
Abstract
Alzheimer's disease (AD) is a chronic neurodegenerative disease that is frequent among the aged and is characterized by symptoms of memory loss. With the development of the disease, cognitive impairment becomes obvious and leads to a heterogeneous spectrum of mental and behavioral problems that put a heavy burden on families and the society. Recent investigations suggest that the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway is important in AD. In this paper, we describe the detailed mechanisms of this pathway that are also involved in AD, and the current research status in terms of the relationship of the effects of acupuncture with it.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease and the most typical kind of dementia, and it is more prevalent in persons aged 65 years and older. 1 It has turned into the world's second greatest health problem. With the epidemiological study by the World Health Organization, the global cases of AD dementia may similarly increase to almost 70 million in 2050. The incidence of AD increases with aging and rises from 5% to 8% in people aged 65 or older to 25% to 50% in people aged 85 or older. 2 The etiology of AD is complicated and variable and has undergone some additional development in its particular mechanism. In addition to typical amyloid-β (Aβ) amyloidosis and the hyperphosphorylation of tau protein,3,4 a host of other components, including impaired acetylcholine, neuroinflammation, oxidative stress, impaired biometal homeostasis, glutamate imbalance, insulin resistance, abnormal gut microbiota, disturbance of cholesterol metabolism, mitochondrial dysfunction, and dysregulated autophagy, might also be implicated in the onset and progression of the disease.5,6 These players might eventually promote neuronal death. The mammalian target of rapamycin (mTOR), a serine-threonine protein kinase in the phosphoinositide 3-kinase (PI) 4-kinase related kinase family, 7 serves as a hub that couples eukaryotic cell growth and metabolism with external stimuli such as growth factors and nutrition. The PI3K/protein kinase B (AKT)/pathway regulates two mTOR complexes and is critical in the regulation of autophagy.7,8 These intricate biological processes encompass cell survival, proliferation, migration, and intracellular transport, which are essential for microtubule transport during axon creation and neuronal polarity in brain development. AD is particularly prevalent in the United States, with a significant prevalence linked to its pathophysiology. 8
PI3K/AKT/mTOR pathway
The protein complex associated with the target site of PI3K is localized on the cellular membrane, potentially leading to the aggregation of normal pleckstrin homology (PH) levels with proteins. Phosphatidylinositol signaling regulates cell polarity and chemotaxis in both of these organisms. Most extracellular stimuli not only regulate cell proliferation and growth but also cause cell migration, inactivate prosurvival signaling pathways, and coordinate gene expression programs that regulate metabolic demand, as well as other tissue-specific functions, in addition to inducing cell migration. To meet these elaborate physiological requirements, the PI3K class I and II were able to cross-link cellular and external signal transduction pathways and to arrange its protein complexes beneath membrane receptors.9,10 Because of their distinct ability to decipher the extracellular environment, class I PI3Ks have been the most widely studied and researched. PI-34,5-P3 is a very powerful second messenger that can be produced from PI-4,5-P2 on the cell membrane,11,12 AKT is a key component of this complexity. Both PI-34,5-P3 and PI-34,4-P can be bound by the PH domain of AKT, and both lipids may be involved in the recruitment of AKT to the membrane as well as the recruitment of the upstream activating kinase PDK1 that is not defined upstream.
In brief the AKT downstream network includes proliferation promotion by suppressing the phosphorylation of Forkhead box O (FOXO) transcription factors and glycogen synthase kinase 3 (GSK3), protein synthesis enhancement through the mechanistic target of rapamycin complex 1 (mTORC1) pathway via phosphorylation of Tuberous Sclerosis Complex 2 (TSC2) and phosphatase and tensin homologue deleted on chromosome Telomerase-Encoding Nuclear RNA and proline-rich AKT substrate of 40 kDa (PRAS40), cell survival promotion via Bcl-2 Associated Death Promoter (BAD) and Murine Double Minute 2 phosphorylation, controlling the association of BAD with 14-3-3 to prevent it from entering the mitochondria to initiate apoptotic signals, leading to mitochondrial stabilization and their further resistance to apoptotic stimuli, and AS160 11 phosphorylate-induced glucose uptake to glut4-containing cells.
