
Abstract
Background:
The metabotropic glutamate receptor 5 (mGluR5) is widely expressed in postsynaptic neurons and plays a vital role in the synaptic plasticity of the central nervous system. mGluR5 is a coreceptor for amyloid-β (Aβ) oligomer, and downregulation or pharmacological blockade of mGluR5 presents the therapeutic potential of Alzheimer’s disease (AD). However, the abnormality of mGluR5 in the pathogenesis of AD and its mechanism of pathology is not clear.
Objective:
In this study, we would like to investigate the expression of mGluR5 in the process of AD and explore the effects and the underlying mechanisms of antagonizing mGluR5 on cognitive function, synaptic structure, and inflammation in 5xFAD mice.
Methods:
mGluR5 expression and interactions with PrPc in 5XFAD mice were detected using western blot and co-immunoprecipitation. The selective mGluR5 antagonist MPEP was infused into 4-month-old 5XFAD mice for 60 consecutive days. Then, cognitive function, AD-like pathology and synaptic structure were measured. Further observations were made in mGluR5 knockdown 5XFAD mice.
Results:
mGluR5 expression was increased with Aβ levels at 6 months in 5XFAD mice. mGluR5 antagonist rescued cognitive disorders, promoted synaptic recovery, and alleviated both the Aβ plaque load and abnormal hyperphosphorylation in 6-month-old 5XFAD mice. Meanwhile, the results were validated in mGluR5 knockdown mice. Blockade of mGluR5 efficiently alleviates AD-like pathologies by inhibiting the PI3K/AKT/mTOR pathway and activates autophagy in 5XFAD mice. Furthermore, antagonism of mGluR5 attenuated neuroinflammation by inactivating the IKK/NF-κB pathway.
Conclusion:
These findings suggest that mGluR5 may be an effective drug target for AD treatment, and inhibition of the mGluR5/PI3K-AKT pathway alleviates AD-like pathology by activating autophagy and anti-neuroinflammation in 5XFAD mice.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive and fatal degenerative disease of the central nervous system that clinically manifests as cognitive and memory function deterioration and impaired social and behavioral functions. AD remains a difficult problem for elderly individuals, and the number of people aged 65 and older with AD in America is projected to reach 7.2 million by 2025 [1]. Between 2000 and 2019, AD deaths increased by more than 145%; in 2020, this trajectory of AD deaths was likely exacerbated by the novel coronavirus disease (COVID-19) pandemic, barring the development of medical breakthroughs to cure AD [2]. The hallmark pathologies of AD are the accumulation of amyloid-β protein fragments (Aβ plaques) outside neurons in the brain and intracellular neurofibrillary tangles composed of abnormally phosphorylated tau protein, which is accompanied by neuronal death and damage to brain tissue [3, 4], followed by microgliosis and astrocytosis, which are typical in AD pathology [5].
Glutamate is a vital excitatory neurotransmitter in the mammalian central nervous system that plays an important role in memory, synaptic plasticity, and neuronal development. Two types of glutamate receptors are expressed: ionic glutamate receptors, such as NMDA and AMPA, and metabotropic glutamate receptors (mGluRs). Type 5 metabotropic glutamate receptor (mGluR5) is a postsynaptic element that is abundant in the hippocampus and expressed at moderate levels in the cerebral cortex [6], therefore, mGluR5 might play an important role in brain diseases associated with cognitive dysfunction, such as AD. Furthermore, the expression of mGluR5 allowed extracellular Aβ to bind to the cellular prion protein (PrPC) and activate intracellular Fyn kinase to damage synapses. Aβo-PrPC-mGluR5 complexes activate mGluR5 signaling and downstream pathways, enabling phospholipase C (PLC) to produce inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG) and release calcium from cellular stores; intracellular calcium stimulates protein kinase C (PKC) and its related downstream signaling pathways [7]. mGluR5 deletion reduces cognitive impairment and AD pathology in mice with AD.
