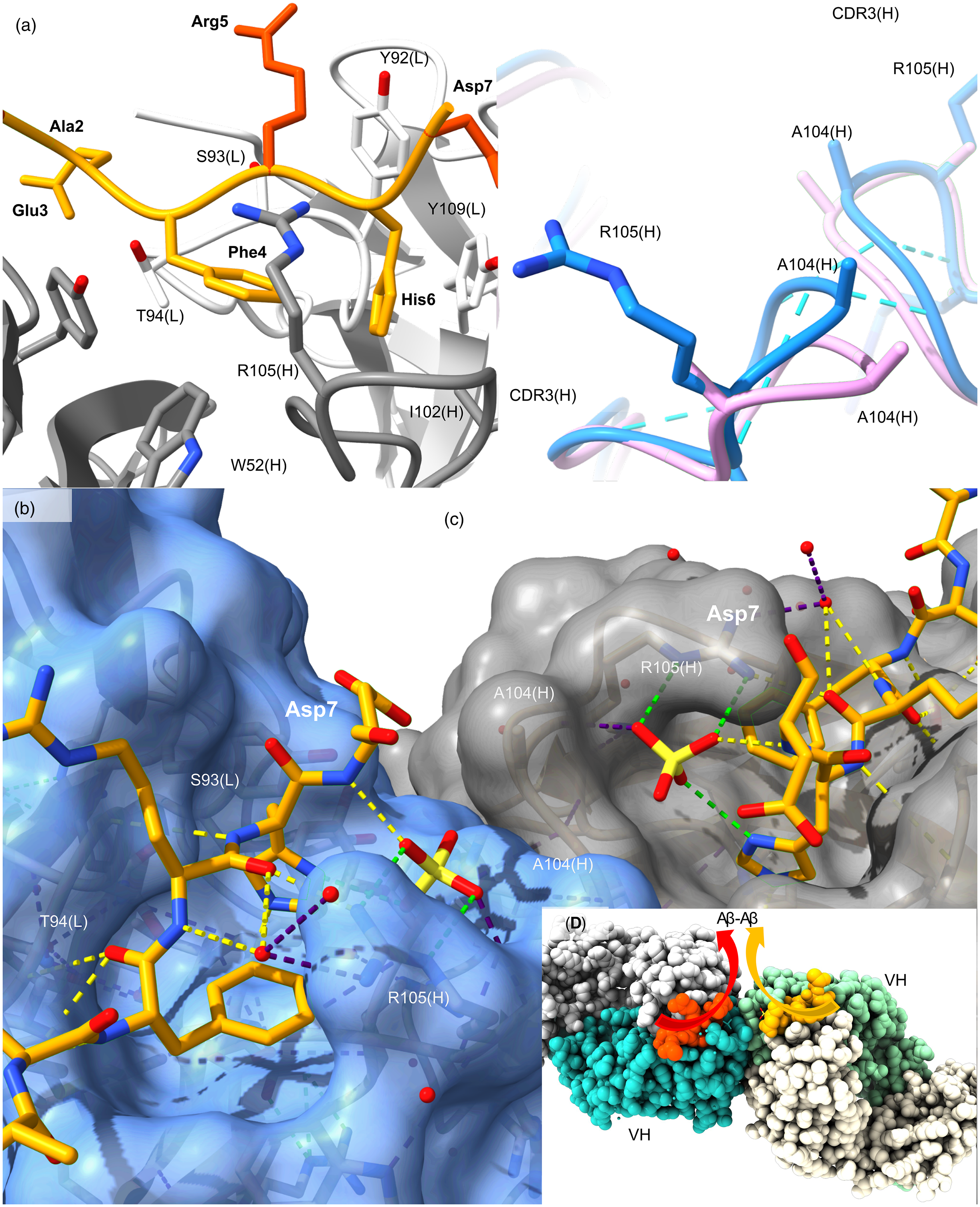
Aducanumab is an IgG1 therapeutic derived from a library of human B-cells. Structures shown are from published X-ray diffraction data (PDB 6co3, 6cnr) reported by Arndt and colleagues. 8 (a) The 2.38 Å aducanumab (Fab) liganded to Aβ1-11 (PDB 6co3). Aβ atoms with high thermal mobility are colored in burnt orange compared to well defined sidechains for Glu3, Phe4 and His6 shown in light orange engaged by aducanumab [white (VL) and gray (VH)]. Thr94(L) is a key H-bonding residue with mainchain Aβ. The tetrapeptide 3-EFRH-6 runs anti-parallel to CDR3 (L) 91-SYST-94. (b) Liganded CDR3(H) of aducanumab is shown in blue and the apo Fab structure is shown in pink. Fab structures align with an 0.3 Å RMSD at Cα positions. Sidechains of Arg105(H) and Arg106(H) are missing in the apo structure indicating mobility in the region prior to Aβ binding. H-bonds (blue dashes) are formed by Fabs when Aβ binds. (c) Crystal symmetry mates dimerize at the ligand binding site. Colored dashes are used to highlight aspects of the H-bonding network including waters and sulphates, notably bridging the Arg105(H) side chain to the Aβ His6 imidazole (green) and Asp7 mainchain. Phe4 and His6 aromatics form a T-shaped pair that contacts the antibody surface. (d) A symmetry-related dimer of Fab-Aβ dimers in deposited structure file (PDB 6co3) is shown. Cartoon arrows have been added to show the projection of Aβ molecules towards the mid-region and C-terminus. The tetrameric assembly structure creates a continuous antigen recognition site across two Fabs for parallel Aβ dimers. More extensive Fab-Fab contacts are described in Supplemental Material.