mTOR is structurally and functionally separated into two multicomplexes, mTORC1 and mechanistic target of rapamycin complex 2 (mTORC2). The mTORC1 consists of three main components mTOR, Raptor, and mammalian Lethal with SEC13 protein 8 (mLST8).13–15 Apart from these three key components, mTORC1 also includes two suppressive elements, PRAS40.16–18 On top of the three components of mTORC2, mTOR, rictor, and mLST8, there is SIN1. Homologue of Rictor, an mTOR binding partner not inhibited by rapamycin. Like its role in mTORC1, mLST8 is necessary for functional expression of mTORC2. 19 The phosphorylation of TSC2 by AKT at serine 939 and threonine 1462 breaks the TSC2 complex,20–22 thereby alleviating its inhibition of Rheb and effectively activating mTORC1. TSC1 (also called hamartin) is the binding partner of TSC2 and a binding partner, which is used to activate the GTPase activating protein (GAP) for the Ras-related small G protein Rheb, which in turn inactivates Rheb. 23 Additionally, mTORC1 signaling was linked to the PRAS40.16,17,24 Akt directly phosphorylates PRAS40 at the T246 site, and this phosphorylation is important for 14-3-3 binding.17,24 PRAS40 associates with mTORC1 and acts to suppress mTORC1 signaling.16,17 mTORC2, in contrast with mTORC1, appears to function primarily downstream of the insulin/PI3K pathway. The mTORC2 subunit mSin1, like most PI3K regulatory subunits, contains a PH domain that binds phosphatidylinositol and is important for regulation of mTORC2 activity by insulin. In the context of no insulin, mSin1's PH domain blocks mTORC2's ability to use its enzyme. Conversely, inhibition is relieved by binding to PIP3 produced by PI3K on the plasma membrane. 25 Moreover, Akt can phosphorylate mSin1, suggesting a positive feedback loop that would amplify the stimulatory effect of Akt partial activation on mTORC2 signaling, leading to full Akt activation via phosphorylation. 26 The complete activation of AKT requires phosphorylation at two of its sites, namely Thr308 in the activation loop and Ser473 in the carboxyl-terminal hydrophobic motif. Although Thr308 is the activation basis of AKT dependent on 3-Phosphoinositide-Dependent Protein Kinase 1 (PDK1), when Ser473 is not phosphorylated, the phosphorylation efficiency of PDK1 for Thr308 decreases. Therefore, the phosphorylation of Ser473 further stabilizes the active conformation of Akt and enables Akt to recognize and phosphorylate its downstream substrates more effectively and stably. 27 At the same time, the dephosphorylation of both Ser473 and Thr308 jointly affects the degradation of AKT, while the single dephosphorylation of either does not affect the degradation of AKT.28,29 Then, the phosphorylation of Ser473 is crucial to the activity of AKT, and its phosphorylation depends on the Rictor-mTOR complex, namely mTORC2. Studies have shown that when rictor is knocked down, the phosphorylation of Ser473 is inhibited. Therefore, rictor is the key kinase for the phosphorylation of Ser473. 27 Surprisingly, mTORC2 signaling is governed by mTORC1 in a manner reminiscent of a classical negative feedback loop that controls insulin/PI3K signaling30,31 (Figure 1, Created with BioGDP.com 32 ). Specifically, mTORC1-mediated phosphorylation of Grb10, a negative regulator of insulin/IGF-1 receptor signaling located upstream of Akt and mTORC2, results in the release of the direct inhibition of Grb10 on insulin/IGF-1 signal, and hence increases Akt activation.33,34 Furthermore, S6K1, an effector downstream of mTORC1, inhibits the activation of mTORC2 by facilitating the phosphorylation-mediated degradation of insulin receptor substrate 1 (IRS1).