The amyloid hypothesis for AD posits a neuron-centric, linear cascade initiated by Aβ and leading to tau pathology, synaptic dysfunction, inflammation, neuronal loss, and ultimately, dementia [8]. Neuroinflammation is a key trigger in AD pathogenesis, as the binding of soluble Aβ oligomers to microglia results in the production and release of inflammatory mediators. Autophagy has been implicated in the pathogenesis of AD, and autophagosomes with Aβ deposits are commonly observed in the brains of AD patients, therefore, activation of autophagy may hinder AD progression [9]. Overactivation of mGluR5 contributes to the inhibition of autophagy and may lead to impaired clearance of neurotoxic aggregates in a variety of neurodegenerative diseases [10]. However, it is unclear whether alterations in these autophagy pathways due to aberrant mGluR5 signaling are also evident in the 5xFADmodel.
The purpose of this study was to investigate the therapeutic effect and underlying mechanisms of the mGluR5 in 5xFAD mice. These findings using different approaches to silence mGluR5 further support the pivotal role of mGluR5 in AD pathogenesis and supply a new mechanism from mGLuR5-autophagy pathway.
MATERIALS AND METHODS
Animals and treatments
Breeding pairs of 5XFAD mice (RRID: MMRRC_034840-MU) and C57/BL6 mice (RRID: IMSR_JAX:00066) were obtained from the Jackson Laboratory (New Harbor, ME, USA). 5XFAD transgenic mice overexpress mutant human amyloid beta (A4) precursor protein 695 (APP) with the Swedish (K670 N, M671 L), Florida (I716 V), and London (V717I) familial Alzheimer’s disease (FAD) mutations along with human presenilin 1 (PS1) harboring two FAD mutations, M146 L and L286 V. Genotyping PCR is used to determine the genotype of the mice (Table 1). B6.129-Grm5tm1Rod/J (Grm5 knockout) mice on a C57BL/6 background were obtained from the Jackson Laboratory (New Harbor, ME, USA, JAX stock: 003558). 5XFAD mice were paired with B6.129-Grm5tm1Rod/J mice and bred in an SPF laboratory animal environment. Then, 5XFAD/mGluR5(+/-) mice were obtained through gene identification (Table 2). The mice were housed in groups of four and had free access to food and water unless otherwise stated. All animal procedures were performed according to protocols approved by our Institutional Animal Care and Use Committee and the PHS Policy on Human Care and Use of Laboratory Animals (revised January 2013); the experiments were approved by the Animal Care and Use Committee of Jinan University. Additionally, this study was not pre-registered. The inclusion/exclusion criteria for this study were the health status of the animals after treatment. All animals that completed treatment correctly and remained healthy were used for continued analysis. In this study, no animals were excluded. To save animals from suffering, technicians examine animals daily for evidence of suffering.
DNA sequences of the primers - 5XFAD
DNA sequences of the primers - Grm5 < tm1Rod>
Mice were housed in an animal center until 4 months of age. All mice were numbered and then correspond to random numbers generated by = Rand () in Excel (each mouse has a unique code number). WT 1-20 (10 female and 10 male mice) and 5XFAD 1– 16 (8 female and 8 male mice) were selected from the beginning of the randomization table, and then the mice were randomly divided into saline injection group or MPEP injection group according to the odd-even principle. The mice were intraperitoneally injected with MPEP (1 mg/kg/d, i.p) or with normal saline (NS) as a control for 60 consecutive days. No mice were found dead over the course of experimentation. The Morris water maze, Novel object recognition, and Passive avoidance tests were carried out successively beginning on Day 30.
After completion of the behavioral tasks, the animals were deeply anesthetized with avertin (600 mg/kg) and trans cordially perfused with 0.1 M phosphate buffered saline (PBS). After perfusion, the brains were removed from the skull immediately. Hippocampus and cerebral cortex were dissected from the left hemisphere, immediately frozen on dry ice, and then stored at – 80°C for biochemical analysis. The complete right hemisphere was mersion fixed in 4% paraformaldehyde in 0.1 M PBS for 24– 48 h for immunohistochemical staining.
Drugs
2-Methyl-6-(phenylethynly)-pyridine (MPEP) was purchased from Tocris (catalogue# 1212, Minneapolis, MN, USA), dissolved in sterile saline and injected i.p. at a dose of 1 mg·kg- 1·d- 1.