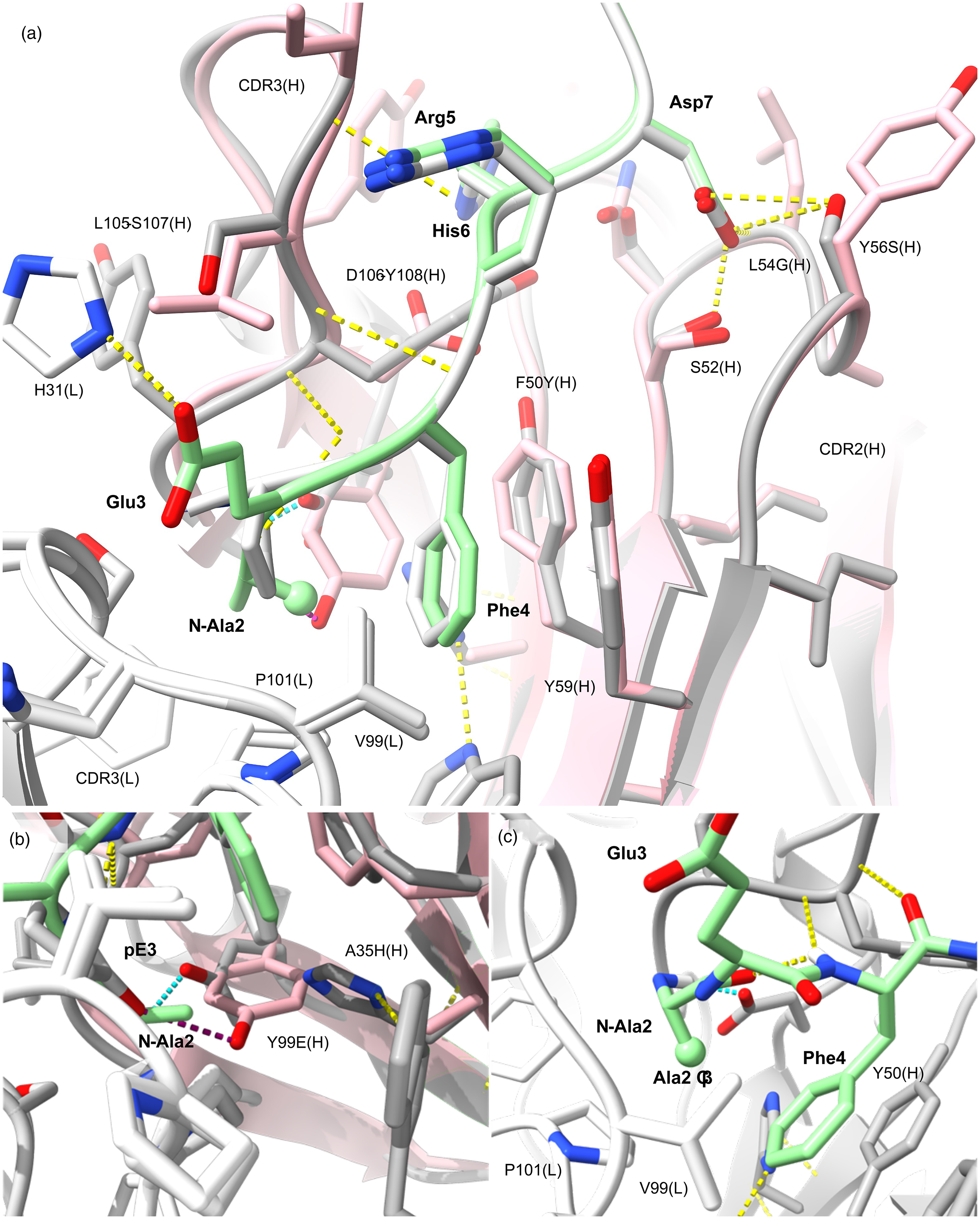
The murine lecanemab m158 comparative model. The structure of m158 [white (VL), gray (VH)] is based on the pE3-Aβ-C#17(Fab) crystal structure (pink) reported by Piechotta and colleagues [PDB 5my4; mAb C#17]. 35 The template and model structures align to 0.1 Å RMSD over all atoms. Mainchain positions only differ significantly in CDR3(H). (a) Contacts in the middle of CDR2(H) are significantly maintained despite differences in sequence, and ends of the CDR are conserved around the ligand binding pocket aromatics Tyr50Phe(H) and Tyr59(H) shown in sticks. The m158 variable region is shown bound to N-Ala2-Aβ (pale green) superpositioned with pE3-Aβ (white). Polar contacts (yellow dashes) show that mainchain contacts are responsible for much of the interface between C#17, and by homology, m158. (b) Tyr99(H) forms a 2.78 Å H-bond (purple dashes) with pE3[OE1] in mAb C#17. His35(H) and Glu99(H) substitute for the Ala35(H) and Tyr99(H) pair and stabilize the pocket but are unable to bond with pE3. Modeling suggests m158 could form a salt bridge (cyan dashes) to an alternative N-Ala Aβ conformation between the amine terminus and the Glu99(H) side chain [(b) and (c)]. A H-bond is evident between the Phe4 amide and the Ala2 carboxyl constraining the conformer. The Ala2 methyl (Cβ, ball) sits between the Pro101(L), Val99(L) contacts and the Phe4 aromatic, filling the role of the Leu101(L) aliphatic in W02/PFA1 structures (Figure 1(d)). A putative H-bond is shown between Glu3 and His31(L) sidechains supporting Phe4 placement in lecanemab in the absence of the pE3 ring. This shallow binding mode allows for binding to unmodified Aβ with significantly reduced affinity.
Results
Several mAbs have been developed to target different epitopes of the Aβ peptide in AD. These antibodies can be grouped based on their primary binding sites. N-terminal: donanemab (Eli Lilly), lecanemab (Biogen and Eisai), aducanumab (Biogen and Neuroimmune) and bapineuzumab (Pfizer and Johnson & Johnson) all target the N-terminal region of Aβ. Dual-epitope: Gantenerumab (Roche) binds the N-terminal epitope but additional reactivity to the central portion has been reported. 27 Mid-region: Solanezumab (Eli Lilly) and crenezumab (Genentech) bind the central region of Aβ incorporating the α-secretase cleavage site of AβPP and the initiating site of amyloid formation.
The immunodominant N-terminal epitope
The immunodominant B-cell epitope of human Aβ in mice is at the N-terminus. 5 This immunodominance may be in-part attributed to Arg5 being one of just three differences between human and murine sequence Aβ (Gly5). The dynamic region is solvent exposed in monomers, nascent oligomers, protofibrils, and fibrils, and is susceptible to protease action and post-translational modification (PTM) in the AD brain. 28 However, targeting this epitope may limit engagement of oligomers and fibrils in vivo if steady state concentrations of freshly synthesized and readily degraded Aβ swamp the antigen binding site.
The earliest structures of anti-Aβ mAbs were raised in mice and are shown in Figure 1 binding Aβ over residues ∼3–7 (N-D1A
Importantly for understanding lecanemab selectivity (described below), the Phe4 aromatic ring in W02 and similar mAbs is positioned against a leucine at position 101 of the light chain (Figure 1(d)). The Phe4 aromatic is otherwise encapsulated by aromatics from the heavy chain (Tyr52/Trp47/His50 in W02) and packed in a T-conformation with Aβ His6. Little to no electron density is evident beyond residues 2–8 due to higher thermal motion outside of the core epitope in each structure.
The short linear epitope (EFRH) defines the core murine immunodominant B-cell epitope for human sequence Aβ, with acidic (Glu3) and basic (Arg5, His6) residues on each side of the buried aromatic Phe4. Notably, Arg5 (Figure 1(b)) and Glu3 (Figure 1(e) and Figure 1(f)) sidechains mediate H-bonds/salt-bridges, and both Glu3 and Arg5 are subject to common post translational modification in the AD brain.17,28,34
PFA1 mAb binds synthetic fibrils 2–3 orders of magnitude more tightly than it binds monomers because of antibody bivalency and antigen multivalency. Avidity at fibril surfaces explains the inability of monomeric Aβ to displace PFA1 from synthetic fibrils during hybridoma screening. 32 This may come into play in vivo where free Aβ in blood and cerebrospinal fluid (CSF) could exceed the capacity of mAbs to engage oligomers centrally. 4
N-truncated and pyroglutamate modified Aβ
pE3-Aβ is a stable modification of N-terminally truncated Aβ where the N-Glu3 is cyclized. The proline-like modification (5-oxoprolinate) may impede further proteolysis of Aβ and thereby accumulate in older cerebral deposits. PFA1 has been shown to also bind pE3-Aβ, the known target of donanemab.31,32 It was anticipated that targeting pE3-Aβ would facilitate clearance of pre-existing plaques by phagocytosis and might lower the incidence of ARIA-H by avoiding Aβ embedded in vascular walls. 12 When bound to PFA1 (Figure 1(e)), pE3 occupies the same position as unmodified Glu3, but without strong polar contacts to the mAb. PFA1 Fab affinity drops from 60–120 nM for intact Aβ to 3000 nM for N-pE3-Aβ as a consequence. 32
PFA1 binds unrelated proteins (Figure 1(f)) bearing similar sequences to Aβ. 31 This demonstrates that antibodies that recognize linear, dynamic epitopes with high monovalent affinity are prone to off-target reactivity in vivo. In the light of clinical trials, the idea of off-target binding can be extended to include Aβ otherwise rapidly turned over in circulation, 4 and reactivity with AβPP.