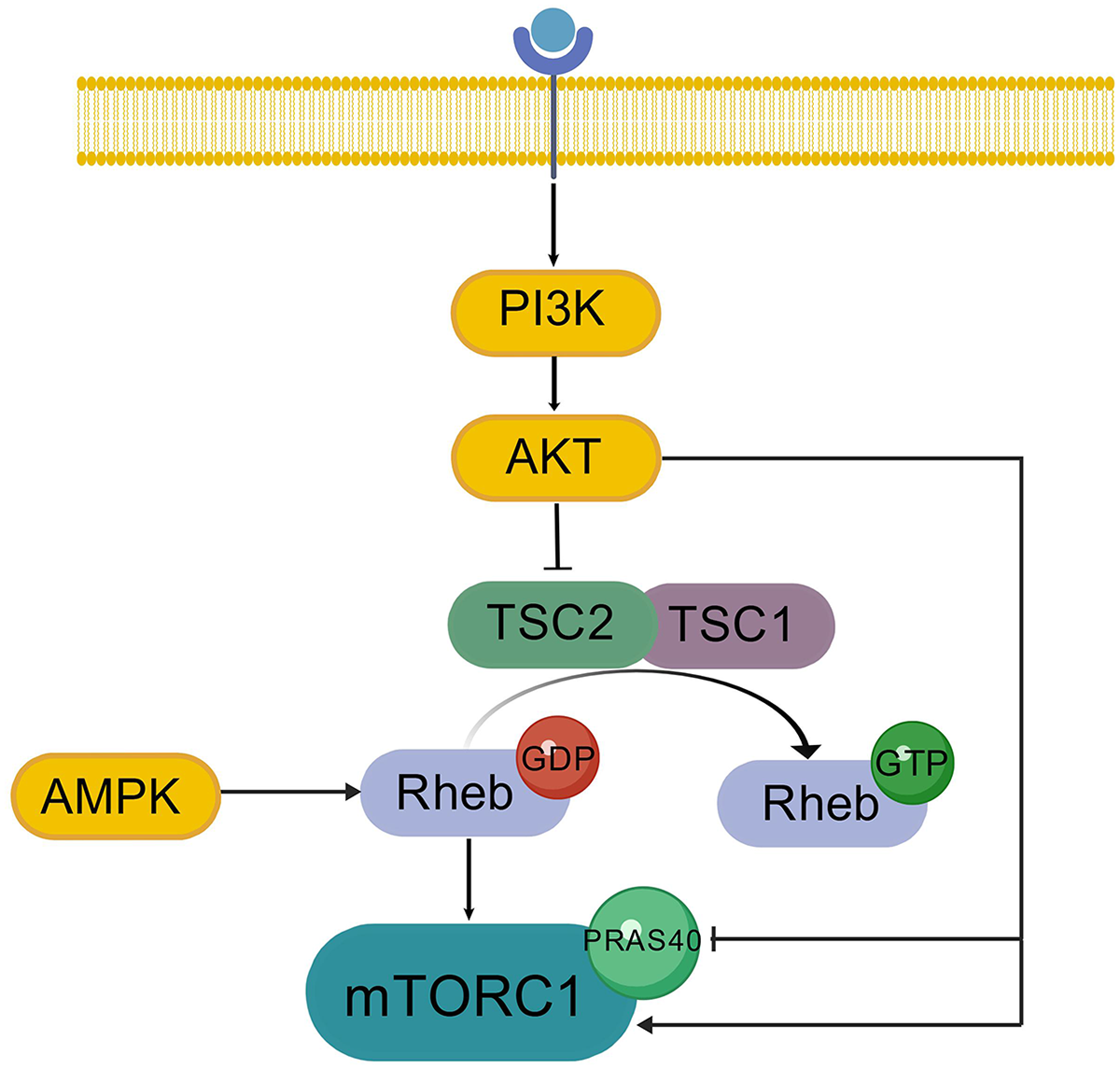
PI3K/AKT/mTORC1 regulatory pathway.
The PI3K/AKT/mTOR signaling route is involved in the pathophysiology of AD
The development of AD is not only related to Aβ deposits and tau hyperphosphorylation as the classic features of disease, but also to disordered autophagy. The PI3K/AKT/mTOR signaling pathway is closely related to autophagy regulation, an evolutionarily conserved catabolic process that degrades and recycles protein aggregates and damaged organelles.35,36 mTORC1 is a negative regulator of autophagy,36–38 and its activity depends on the nutritional status of the cell. 39 Key early events in autophagy induction include activation of the Unc-51-like autophagy activating kinase 1 (ULK1), which associates with autophagy-related proteins (ATG)13, ATG101, and focal adhesion kinase family interacting protein of 200 kDa (FIP200) to promote autophagosome formation (Figure 2, Created with BioGDP.com 32 ). Under glucose starvation conditions, the energy sensor AMP-activated