Morris water maze
The Morris Water Maze test was used to evaluate long-term learning and memory. The test was performed in a circular white pool (97 cm in diameter and 60 cm in height) filled with nontoxic white dye– tinted water maintained at room temperature (23±1°C). The maze was designed with four equal quadrants, the end of each line indicated the four cardinal points, and a platform (10×10 cm) was submerged 1 cm below the water surface in the middle of one of the quadrants. The Morris water maze test was composed of two parts: the place navigation test and the spatial probe test. An automated video tracking system was used to record the swim path, velocity, and time taken to reach the platform (latency) or the time spent in each zone.
In the positioning navigation test, the mouse was placed into the pool in the predetermined quadrant on that day, and the movement track for 60 s was recorded. Then, if a mouse failed to find the platform, it was gently guided to the platform, left on the platform for 20 s, removed, dried, and returned to its home cage. When all mice had completed the training in the quadrant, they were trained in the next quadrant until all four quadrants had been rotated. In the place navigation test, each mouse performed four trials for six consecutive days to find the hidden platform from semi-random start positions. To examine spatial memory, a spatial probe test was administered 24 h after the last training session. During the probe test, the platform was removed from the pool, and the mouse was placed in the quadrant opposite the platform. The mouse was allowed to swim freely for 1 min, and the number of times the platform was crossed and the stay time in each quadrant were recorded.
Passive avoidance test (PAT)
The Passive Avoidance Test is a behavioral experiment used to test short-term memory that consists of a dark room, a bright room, and a gate in the middle that can be opened and closed freely. When the gate is opened, the mouse can freely shuttle between the dark room and the bright room. The bottom of the dark room was equipped with electrified iron bars. In the absence of external stimulation, the mouse entered the dark room because of its innate habit of preferring the dark; however, when the mouse was stimulated with electricity in the dark room, it quickly escaped to the bright room. On Days 1 and 2, the gate was opened and there was no electricity, and each mouse was placed into the bright room. The mice were acclimatized to the environment and shuttled freely between the bright room and dark room for 5 min. On Days 3 and 4, the device was powered. The time was set as 5 min, and the voltage was 32 V. The camera was turned on to monitor and record the time (escape latency) of the experiment to determine when each mouse entered the dark room for the first time and received an electric shock and the number of times (error times) each mouse entered the dark room and received an electric shockwithin 5 min.
Novel object recognition (NOR)
The novel object recognition test is used to evaluate learning and memory ability by evaluating the differences in the exploration times of novel and familiar objects [11]. The testing apparatus is a classic open field (i.e., a square arena, 50×50 cm, with walls 40 cm high). In brief, mice from each cohort were acclimatized to the testing environment for 2 min. After acclimatization, the mice were returned to a separate holding box for a 1-min rest period, and two identical control objects of the same were placed in diametric quadrants of the activity box. Mice were reintroduced, and the ratio of both the number of investigations and the time spent at each object was recorded for 8 min. Following training, the mice was removed to a separate holding box for 20 min, and one object was replaced with another novel object, the novel object was not presented in the training session. The testing phase consisted of an 8-min exposure to one control object (same object from the training session) and a novel object. The mice were reintroduced and the number of investigations and duration per object was recorded as before. Data collection was performed using a video tracking system. The number of investigations/duration of the novel object was divided by the number of investigations/duration of the control object to generate the discrimination index [12].
Immunostaining
Immunostaining was performed on free-floating brain sections with clear hippocampal structures in phosphate-buffered saline (PBS) containing 1% Triton X-100 for 30 min. The sections were then incubated in 3% bovine serum albumin (BSA) for 60 min at 25°C to block nonspecific antibody binding. The sections were incubated with the primary antibodies listed in Table 2 at 4°C for 24 h and then incubated with the secondary antibodies at 37°C for 2 h in the dark. The following secondary antibodies were used: Alexa 488-conjugated goat anti-mouse IgG antibody (1:500, Molecular Probes; Carlsbad, CA, USA) or Alexa 594-conjugated donkey anti-rabbit IgG antibody (1:200, EMAR; Emarbio, Beijing, China). Fluorescence images were acquired with a confocal laser microscope (Leica TCS SP8 MP, Germany) using the sequential scanning mode.