Structures for antibodies raised against the hexapeptide pEFRHDS have been published. 35 The studies reveal that pE3 and Phe4 ring structures form a bulky moiety, and two distinct binding modes were identified. A mAb called C#24 buries this moiety deeply in the antibody, whereas another mAb called C#17 captures the pair in a shallow groove at the solvent interface. We find that donanemab and lecanemab are variable domain homologues of mAb C#24 and mAb C#17, respectively (described below).
Dimerization of aducanumab enables detection of Aβ oligomerization
Aducanumab activates microglial clearance functions and is more effective with diffuse Aβ deposits than with cored plaques in AβPP/PS1 mice. It also induces strong transcriptional changes in genes related to antigen presentation in that model. Intriguingly, microglial responses to re-accumulating Aβ are blunted in mice after aducanumab withdrawal compared to untreated controls, 9 suggesting treatment either incapacitates microglial metabolic processes further or Aβ clearance exposes another underlying cause of inhibited microglial response to plaques. An unusual or atypical profile for aducanumab action in vivo might be expected as the antibody exhibits remarkable properties in vitro.1,8
We identified a novel mechanism for aducanumab binding to Aβ based on crystallographic data that is consistent with atypical aducanumab reactivity. The published structure of aducanumab in Figure 2(a) shows that it binds residues 3–6 of 1–11 Aβ (N-D1A
Importantly, aducanumab exhibits low μM affinity for the epitope unless multiple copies of Aβ are present at high density. Affinity for monomers and oligomers is around 9000 nM and ∼ 1 nM, respectively, driven by a striking 1000-fold increase in the association rate constant.1,8 Aducanumab exhibits just a 1.4-fold loss in apparent affinity (mAb, ELISA, EC50) for Aβ3−42 compared to Aβ1-42, but a 31-fold loss for pE3-42 Aβ.1,8 Lack of affinity for common Aβ in circulation suggest that the pE3 modification in heterogeneous aggregates could still contribute to target engagement in vivo. A further loss of affinity is seen with subsequent truncations of the terminus.
Direct comparison in vitro shows that aducanumab binds to diffusible fibrils at a ∼10-fold higher density of mAb on the Aβ substrate than versions of bapineuzumab and gantenerumab, while solanezumab shows little to no affinity for the fibrillar substrate. These reports suggest that aducanumab binds Aβ by avidity with atypical packing at the fibril interface.1,8
Re-examination of published crystal data reveals a symmetry-related interface that mediates Aβ binding to coordinated Fabs in a dimer of Fab-Aβ dimers. The crystallographic assembly shown in Figure 2 readily explains aducanumab reactivity in vitro. Crystals were produced with apo Fab then Aβ1-11 was soaked into the crystals. Figure 2(b) shows conformational change in CDR3(H) on liganding Aβ in situ. Thermal factors drop and the Arg105-Arg106 sidechains of aducanumab that are not evident in the apo crystal form, adopt well-ordered conformations upon ligand binding. Arg100(H) and Tyr110(H) sidechains H-bond to stabilize the Fab dimer conformation we have identified (expanded in Supplemental Figure 4). Arg105(H) caps the binding site when occupied by Aβ with H-bonds to Aβ in an antiparallel sheet with Tyr92-Ser93-Thr94 of CDR3(L). Thr94(L) is the hub for H-bonding Aβ at Glu3 and Phe4 (Figure 2(a)). The change cements the Aβ in place between VH and VL domains and strengthens Fab-Fab contacts at the ligand binding site. This creates an extended ligand binding site (see Figure 2(b)) that is stabilized by two appropriately spaced copies of Aβ. This would result in Aβ-oligomer specificity and permit daisy-chain binding to other oligomers in flux or dense packing at surfaces where the epitope is repeated at regular intervals. The novel motif explains high affinity for cross-linked Aβ1-15 tetramers in contrast to little affinity for monomers and artificially cross-linked dimers, 8 and the unique ability of aducanumab to dramatically reduce the flux of Aβ oligomers compared to solanezumab, bapineuzumab and gantenerumab. 1 This might also explain new biophysical evidence that aducanumab stabilization on binding to dimers is dependent on the Fc region. 36
More extensive inter-molecular polar contacts are evident in the Aβ-aducanumab crystal structure presented in Supplemental Figure 4 without further refinement, including Gly103(H)-Gly103(H), Arg100(H)-Ser31(H) [OG and O], and Tyr49(L)-Arg106(H). The interface is heavily populated by Arg/Tyr residues with additional interfacing residues in CDR1(H), CDR2(L) and CDR3(H). The structure places Asp7 carboxyl groups of adjacent Aβ residues 12.6 Å apart at the interface suitable for contacts between Aβ peptides further along the sequence, but not visualized with the truncated Aβ1-11 ligand used in those studies. 8
The lack of contact with residues at the N-terminus and Arg5 sidechain might also be reconsidered in the context of recognition of N-terminally truncated Aβ species and citrullinated Aβ affecting a third of pyroglutamate modified Aβ in the AD brain.28,34 The interface between Fab dimer and Aβ dimer is in the order of 674 Å2, with 18 putative H-bonds and two salt bridges between aducanumab and the ligand [Glu3-Lys65(H)], not taking sulphate mediated contacts into account. The proposed dimer of Fab-Aβ dimers explains mAb specificity for Aβ that retains low nM avidity and specificity after N-terminal truncation and pyroglutamate modification to an otherwise small, linear, and dynamic epitope. The atypical assembly structure might also communicate unanticipated IgG1 Fc-mediated signals to the immune system via complement factor C1q and Fcγ receptors at a distance from plaques.
Lecanemab targets a labile epitope in aging Aβ
Here we find that m158 is a light chain variable domain homologue of the N-terminal antibody PFA1 (96.4% identity) and the pE3-specific mAbC#17 (92% identity; 97% similarity). mAbC#17 bound to pyroE3-12 Aβ (N-pE3FRHDSGYEV12-poly ethylene glycol) [PDB 5my4] is the best of seven templates in the PDB ranked as suitable for VH-VL hetero-dimer (VH-VL) homology modeling in Swiss-model Workspace (SMW). 35 We have used comparative analysis to explore homology between these templates and lecanemab, described in the Supplemental Material.