protein kinase (AMPK) phosphorylates and activates ULK1 at Ser317 and Ser777, resulting in autophagy stimulation. In contrast, in the presence of nutrients, mTORC1 suppresses autophagy by directly phosphorylating ULK1 at Ser757 (Figure 3, Created with BioGDP.com 32 ), thereby disrupting the interaction between ULK1 and AMPK. The mTORC1 activator Ras homolog enriched in brain (Rheb) overexpression decreases the coimmunoprecipitation of ULK1 and AMPK, and the mTOR inhibition of Rheb. 40 Here malnourished neurons can be observed displaying an accumulation of autophagic vacuoles. This is linked to Aβ/APP-βCTF, indicating that pathological autophagic clearance is dramatically compromised and closely correlated with amyloid pathology.41,42 Autophagic defects in AD coordinate with breakdown of proteostasis network (Aβ generation and extracellular secretion, abnormal accumulation of tau protein), damage-accumulation (e.g., dysfunctional mitochondria). 42
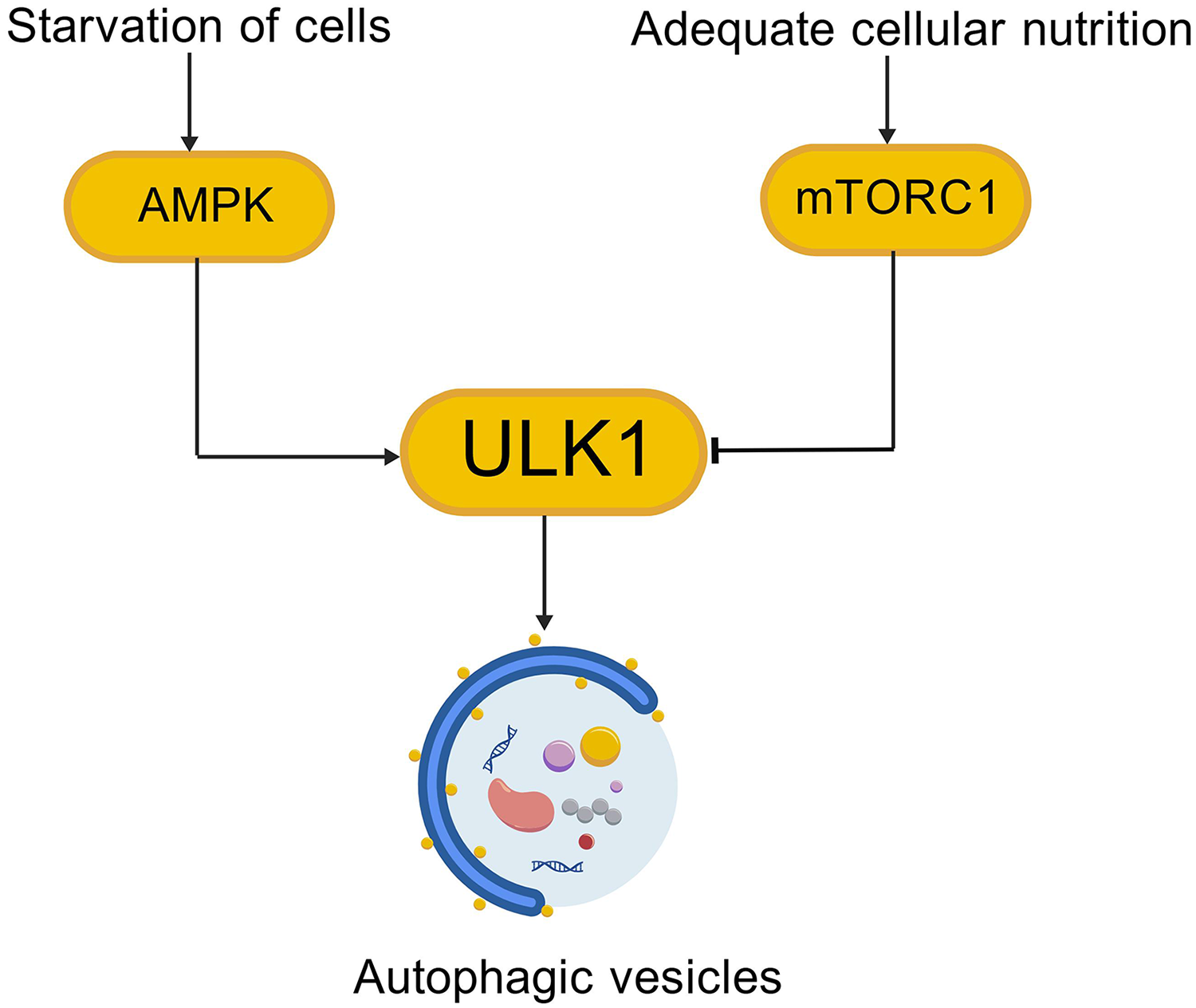
Autophagy regulation pathways.