Thioflavin S staining
Free-floating brain sections were washed with PBS 3 times for 5 min and then incubated with 0.3% thioflavin S (dissolved in 50% ethanol) at room temperature for 10 min. The sections were then decolorized in 50% ethanol 3 times for 5 min, washed in PBS, and subsequently stained with DAPI for 5 min.
Western blot
Cerebral cortex tissue was rapidly dissected and homogenized at 4°C in a buffer containing 50 mM Tris, 0.25% SDS, 150 mM NaCl, 1% NP-40, and 1 mM EDTA and a protease inhibitor (pH 7.4). Then, the sample was placed into a tissue homogenizer and ground 5 times for 120 s each time at intervals of 10 s and a frequency of 60 hertz. Next, the homogenates were centrifuged at 4°C at 12000 x g for 10 min, and the supernatants were mixed with 4×SDS loading buffer, heated in boiling water for 10 min, and stored at – 20°C until western blot analysis was performed. The protein concentrations in the supernatants were estimated using a BCA protein assay kit (Cat: P0012, Beyotime, Jiangsu, China) according to the manufacturer’s instructions. The samples were subjected to 8% or 10% SDS– PAGE and transferred onto Immobilon-NC membranes (Cat: HATF00010, Millipore, USA). The membranes were blocked with 5% skimmed milk (Cat: 1148GR100, Beyotime, Jiangsu, China) and were then incubated with primary antibodies (Table 3) at 4°C overnight. The blots were washed three times for 10 min in Tris-buffered saline supplemented with 0.25% Tween (TBST) and developed with the corresponding HRP-conjugated secondary antibody for one hour at 37°C. Protein expression was then visualized with an enhanced chemiluminescence (ECL) kit (Cat: WBKIS0100, Millipore, Bedford, MA, USA), and densitometry analysis was performed with ImageJ (NIH v1.51 n).
Primary antibodies used in this study
WB, western blot; IF, immunofluorescence; mAb, monoclonal antibody; pAb, polyclonal antibody.
Transmission electron microscopy
The hippocampus was quickly fixed in electron microscopy fixative solution (Cat: G1102, Servicebio, Wuhan, China) at 4°C for 3 h, rinsed with 0.1 M phosphoric acid buffer PB (pH 7.4) 3 times for 15 min each. Then, 1% osmium (0.1 M) and phosphoric acid buffer PB (pH 7.4) was added and incubated at room temperature (20°C) for 2 h; then, the tissue was rinsed 3 times in 0.1 M phosphoric acid buffer PB for 15 min each. Then, the tissues were dehydrated in alcoholic solutions of 50%, 70%, 80%, 90%, 95%, 100%, and 100% acetone. Next, the tissue was placed into a solution of acetone and 812 embedding agents (Cat: 90529-77-4, SPI Supplies, USA) (1:1) for 3 h and a solution of acetone and 812 embedding agents (1:2) overnight for infiltration. D 2, 812 embedding agent was used for 7 h, 812 embedding agent was added to the embedding plate, and the sample was inserted into the embedding plate and placed in a drying box at 37°C. On Day 3, the samples were placed in an oven and polymerized at 60°C for 48 h, cut into slices of 60– 80 nm using an ultrathin slicer (Leica, UC7, Germany), stained with 2% uranium-acetate saturated alcohol solution and lead citrate, and dried overnight at room temperature. Finally, images were taken by using a HITACHI transmission electron microscope.
Co-immunoprecipitation (Co-IP) assay
Co-IP assay was performed using an immunoprecipitation (IP/Co-IP) Kit (Cat: abs955, Absin, China). All procedures were performed according to the manufacturer’s instructions. Five microgram of appropriate antibody was incubated overnight at 4°C with 5 mg of brain lysate. Add 5μl Protein A and 5μl Protein G and incubate the mixture overnight at 4°C. Centrifuge at 12,000 g for 1 min at 4°C, save the pellet and wash 3 times with 500μl wash water. Add 40μl of 1xSDS sample buffer to resuspend the pellet, heat to boil at 100°C for 5 min, then briefly centrifuge at 14,000 g for 1 min and remove the supernatant. Immune complexes were then separated by SDS-PAGE and immunoblotted with appropriate antibodies.