C#17 is predominantly a heavy chain contacting antibody, with only two light chain residues contacting Aβ, namely Val99 and Pro101: both specifically contact N-pyroglutamate in C#17 and are conserved in m158. VL sequence alignments are shown in Supplemental Figure 1. A summary of heavy chain homology is shown in Supplemental Figure 2, including sequences for m158, C#17 and other VH templates used for structure calculations. The final energy minimized model of m158 bound to the core epitope of Aβ3-8 (N-
Figure 3(a) (expanded in Supplemental Figure 2) shows that m158 binds predominantly at CDR2(H) and CDR3(H). Contacts with CDR3(H) are significantly mediated by mainchain elements as indicated by H-bonding (Figure 3(a)). A volume sparing Gly100-Gly101 helix insertion at the beginning of CDR3(H) in m158 (see Supplemental Figure 2) is at a solvent-exposed position, away from the antigen binding surface. The glycine pair preserve the position of Tyr101(H) (Tyr103(H) in m158), the sole pi-pi contact to Aβ (at His6) identified in complex with C#17. 35 m158 also conserves VH aromatic contacts that surround pE3 in C#17 at the base of CDR2(H), including Trp47, Phe50/Tyr50, and Tyr59. The central loop conserves polar contacts and an additional H-bond is seen from Tyr50(H) to Glu99(H), where Tyr50 is Phe50 in C#17. Figure 3(a) and Supplemental Figure 2(c) show Leu54Gly(H) and Ala55Ser(H) differences are at the top of CDR2(H) and conserve mainchain contacts at those positions. Tyr56Ser(H) results in an additional H-bond to the Asp7 sidechain of Aβ, and to the central Ser52(H) that mediates Asp7 engagement in both template and target structures. Ser57Thr also slightly increases hydrophobic contact with Aβ at Arg5(Cβ). This enhanced motif in CDR2(H) imparts affinity for the Asp7 side chain of Aβ and may allow for alternative binding motifs nearer the N-terminus, while conserved hydrophobics Tyr50(H)/Tyr59(H) and neighboring isoleucine residues maintain CDR2 structure and engagement of Phe4 (see Figure 3(a)).
In CDR3(H) (see Figure 3 and Supplemental Figure 2(d)), central residues of the loop may contribute to interactions outside of the core immunogen binding site. The Aβ liganding residue Asp106(H) in C#17 is Tyr108(H) in m158. Polar contact mediated by water from Asp106(H) to Asn53(H) in C#17, is replaced by a direct H-bond between the OH group of Tyr108(H) in m158 and OG of Ser53(H). This change fortifies CDR-CDR contacts and enhances van der Waals contacts with Aβ via the aromatic insertion and is positioned by the Gly-Gly helix at the start of the CDR.
Leu101(L) is conserved in PFA1 and homologues (see Figure 1 and Supplemental Figure 1) as the key light chain ligand of Aβ Phe4, where the aliphatic side chain projects into the aromatic ring face. Pro101 is a signature difference between the otherwise highly conserved PFA1 and C#17 light chains. Conservation of both Val99(L) and Pro101(L) in m158 suggests pE3 engagement, but critically, Tyr99 in the heavy chain is occupied by glutamic acid in m158/lecanemab (Y99E(H) in Figure 3(b)). This Tyr99Glu difference in the hypervariable CDR3(H) is highly consequential for ligand binding and significantly distinguishes m158 from the pyroglutamate-specific template C#17. Tyr99(H) in C#17 anchors pE3-Aβ via a 2.78 Å H-bond between the hydroxyl group and OE of pE3. The shorter Glu99 side chain in m158 cannot make these contacts, but rather forms polar contacts with His35(H) in CDR1. Notably, Ala35His(H) is the only non-conservative difference in CDR1(H) between m158 and C#17 (see Supplemental Figure 2(a) and Supplemental Figure 2(b)). This change compensates for the other, Tyr99Glu, where small Ala35 side chain allows for the bulk of Tyr99(H) in C#17, a volume alternatively occupied in the binding pocket by the pair of mid-sized, Glu99(H)-His35(H) residues in m158. These differences maintain the volume for seating pE3 but lack electrostatic and hydrophobic contacts to the pE3 terminus. This analysis suggests that like the template antibody C#17, m158-lecanemab shows preference for N-terminally truncated Aβ but unlike C#17, they do not possess the contacts that drive monovalent affinity due to cyclisation of the Glu3 terminus. 35 We then considered alternative N-terminal binding modes likely to confer selectivity.
mAb C#17 exhibits micromolar (3120 nM) affinity for unmodified Aβ, and 10-fold preference (∼300 nM affinity) for N-terminally truncated forms (N-Ala2-Aβ and N-Glu3-Aβ) with no affinity for N-Phe4 Aβ. Importantly, C#17 shows ∼1 nM affinity for pE3-Aβ on 1:1 binding. 35 In contrast, PFA1 binds pE3-Aβ with a 77-fold loss in affinity compared to unmodified Aβ. 32
Mutation of pE3 in the structure to Glu3 revealed potential H-bonding from the Glu3 sidechain to His31(L); like that seen in W02. This also reveals additional H-bonding to key positions of difference in m158 and the template C#17, including Ser107(H), Tyr108(H), and Gly96(L). Extending the terminus to include Ala2 revealed additional H-bonds from the amine terminus to Tyr109(H) and the key liganding residue Glu99(H) which forms a side chain mediated salt bridge to the terminus. Importantly, the Ala2 methyl occupies the position of the Leu101 side chain between the face of Phe4 and the signature Pro101(L) residue. The hydrophobic pocket is filled out by a Phe94(L), uniquely occupied by serine in the light chains aligned in Supplemental Figure 1. These constitute nine H-bonds with three salt bridges within the core antigen interface of 561.9 Å2. Extension to unmodified Aβ would require detachment and loss of the N-Ala2 bridge to accommodate the solvent exposed terminus and could account for the weak affinity reported for 1:1 binding intact Aβ. The dominance of VH contacts including 8 of 9 H-bonds, 1 of 3 salt bridges and over two thirds of the interfacing area could compensate sufficiently to support avidity for unmodified synthetic Aβ fibrils by lecanemab in the absence of high monovalent affinity for the peptide. 37 Apparent affinity should diminish as the proportion of pE3, and further truncated species increases. This is a possible point of distinction for lecanemab in avoiding both fresh monomers or nascent oligomers in solution, and insoluble bodies that are more extensively truncated and modified, giving the mAb a preference for transient and soluble oligomeric Aβ species broadly referred to as protofibrils.
Our analysis suggests that copy number of N-Ala2 can anchor avid capture of larger oligomers of heterogeneous Aβ or “protofibrils” by lecanemab. This mechanism also supports engagement of fibrils and stimulation of phagocytosis at surfaces, but it is not known if this is necessary to slow decline in AD. Mass spectra have now been published where lecanemab immunoprecipitation was used to examine soluble extracts of AD temporal cortex. 38 The study shows the material is predominantly N-terminally truncated and modified Aβ42, including 5% N-Ala2, 2.5% N-Glu3 and 33.5% N-Phe4 Aβ, consistent with avid recognition of the modeled epitope likely anchored by N-Ala2. The material was also comprised of 7.7% N-pE3-42 Aβ.