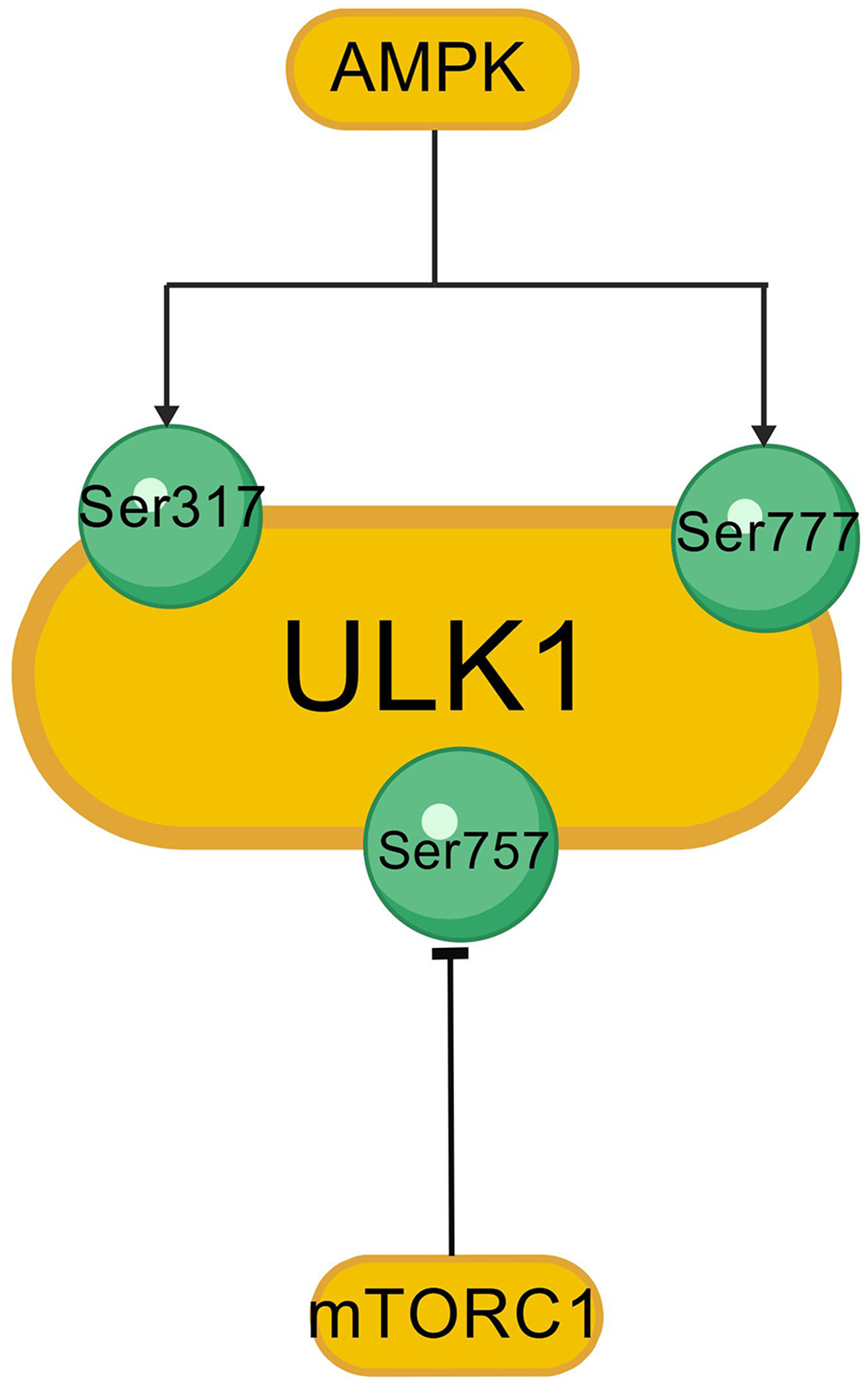
Protein sites of ULK1 regulated by AMPK and mTOR.