Statistical analysis
Statistical analyses were conducted using SPSS version 22.0 (\copyright SPSS Inc., IL, USA) and GraphPad Prism version 8.0 (GraphPad Software Inc.; La Jolla, CA, USA). The data are presented as the mean±standard error of the mean (S.E.M.). The normality of the data was determined by using the Kolmogorov– Smirnov test. Within-group comparisons were performed using Unpaired two-tailed t tests. For comparisons among more than two groups, one-way ANOVA was used with Tukey’s post hoc tests for multiple comparisons. Two-way ANOVA was used for comparisons among more than two groups to investigate the relative effects of treatment and genotype. p < 0.05 were considered statistically significant.
RESULTS
The expression of mGluR5 increased with Aβ in 5XFAD mice
To investigate the role of mGluR5 in different stages of AD, we measured the expression of Aβ in 1-, 4-, 6-, and 12-month-old 5XFAD mice by immunostaining (Fig. 1A). We found that the number of Aβ plaques increased after 4 months in 5XFAD mice (Fig. 1B). With the increase in Aβ, the protein expression of mGluR5 in 6-month-old 5XFAD mice and 12-month-old 5XFAD mice was higher than that in WT mice at the same age (Fig. 1C, D). The binding of Aβ and Prion protein (PrP) activates mGluR5 downstream pathways, and we used immunoprecipitation to examine the binding of mGluR5 and PrP in 6-month-old WT mice and 5XFAD mice. The results showed that the binding of mGluR5 and PrP was enhanced in 6-month-old 5XFAD mice relative to WT mice. (Fig. 1E).
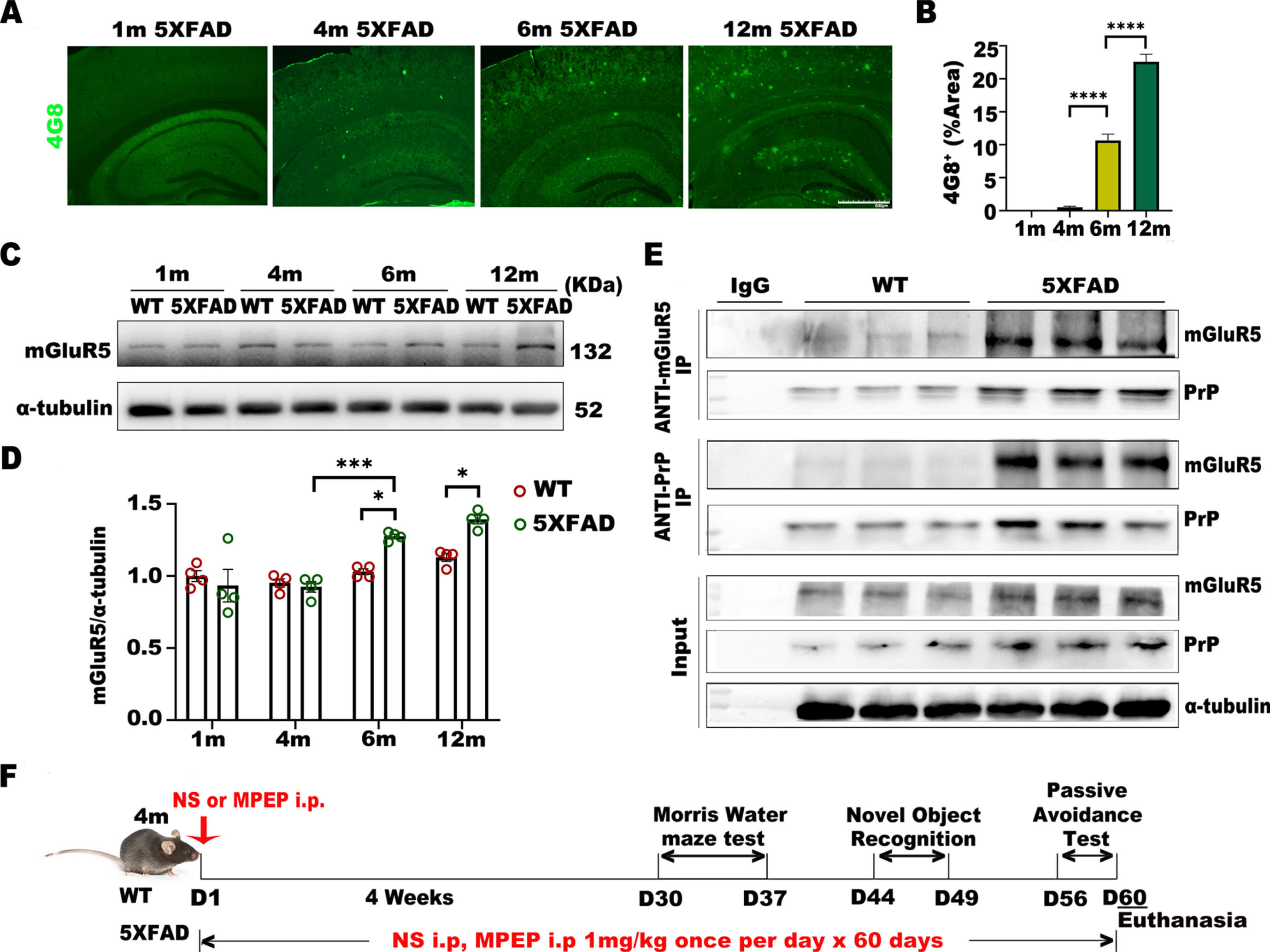
mGluR5 expression increases with amyloid plaque deposition in 5XFAD mice. A, B) Representative images of brain plaque deposition (4G8) in 1-month, 4-month, 6-month, and 12-month-old 5XFAD mice. Scale bar: 500μm. One-way ANOVA with Tukey’s post hoc test, F (3,8) = 587.5, p < 0.0001. 1-month versus 4-month, p = 0.8363; 4-month versus 6-month, p < 0.0001; 6-month versus 12-month, p < 0.0001. C, D) Western blot analysis of mGluR5 in 5XFAD mice and WT mice. Densitometric quantification of blots after normalization to α-tubulin. Two-way ANOVA with Tukey’s post hoc test. Interaction, F (3, 24) = 6.582, *p = 0.0021; Month, F (3, 24) = 20.31, *p < 0.0001; Genotype, F (1, 24) = 9.258, *p = 0.0056. 