Donanemab buries the pyroglutamate-3 modified N-terminus
The detailed homology structure of murine donanemab mE8 bound to the core epitope pyroE3-8 Aβ (N-p

The murine donanemab mE8 comparative model. (a) and (b) show top and side views of mE8 superpositioned with the 1.49 Å template antibody crystal structure [PDB 5myx] of C#24 in complex with pE3-18 Aβ. 35 The core antigen pEFRHDS is shown as blue surfaces. All Aβ contacting residues of the template (blue sticks) and target mAbs are shown with few differences evident. Arrows indicate the positions of the four non-identical residues that recognize the immunogen used to raise C#24 in mice. Tyr99(L) in donanemab is Phe99(L) in the C#24 template, Tyr33(H) in donanemab is Trp33(H) in C#24, and Trp50(H) in donanemab is Gln50(H) in C#24. Position 101(H) in mE8 (b) is the sole contacting residue that is different in each of the three antibodies (donanemab, murine mE8, and C#24). Position 101(H) is tyrosine in mE8, isoleucine in donanemab and glutamic acid in C#24. The lower panel (c) shows the structure of N-pE3-9 Aβ (blue) seated in the mE8 antibody surface. H-bonds are shown as yellow dashes, and green dashes show the putatively conserved Lys35(H) salt bridge to Aβ Asp7. Gly9 of Aβ has been included in structure calculations as pE3-8 Aβ antigen was fused to a carrier protein and hence mainchain elements may be conserved, such as H-bonding to Tyr33(H) shown.
16 out of 20 Aβ-contacting residues lining the deep ligand binding cavity of mAb C#24 are identical in donanemab. Two of the four differences are conservative [F99Y(L) and W33Y(H)]. Gln50(H) of C#24 is Trp50 in mE8 and donanemab, while Tyr101(H) is Glu101 in mE8 and Ile101 in donanemab; the latter seemingly introduced to promote van der Waals contacts with Aβ. PISA server identified a 750 Å2 Aβ buried surface area at the interface with 16 putative H-bonds, including a conserved salt bridge at the end of this epitope, from Lys35(L) to Asp7, and two direct bonds from Glu99(H) to the pE3 residue. H-bonds are made with both VH and VL domains at Asn35 (H), Glu99 (H), and Thr97(H) in tension with Asn39(L) and Gly96(L). Glu99(H) is the core residue that protrudes into Aβ and isolates the bulky pGlu3-Phe4 head group in a predominantly hydrophobic cage from the remainder of the peptide that adopts an (i) to (i + 3) H-bonded turn (Arg5 to Ser8). The N-terminal moiety is also engaged by polar contacts at Asn35(H), and Asn39 (L) sidechains conferring specificity in the dimer interface. Further polar contacts are evident between pE3, Phe4, Arg5 and Asp7, engaged by the dipeptide Gly96(L)-Thr97(L) and a strand over residues 35-KTYLN-39 of CDR1(L) with alternating polar residues in contact with Aβ. Notably residues Asn35(H), Glu99(H), Asn39(L), Tyr37(L), Lys35(L), and Gly96(L) are identical in mAb C#24 and donanemab while differences at Aβ contacting residues are on the periphery of the cavity (see Figure 4(a) and Figure 4(b)). Furthermore, the conservative Phe99Tyr(L) difference produces another polar contact between the Tyr99 hydroxyl and Arg5 guanidinium pharmacophore, enhancing specificity for the epitope in donanemab. This is a reliable structure for donanemab core epitope engagement but contacts outside of this core have not been considered.
The template mAb C#24 exhibits high monovalent affinity for pGlu3-18Aβ (2.0–6.9 nM) but does not bind N-terminally unmodified Aβ, N-Ala2-18 Aβ, murine pE3-18 Aβ (position 5 is Gly), or human N-Phe4-18 Aβ, and data shows >500 nM affinity for the truncated but not pyroglutamate modified N-Glu3-18 Aβ form. Interestingly, the rotamer adopted by Glu3 in Aβ captured by bapineuzumab closely resembles the structure of pyroglutamate, 16 which would account for this limited reactivity. Donanemab is likewise exquisitely specific for the free pyroglutamate modified N-terminus of human Aβ as a monovalent epitope. The effects of avidity can be expected to be modulated by the availability of the N-pE3 (and perhaps N-E3-Aβ) epitope alone as opposed to other antibodies where the terminus is in a shallow groove and solvent exposed to accommodate N-terminal extension or including lecanemab or PFA1 compared in Supplemental Figure 5. The model suggests that donanemab is reactive to any pool of Aβ that incorporates pE3-Aβ unless the terminus is somehow occluded. 12 The deep and shallow binding modes of donanemab and lecanemab are compared in Supplemental Figure 5 which contrasts the shallow (m158) and buried (mE8) topology that distinguishes donanemab as uniquely specific for pE3-Aβ to the exclusion of direct binding to other Aβ epitopes.