Abnormal autophagy is associated with the two classic pathogenic pathways, mainly by abnormal insulin signaling, disrupted neurotrophic factor pathways, and neuroinflammation signals. 43 The PI3K-Akt signaling cascade is one of the key signaling pathways of insulin. In the AD brain, it has been reported the reduction of IRS-related PI3K activity and Akt kinase phosphorylation,44,45 leading to a reduced Akt-induced inhibition of GSK3 kinase activity, GSK3β phosphorylates Phosphatase and Tensin Homolog deleted on chromosome Ten (PTEN), activates PTEN, inhibits PI3K/Akt and Extracellular Signal-Regulated Kinase1/2 functional activity, leads to cognitive impairment in animal models, and indirectly promotes tau protein phosphorylation. Activated GSK3β can inhibit the Wnt/β-catenin signaling pathway, thereby reducing β-catenin stability and impairing synaptic formation, dendritic spine development, and long-term potentiation-processes that underlie the pathological mechanism of long-term memory loss in AD. Concurrently, this activation may contribute to maintain high Protein Methylesterase-1 levels and interfere with methylation homeostasis, thereby promoting demethylation and inactivation of Protein Phosphatase 2A, 45 that ultimately favor tau protein phosphorylation and Aβ production.46–48 The PI3K/AKT/mTOR signaling pathway regulates protein synthesis, which is involved in the synthesis of different ligands of Wnt in presynaptic synapses. mTORC1 is a downstream target of Akt and it connects insulin signaling to the autophagy 49 system. It can be regulated by IRS1 inhibitory phosphorylation, and upon overactivation, impairs synaptic protein synthesis, synaptic plasticity and autophagy control, leading to pathological protein deposition and cognitive deficits in AD. Inhibition of excessive mTORC1 activation, could promote autophagy, reducing Aβ and hyperphosphorylated tau.50,51 In an elevated inflammatory state, cytokines such as TNF-α and NF-κB trigger oxidative stress, which contributes to inhibitory serine phosphorylation of IRS-1, which impairs insulin signaling in the brain, contributing to AD neurologic dysfunction.52,53 Meanwhile, these aforementioned mediators and abnormal proteins such as aggregated Aβ or hyperphosphorylated tau activate microglia to propagate downstream pathways, including MAPK, NF-κB, and PI3K/Akt. Activated microglia transit to senile plaques, englut Aβ, and express enzymes that act to degrade Aβ. 54 Eventually, they may process Aβ less efficiently and continue to produce proinflammatory cytokines. In microglia, Aβ also triggers the formation and activation of NLRP3 inflammasome and the release of ASC specks that bind Aβ, facilitating its aggregation and spreading.55,56 In later stages of AD, microglia-tau interaction may stimulate tau phosphorylation and exosomal tau release, promoting tau propagation.57–59 NF-κB, co-regulated by pathways such as MAPK and PI3K/Akt, then upregulates its own transcriptional activity to promote pro-NLRP3 inflammasome component expression. 60 In AD, the action of neurotrophic factors like nerve growth factor, and brain-derived neurotrophic factor (BDNF) occurs via receptors like Trk and p75NTR. BDNF downregulation also weakens BDNF signaling, 61 which activates the JAK2/STAT3 and C/EBPβ pathways while deactivating the Akt signal protein,62,63 thereby activating AEP (or δ-secretase) and thus amyloid-β protein precursor (AβPP) and tau protein cleavage. 61 The tau fragments then bind to TRKB receptor, inducing neuronal apoptosis. 64 Impaired BDNF signaling can reciprocally upregulate AβPP and PS1 expression, thus exacerbating amyloidosis. 65 Moreover, Aβ damages neuroprotection pathways activated by BDNF binding to the TRKB receptor, including Raf-MAPK/ERK and P13K-Akt, leading to dysfunction in cortical neurons.
The PI3K/AKT/mTOR pathway preferentially on the whole increases autophagic vesicle formation in neurons through the disordered autophagy, exacerbates AD pathology. Besides, Aβ and hyperphosphorylated tau protein may regulate the PI3K/AKT/mTOR pathway via alternative mechanisms to suppress mTOR, and then to exacerbate the disturbances of autophagy. This implies a possibility of a cascade phenomenon in the AD pathological mechanism.