6-month-old 5XFAD versus 6-month-old WT, *p = 0.0385; 12-month-old 5XFAD versus 12-month-old WT, *p = 0.0210; 6-month-old 5XFAD versus 4-month-old 5XFAD, *p = 0.0008; 12-month-old 5XFAD versus 6-month-old 5XFAD, p = 0.9485. E) The specificity of mGluR5 interactions with PrP was examined by coimmunoprecipitation. F) Experimental scheme. 5XFAD or WT mice were treated with MPEP or normal saline for 60 consecutive days beginning at 4 months of age. All mice were subjected to a Morris water maze test, novel object recognition test, and passive avoidance test consecutively from Day 30 and were euthanized at 6 months. Half of the cerebral hemisphere homogenate was used for biochemical analysis, and half of the cerebral hemisphere was used for immunostaining. Data were analyzed by one-way or two-way ANOVA and were shown as Mean±SEM. n = 4 mice/ per genotype / per month. *p < 0.05, **p < 0.01, ***p < 0.001.
To explore how antagonizing mGluR5 affects cognitive function and pathological changes in 5XFAD mice, we infused MPEP (the negative allosteric modulator of mGluR5) or normal saline into 4-month-old 5XFAD and WT mice for 60 consecutive days. Behavioral tests were performed beginning on Day 30, and all mice were euthanized after 6 months of age (Fig. 1F).
The mGluR5 antagonist reversed cognitive deficits in 5XFAD mice
To determine whether antagonizing mGluR5 can improve cognitive function in 5XFAD mice, we examined long-term learning and memory with the Morris water maze. During the spatial probe test, it was observed that the incubation period in each group decreased gradually with training. On Day 6 of training, the latency of 5XFAD mice was significantly longer than that of WT mice, and the latency of 5XFAD mice injected with MPEP was significantly shorter than that of 5XFAD control mice (Fig. 2A). In the place navigation test, 5XFAD mice spent less time in the target quadrant (Fig. 2B) and had fewer target crossings (Fig. 2D) than WT mice, whereas 5XFAD+MPEP mice had more target crossings than 5XFAD mice. The classic trajectory diagram is shown in (Fig. 2C).