Bapineuzumab buries the unmodified N-terminus of Aβ
The atomic structure of mE8 reported here indicates that donanemab is the quintessential mAb for pE3-Aβ selection to the exclusion of other species. This is akin to the structure we determined for bapineuzumab binding to unmodified N-terminal (N-
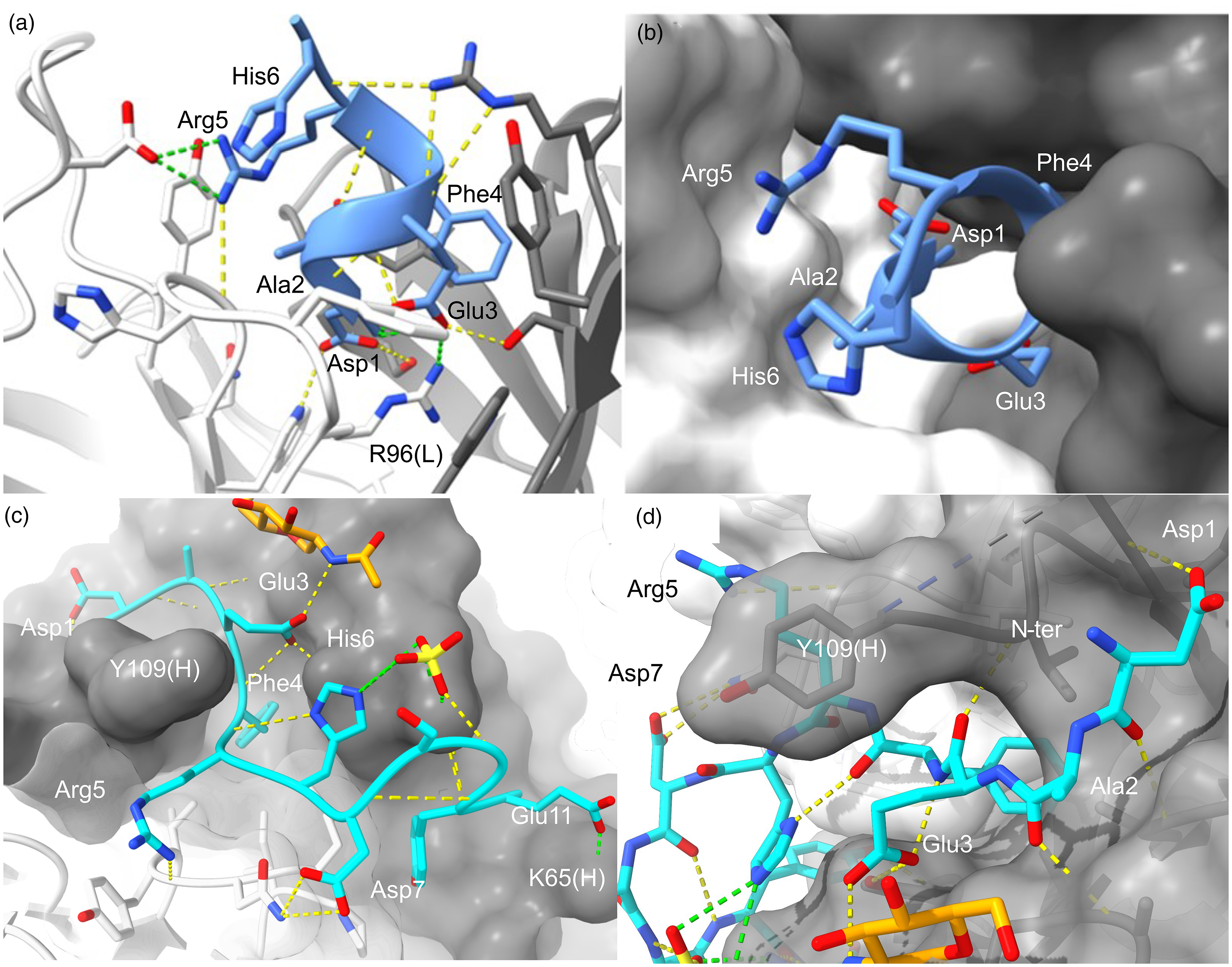
Buried and shallow capture of unmodified Aβ by bapineuzumab and gantenerumab. N-terminal Aβ is recognized by bapineuzumab [(a) and (b)] and gantenerumab [(c) and (d)]. H-bonds and salt-bridges are shown as yellow and green dashes, respectively. VL and VH are white and gray, respectively. The structure we reported for bapineuzumab [PDB 4hix] reveals Aβ is engaged as a helix over residues 1-DAEFRH-6 with the terminus buried (blue cartoon, (a) and (b)).
16
The surface area of Aβ buried in the interface is 580 Å2. (c) and (d) The gantenerumab structure shown (PDB 5csz; 1.80 Å) was obtained from crystals of Fab soaked with Aβ1-11 peptide (light blue). Sidechain mediated salt-bridges are evident at each end Asp1(OD1)-Lys100(H)[NZ; 3.9 Å] and Glu11(OE1)-Lys65(H)[NZ; 2.9 Å] and the amine terminus (N-ter) is solvent exposed. Sidechain H-bonds are seen from Asp1, Glu3 and Tyr10 to VH, and from Arg5 and Asp7 to VL. The core 3-EFRH-6 epitope contacts Tyr109(H) that immediately precedes missing residues of CDR3(H) in the data. CDR3(L) [white surface (
Bapineuzumab was the first anti-Aβ-mAb evaluated in phase 3. Bapineuzumab slowed Aβ accumulation and ARIA first emerged during those trials. 20 ARIA with edema increased with dose and APOE ε4 allele number leading to discontinuation of the 2.0 mg/kg dose. Another version of 3D6 with reduced Fc-effector function was tested and required higher doses to bring about ARIA but the mAb did not show significant changes in AD biomarkers. 39 These studies show that higher dosing enabled bapineuzumab to engage brain and cerebrovascular Aβ despite reactivity for Aβ monomers in the periphery. Strong effector function was required for bapineuzumab to shift central AD biomarkers, and only in APOE4 carriers, 20 yet both formulations induce ARIA. This points to a high threshold for microglial activation to reduce brain Aβ deposits detected by PET. 39
Affinity matured mAb gantenerumab preferences N-terminally unmodified Aβ
Gantenerumab is an affinity matured IgG1 antibody engineered from human antibody fragments that induces phagocytosis. 27 It binds N-terminal (N-D1AEFRHDSGYE11) and mid-region (18-VFFAEDVGSN-28) epitopes of Aβ, where Aβ1−11 appears to be the primary epitope, and the only liganded structure reported for the mAb which is shown in Figure 5(c) and Figure 5(d) for comparison with bapineuzumab. 27 Seven residues (102-NTHKPYG-108) are missing from the structure, which might be expected to coordinate unseen interactions with larger Aβ ligands. Phe4 and Tyr11 protrude into the CDR surface with hydroxyl-mediated H-bonds at Tyr11. A sulphate attached to His6 and a glycan of Asn52(H) H-bonding Glu3 add to the complexity of the epitope and may contribute to discordant results from binding studies.1,27,37 Gantenerumab binds a long epitope stretching 28 Å from Asp1[OE1] to Glu11[OE1] with these peripheral atoms forming salt-bridges to Lys100(H) (3.91 Å) and Lys65(H) (2.91 Å), respectively.
Eight of 12 putative H-bonds between Aβ and gantenerumab are mediated by N-terminal residues: four to Asp1, one to Ala2, and two to Glu3 with a third bond to the Asn52(H)-glycan. In this way, gantenerumab exhibits preference for intact Aβ over N-terminally truncated (and pE3-modified) species, but it is not as specific as bapineuzumab where the terminus is buried. This is consistent with findings that 3D6-bapineuzumab and chimeric gantenerumab have similar mechanisms of action when binding synthetic Aβ1-42. 1
Genuine gantenerumab binds Aβ1-40 monomers with an equilibrium KD of 17 nM compared to 1.6 and 0.6 nM for binding to oligomers and fibrils “under conditions that monitor the bivalent binding”. 27 The gantenerumab structure shows why it is amenable to reactivity with N-terminally truncated species which supports avid binding to homogeneous oligomers and heterogeneous bodies such as protofibrils and plaques. Gantenerumab is therefore somewhat like lecanemab but with preference for N-terminally intact Aβ, including Aβ in the periphery. In clinical trials of gantenerumab or solanezumab, dosing of gantenerumab was increased from 225 mg (sub-cutaneous) every 4 weeks which was later increased to 1200 mg based on a futility analysis. Neither gantenerumab nor solanezumab demonstrated a beneficial effect on cognition in the DIAN-TU-001 study. 40 An interesting prospect is that the antibodies might perform better used in combination to exploit the ability of solanezumab to effectively sequester free Aβ in the periphery with slow dissociation kinetics. 17
Solanezumab and crenezumab sequester monomers at the α-secretase cleavage site
Solanezumab is an IgG1 humanized mAb derived from the murine m266 and developed by Eli Lilly.27,41 Genentech developed IgG4 crenezumab as safer alternative to IgG1 subclass antibodies by avoiding over stimulation of microglia implicated in ARIA.