Regulation of acupuncture on autophagy
The study demonstrates that acupuncture's modulation of autophagy varies considerably at different stages of traumatic brain injury (TBI). 66 Three days after TBI, acupuncture increases neuronal autophagy, which in turn removes the impaired cellular components. By contrast, acupuncture inhibits neuronal autophagy on days 7 and 14, alleviating excessive autophagy and the resulting neural impairment. Acceleration of the crude neuron nuclear edge, production of heterochromatin, clusters of chromatins at the nuclear edge, rising in cytoplasmic electron density, swelling of mitochondria, and activation of several autophagic processes can be achieved during the early stage of nerve injury by acupuncture treatment. Meanwhile, the elevated p62 protein levels, generated by TBI, was diminished, whereas Microtubule-Associated Protein 1 Light Chain 3 (LC3)II/I protein expression was enhanced with acupuncture. During the middle and late stages of neuronal damage, acupuncture inhibits neuronal degeneration, especially decreasing autophagic vesicles in chromatin. The proportion of LC3-positive neurons is markedly reduced by acupuncture intervention, and the structural improvement is obvious by day 14 post-TBI. Also, acupuncture enhances p62 protein expression but decreases the level of LC3 II/I expression. Acupuncture regulates mTOR through both “yes” and “no” way. First, it contributes to the early autophagy by reducing the p-mTOR/mTOR and p-ULK1 Ser757/ULK1 ratios on early neurological damage. In contrast, acupuncture subsequently promotes the expression of p-mTOR/mTOR and p-ULK1 Ser757/ULK1, thus promoting cell proliferation. That acupuncture can modulate the early stage of autophagy including initiation and nucleation via mTOR/ULK1 signaling pathway.
Examples of this study 67 have demonstrated that AMPK/mTOR-mediated autophagy is activated by electroacupuncture on spinal cord microglia. This stimulus results in a decrease in AMPK activity and an increase in AMPK phosphorylation that activates AMPK and mTOR phosphorylation. After that, the result is lowering the amount of phosphate on ULK1, which causes autophagy. Additionally, electroacupuncture significantly increases the amount of autophagic process that is upregulated by increasing autophagy markers LC3-II and Beclin-1, which also increases p62 degradation. Activated microglial cells migrate to the neuropil of phagocyte Aβ and neuronal senile plaques, which in turn cause the Aβ protein to degrade enzymatically. According to this study, electroacupuncture controls microglia by inhibiting mTOR, which then promotes autophagy and lowers Aβ proteins in neurons.
Li-da Zhang 68 applied moxibustion to the Baihui(GV20), Fengfu(GV16) and Dazhui(GV14) acupoints in AD mice. Observations indicated an increase in autophagic vacuoles and increased expression of autophagy-related proteins compared to AD model mice, suggesting increased autophagy levels and cellular autophagic capacity after moxibustion. Further investigation showed a reduction in PI3K, p-Akt, and p-mTOR levels, indicating that moxibustion increases cellular autophagy by blocking the PI3K/AKT/mTOR pathway. This inhibition decreases Aβ deposition and alleviates AD symptoms. The result shows that moxibustion, akin to acupuncture, modulates the PI3K/AKT/mTOR pathway through different cellular stages to ameliorate abnormal autophagy.
The results of the Baihui and Shenshu acupoints in AD rats were examined by Qing Zheng. 68 Following therapy, these rats showed significant behavioral improvements, indicating improved cognitive function. Furthermore, there was a noteworthy rise in autophagic cells and neuronal activities in the hippocampus. The expression of PI3K, AKT, and mTOR decreased in PHF-1, which changed from being positive to weakly positive. It also suggests that acupuncture could be useful to treat AD. The PI3K/AKT/mTOR route is probably involved in the process, whereby acupuncture increases autophagy by decreasing neurofibrillary tangles. According to this study, decreasing the PI3K/AKT/mTOR pathway and increasing autophagy might result in decreased paired helical filament-1 expression, decreased tau phosphorylation, and decreased neurofibrillary tangles.