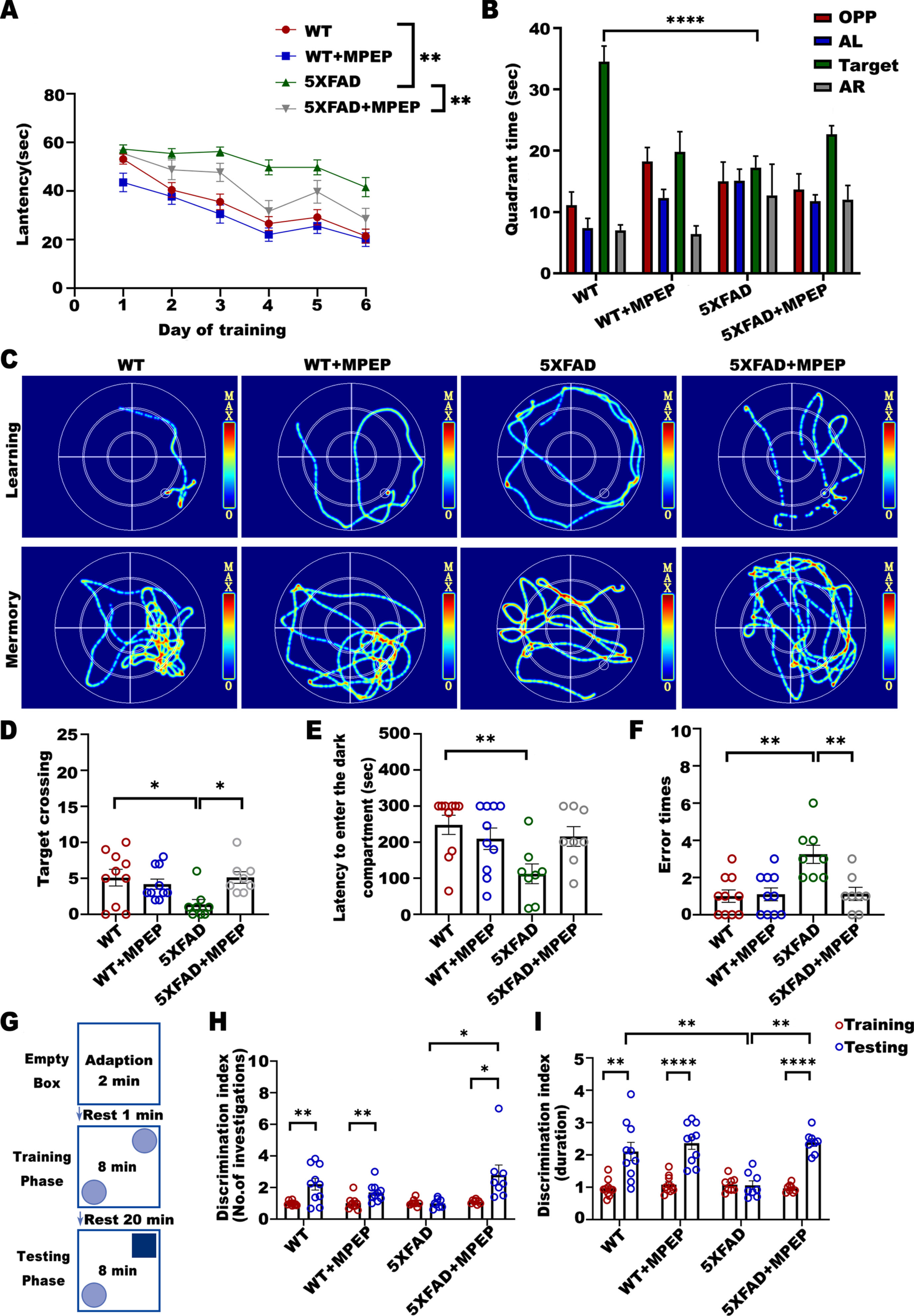
MPEP improves cognitive performance in 6-month-old 5XFAD mice. The Morris water maze test (A-D), passive avoidance test (E, F), and novel object recognition test (G-I) were carried out consecutively from Days 30 to 60. A) Escape latency on day 6 of MWM training. One-way ANOVA with Tukey’s post hoc test, F (3, 140) = 6.833, p = 0.0002. WT versus 5XFAD, *p = 0.0016; 5XFAD versus 5XFAD + MPEP, *p = 0.0043. B) Target quadrant time during probe trial phase of MWM task. One-way ANOVA with Tukey’s post hoc test, F (3, 32) = 11.19, p < 0.0001. WT versus 5XFAD, *p < 0.0001; 5XFAD versus 5XFAD + MPEP, p = 0.4069. C) Representative images of the swimming paths. D) The number of crossings the target. One-way ANOVA with Tukey’s post hoc test, F (3, 32) = 3.585, p = 0.0243. WT versus 5XFAD, *p = 0.0307; 5XFAD versus 5XFAD + MPEP, *p = 0.0416. E) Latency to enter the dark compartment in the Passive Avoidance Test. One-way ANOVA with Tukey’s post hoc test, F (3, 32) = 4.108, p = 0.0142. WT versus 5XFAD, *p = 0.0095; 5XFAD versus 5XFAD + MPEP, p = 0.0865. F) Error times in the Passive Avoidance Test. One-way ANOVA with Tukey’s post hoc test, F (3, 32) = 7.653, p = 0.0005. WT versus 5XFAD, *p = 0.0012; 5XFAD versus 5XFAD + MPEP, *p = 0.0038. G) Novel object recognition experiment setup. H) Discrimination index in the testing phase was improved in 5XFAD+MPEP mice. Number of contacting: Two-way ANOVA with Tukey’s post hoc test. Interaction, F (1, 32) = 10.11, *p = 0.0033; Treatment, F (1, 32) = 3.024, p = 0.0916; Genotype, F (1, 32) = 0.01972, p = 0.8892. WT versus 5XFAD, p = 0.1085; 5XFAD versus 5XFAD + MPEP, *p = 0.0121. Statistical number of explorations before and after training by unpaired t test: WT, *p = 0.0040, t = 3.294, df = 18; WT+MPEP, *p = 0.0068, t = 3.056, df = 18; 5XFAD, p = 0.6906, t = 0.4063, df = 14; 5XFAD+MPEP, *p = 0.0198, t = 2.630, df = 14. I) Duration: Two-way ANOVA with Tukey’s post hoc test. Interaction, F (1, 30) = 5.431, *p = 0.0267; Treatment, F (1, 30) = 12.08, *p = 0.0016; Genotype, F (1, 30) = 5.255, *p = 0. 