42
We reported the 2.4 Å structure of solanezumab bound to Aβ12−28 (N-V12
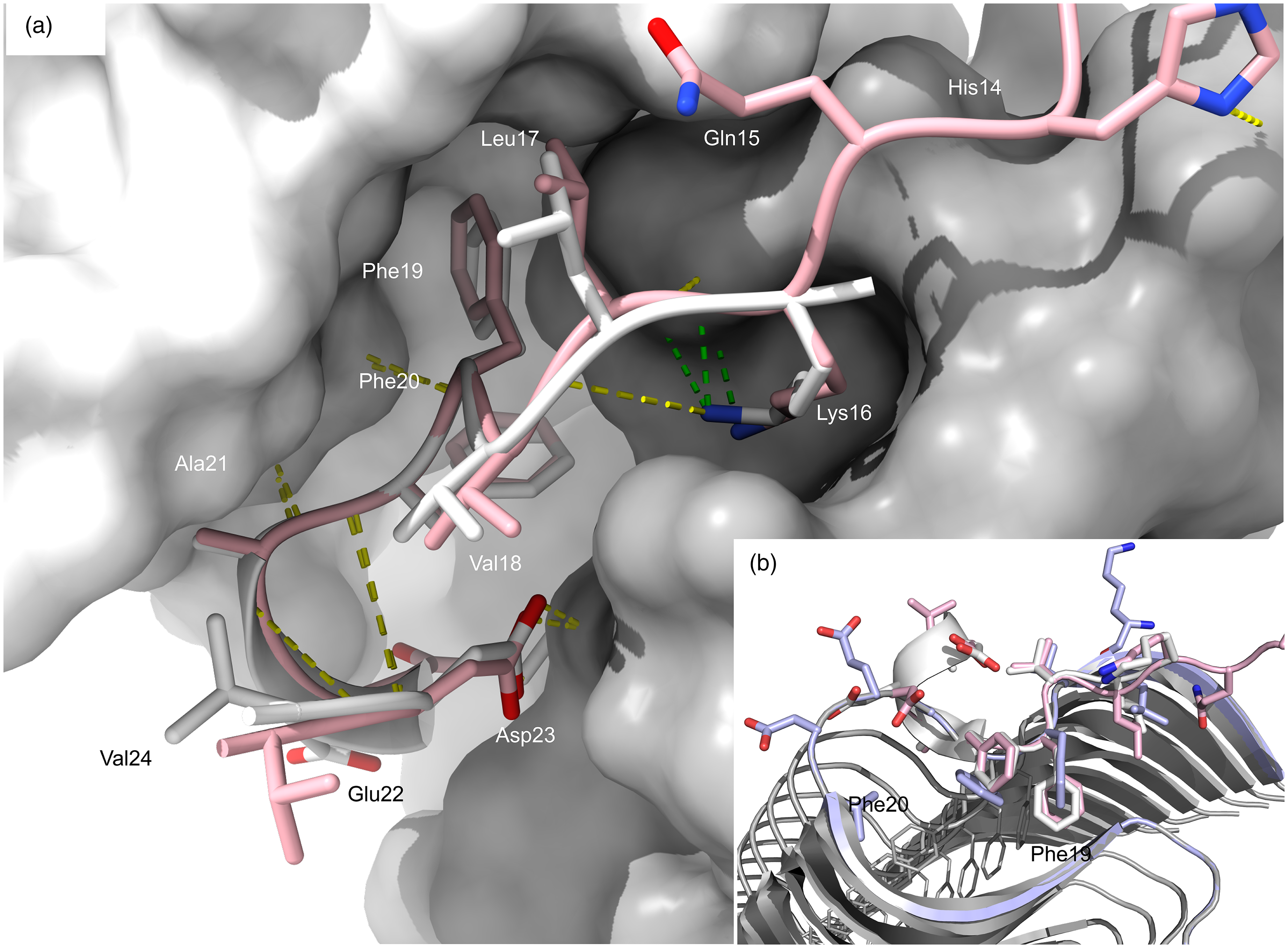
Solanezumab and crenezumab engage the linear KLVFFA mid-region epitope. (a) The conformation of monomeric Aβ captured in our structure of solanezumab to 2.4 Å resolution [PDB 4xxd] is shown super positioned with crenezumab-Aβ, resolved to 2.3 Å [PDB 5vzy].15,43 Aβ bound to crenezumab is shown in pink cartoon and sticks. The crenezumab VH (gray) and VL (white) surfaces are shown. Lys16 is buried with green dashes indicating salt-bridges to Asp101(H) in both mAbs. Ser33(H) and Phe36(L) were identified in our previous comparative study as Aβ-contacting residues in the solanezumab crystal structure that were not identical in crenezumab (Gly33(H) and Tyr36(L) in crenezumab). 15 A helix is evident in the solanezumab crystal structure, partially constrained by H-bonds (yellow dashes) between Asp23 and Ser33(H) not conserved in crenezumab [Gly33(H)]. (b) Conformations of Aβ when liganded by solanezumab (gray) and crenezumab (pink) bound are superpositioned with a terminal strand in a fibril structure of synthetic Aβ1-42 [PDB 2mxu]. 43 Phe19-Phe20 sidechains buried by the mAbs are shown to be fully occluded in the fibril body.
The structure of solanezumab reveals key interactions are mediated by Lys16, Phe19, Phe20 and Asp23, where the antibody recognizes an amphipathic epitope with 960 Å2 of Aβ buried in a long, deep cavity between heavy and light chains. Solanezumab salt-bridges Lys16 from the very short hypervariable CDR3(H) (94SGDY97) that is identical in crenezumab. The crenezumab structure shows His13, His14, and Gln15 where contact is made to non-CDR residues and may be a consequence of different crystallization conditions. Capture of the highly hydrophobic central epitope reveals why solanezumab selects monomers 15 or deposits not fully integrated into oligomeric/fibril sheet forms. The epitope appears to be in transition between a membrane spanning helix and sheet structures observed in fibrils, shown in Figure 6(b) where the Phe-Phe dipeptide recognized by solanezumab and crenezumab is occluded.
Solanezumab forms a stable immunocomplex with monomeric Aβ and clearance of the complex seems to be less efficient than endogenous Aβ clearance pathways in vivo, reflected in a 300–500-fold sustained increase in blood Aβ, likely to be similar in CSF at higher doses.4,18 Poor brain penetrance resulted in molar ratios (mAb:Aβ) of 0.26 in CSF (Expedition 3) indicating that antibody levels were insufficient to counter Aβ production. 4 Strong binding to an epitope displayed by monomers has several implications. It means that each molecule of mAb making it into the brain can be spent with the removal of just two Aβ molecules, and clearance of the immunocomplexes is reliant on endocytosis as opposed to bulk removal by phagocytosis. The binding mode might also preclude recognition of Aβ in complex with important biological ligands such as ApoE. 21 The mAbs might inhibit endogenous protease breakdown of Aβ, and it is plausible that cross reactivity with AβPP at the membrane interface could lead to α-secretase inhibition in vivo and increased AβPP processing along amyloidogenic pathway. The sustained increase in plasma Aβ immunocomplexes by solanezumab and crenezumab is likely to be parallelled in CSF at higher doses.