Discussion
The PI3K/Akt pathway is involved in the regulation of a variety of key cellular processes, including cell adhesion and cytoskeletal organization. This pathway generates the second messenger PI-34,5-P3, which recruits proteins with PH domain, such as Akt, to the cell membrane, thereby activating the downstream signaling network. These signals not only regulate cell survival, proliferation and metabolism, but also affect cell migration, polarity establishment and microtubule transport, which are essential for neuron axon development and cytoskeleton dynamics. Furthermore, PI3K signaling coordinates chemotaxis and cell motility in response to extracellular stimuli, suggesting an important role in cell adhesion and cytoskeletal rearrangement. Aβ deposition and tau protein phosphorylation are currently recognized pathological diagnostic criteria for AD, but they cannot be simply regarded as separate. At the same time, the PI3K/AKT/mTOR pathway also has an impact on both. Current research 68 indicates that the abnormal cleavage of AβPP and the subsequent formation of Aβ plaques are the main initiating basis of AD, which in turn triggers downstream cascade reactions leading to tau pathology, resulting in synaptic fibrillary tangles in neurons. Therefore, the deposition of Aβ protein creates a toxic environment, making tau protein pathological and spreading, destroying the tubulin in the synaptic fibers of neurons, and leading to the deposition of a large number of autophagosomes, thereby affecting the PI3K/AKT/mTOR pathway. Meanwhile, the PI3K/AKT/mTOR pathway is crucial for protein synthesis, with ULK1, a downstream effector of mTORC1, playing a significant role in autophagy. ULK1 is inhibited by mTORC1 but activated by AMPK, which is itself activated by AMP. This study observed an increase in AMP expression under cellular starvation. Inhibition of IRS phosphorylation suppresses the PI3K/AKT/mTOR pathway, thereby enhancing autophagy. In AD pathology, neurons accumulate numerous autophagic vacuoles, potentially linked to Aβ deposition and excessive tau phosphorylation, which lead to neurofibrillary tangles and disrupted autophagic vesicle transport. Additionally, inhibition of the PI3K/AKT/mTOR pathway may result in aberrant autophagy. Most studies have shown that acupuncture can down-regulate PI3K/AKT/mTOR pathway and enhance autophagy. However, the study found acupuncture can modulate autophagy by upregulating or downregulating the PI3K/AKT/mTOR pathway depending on cellular conditions and may influence this pathway through other mechanisms. For example, enhancing insulin signaling by regulating blood sugar, improving the blood-brain barrier via head acupuncture to aid in brain waste clearance, and using acupuncture to modulate intestinal flora can collectively improve blood sugar levels. This suggests acupuncture could play a role in comprehensive AD treatment. Acupuncture also may enhance microglial autophagic function through the PI3K/AKT/mTOR pathway, enabling clearance of abnormal proteins in central neurons. The mechanism of acupuncture in AD treatment also may involve regulation of the PI3K/AKT/mTOR pathway by modulating neurotrophic factors, insulin signaling, and immune factors, thereby disrupting crosstalk between the PI3K/AKT/mTOR pathway and other pathways and alleviating the cascade of autophagic abnormalities. According to the existing research on acupuncture and moxibustion for AD in this paper, scalp acupuncture based on GV20 is the main acupuncture point for AD. Our team used Sun Shen-tian's transcranial repeated acupuncture 69 to conduct relevant clinical and basic research, which proved the efficacy and safety of transcranial repeated acupuncture in the treatment of AD, and found that it can regulate the PI3K/AKT/mTOR pathway. Moreover, according to the article, acupuncture has a bidirectional regulation on autophagy, so we suggest that acupuncture can be combined with the whole process of AD treatment. When cognitive impairment occurs, acupuncture should be applied to the relevant acupoints of the kidney meridian. This study expands the theoretical understanding of the mechanism of acupuncture treatment for AD based on the PI3K/AKT/mTOR pathway.
Conclusion
PI3K/AKT/mTOR pathway plays a pivotal role in the pathogenesis of AD, which not only regulates autophagy, but also connects multiple links such as insulin resistance, neuroinflammation, tau pathology and so on. Acupuncture and moxibustion can regulate this pathway in a multi-scale and context-dependent manner and exert a two-way regulation of autophagy. Its mechanisms include improving metabolism, enhancing protein clearance, and protecting neuronal function. However, the clinical promotion of acupuncture and moxibustion in the treatment of AD faces double challenges. First, the therapeutic manipulation and acupoint selection scheme are highly dependent on the individual experience of acupuncture-moxibustion practitioners, and lack of standardization and repeatable operation. Second, it is difficult for patients to adhere to treatment due to the long course of treatment, inconvenience to see a doctor, psychological resistance and other factors, which seriously affects the stability and evaluation of curative effect. Therefore, a large number of high-quality clinical studies should be conducted to determine the influence of acupuncturists’ manipulations on AD, and the acupoint scheme for the treatment of AD based on data mining technology should be determined, so as to improve the acceptance of acupuncture and moxibustion in patients. This study expands the theoretical basis of acupuncture for AD based on PI3K/AKT/mTOR pathway and supports its potential as a comprehensive intervention strategy.
Footnotes
Acknowledgements
We gratefully acknowledge all contributing authors. Their scholarship on Alzheimer's disease psychosis has significantly propelled progress in this complex area.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Number:82174509); Postgraduate of Heilongjiang University of Traditional Chinese Medicine (Grant Number:2024yjscx009).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.