Discussion
This structural review of anti-Aβ immunotherapies for AD highlights the complex structure-function relationships of various antibodies and their implications for treatment efficacy and safety.
Bapineuzumab is uniquely specific for N-terminally intact and unmodified “new” Aβ, requiring Asp1 being buried in the antibody surface. The epitope is common in circulating Aβ but diminishes in aging deposits, thereby limiting efficiency in targeting brain derived and aged Aβ. Bapineuzumab showed evidence of slowing the accumulation of Aβ in the brain, as demonstrated by PET imaging studies, but without effect on cognition and it revealed ARIA as the major safety concern of anti-Aβ therapies.
Donanemab exhibits high specificity for pyroglutamate-3 modified Aβ (pE3-Aβ) making the antibody highly efficient at removing pre-existing aged Aβ deposits by phagocytosis, as designed. pE3-Aβ is also present in protofibrils engaged by lecanemab ex vivo. The binding mechanism involves high monovalent affinity for the N-pE3Aβ. We find that it does so by burying the terminal moiety in the Fab surface with little capacity for cross reactivity. It is structurally similar to bapineuzumab, but bapineuzumab and donanemab recognize mutually exclusive epitopes and as such are ideal comparators to investigate the causes of ARIA and the contributions of the pyroglutamate-Aβ cascade in AD. The innovative approach exploits a specific post translational modification to maximize microglial recruitment to plaques and aims to avoid intact Aβ that accumulates in blood vessels. Targeting PTMs including truncation is now a validated approach to treating proteinopathies with antibodies which may obviate the need in some applications for elusive conformation-specific antibodies.
Lecanemab preferentially binds to labile Aβ epitopes in aggregates. Homology models suggest the antibody selects for partially degraded species lacking Asp1. Humanized IgG1 bivalency and the ability to preference a limited range of N-terminally truncated/modified Aβ species contributes to its ability to shift PET-Aβ in AD and bind soluble heterogeneous oligomers. Lecanemab has uniquely significant effects on cognition that may be derived from engagement of species that emerge early in the Aβ cycle on-path to plaques imaged by PET. From another perspective, the homologue of lecanemab (C#17) shows no binding to N-Phe4 and further truncated Aβ species. An advantage to lecanemab might stem from its ability to avoid these protease-resistant species to better preserve the fitness of microglia and their response to more biologically active Aβ species.
Gantenerumab is human derived and engages intact Aβ with decreasing affinity for N-truncated forms. The long epitope may also affect clinical outcomes by engaging Aβ further along the sequence than other N-terminal antibodies, closer to the histidine residues at positions 13 and 14 implicated in membrane binding. In addition, the mid-region epitope recognized by gantenerumab could further attenuate how the antibody engages Aβ in association with other important biological ligands such as ApoE.
A modified version (trontinemab) developed to overcome blood-brain barrier exclusion might exhibit more effective engagement of the earliest aggregates in the AD brain including nascent oligomers. It is not clear if cross reactivity with Aβ monomers will significantly accumulate immunocomplexes in CSF that might complicate clinical outcomes as seen at very high doses of solanezumab in the recent A4 trials. 18
Aducanumab is the first-in-class therapeutic derived from a human autoantibody. The Fab barely recognizes the monovalent epitope shown in the crystal structure reported by Arndt et al. It looks like a human antibody raised against the murine version of Aβ, showing no constraints on the core Arg5 guanidinium pharmacophore that dominates the murine response to human Aβ. This enables reactivity with common modifications like citrullination and truncation found in the AD brain. Loss of the first two amino acids and even the loss or modification of Glu3 are accommodated by the antigen binding mode. We propose that the dimer of Fabs seen in crystal structures constitutes a single antigen recognition site for the detection of multiple Aβ species, fresh and modified, but only as they form oligomers.
The binding mode presented raises the possibility of Aβ-mediated oligomerization of bivalent IgG in vivo. This might account for atypical immune responses recently reported in murine models using an aducanumab-like antibody to prevent or remove pre-accumulated Aβ. The requirement of a dynamic ternary complex of mAbs and Aβ oligomers might also account for efficacy only at higher doses in vivo. The conditional reactivity and the underlying mechanism we describe for aducanumab suggests evolution of the immune response from IgM origins, proposed for similarly complex natural antibodies to neutralize HIV. 47 The IgG1 format might not fully recapitulate the protective adaptive immune response that might confer cognitive resilience.
Solanezumab and crenezumab are the rational attempts to prevent Aβ aggregation, but stoichiometry involved in targeting monomers is unfavorable in vivo, where one mAb molecule theoretically removes just two Aβ molecules and the epitope is exposed by Aβ in other tissue, including blood. This might also result in excessive immunocomplex accumulation in CSF as seen in plasma. 4 By targeting a simple linear epitope with high affinity, solanezumab might also interfere with normal AβPP processing. The epitope is at the membrane interface but at very high doses this is a plausible explanation of worsened cognitive measures. Comparison of IgG1 solanezumab and IgG4 crenezumab may therefore provide insights into off-target effects and the potential for Fc-mediated toxicity of immune therapies in the CNS if the lower affinity of crenezumab is taken into account.
Conclusions
We might not yet be fully realizing the protective potential of anti-Aβ therapy. The use of post translational modifications as proxies for oligomer specific drugs is a significant advancement. N-Ala2-Aβ, N-Glu3-Aβ and N-pE3-Aβ are only a fraction of circulating pooled protofibrils but they might variably present specific epitopes to select AD-relevant, biologically active Aβ oligomers.
The use of human/humanized IgG1 antibodies to target antigens on surfaces that we seek to preserve is also problematic. Human IgG1 class antibodies have Fc potency and have the ability, in complex with some antigens to hexamerize and activate compliment cascades which we speculate might contribute to treatment-associated brain atrophy. Fc modifications to modulate compliment pathways may improve both safety and efficacy of passive immune therapies and such controls strongly support the prioritized development of passive immunotherapies above immunization. However, these monoclonal antibody therapies have no benefit in the absence of accumulating Aβ yet may be of most value soon after the onset of prodromal disease. It is therefore essential that robust, non-invasive and cost-effective methods for early detection of disease-relevant Aβ be developed to help prevent AD and CAA.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251361049 - Supplemental material for The structural foundations of anti-amyloid-β immunotherapies: Unravelling antibody-antigen interactions in Alzheimer's disease treatment
Supplemental material, sj-docx-1-alz-10.1177_13872877251361049 for The structural foundations of anti-amyloid-β immunotherapies: Unravelling antibody-antigen interactions in Alzheimer's disease treatment by Luke A Miles and Colin L Masters in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
Thank you to Dr Laura Ellett for editorial advice.
Author contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
LAM declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. CLM reports Ad Hoc consultancy speaking engagements and scientific advice with Actinogen, Acumen, Alterity, Biogen, Eisai, Eli-Lilly, Roche.
Data availability statement
All data are available in the main text or the Supplemental Material. Structure files for mE8 and m158 are available on request from the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.