0291.WT versus 5XFAD, *p = 0.0094; 5XFAD versus 5XFAD + MPEP, *p = 0.0038. Statistical duration before and after training by unpaired t test: WT, *p = 0.0012, t = 3.836, df = 18; WT+MPEP, *p < 0.0001, t = 6.095, df = 18; 5XFAD, p = 0.9124, t = 0.1121, df = 14; 5XFAD+MPEP, *p < 0.0001, t = 8.742, df = 10. Data were shown as Mean±SEM. n = 10 for WT; n = 10 for WT + MPEP; n = 8 for 5XFAD; n = 8 for 5XFAD + MPEP. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we examined learning and memory ability with the passive avoidance test. The latency to enter the dark compartment in the 5XFAD group was shorter than that in the WT group. 5XFAD mice injected with MPEP had significantly improved memory function, and the error times were lower than 5XFAD mice (Fig. 2E, F).
Novel object recognition was also performed to test cognitive function (Fig. 2G). No statistical differences were observed among the groups when the mice explored the two objects during the training period. During the testing phase, both the number of investigations and duration by 5XFAD mice was less than that by WT mice; whereas MPEP-treated 5XFAD mice were more inquisitive towards the novel object during the testing phase, as did the contact duration (Fig. 2H, I).
Antagonizing mGluR5 reduced damage to synaptic structure in 5XFAD mice
Synaptic defects are characteristics of AD pathology and are directly related to cognitive function [13]. To explore whether and how MPEP affects synaptic function, we used western blot to measure related synaptic proteins and neuron-associated proteins (Fig. 3A). The protein expression of PSD95 and synaptophysin in 5XFAD mice was significantly lower than that in WT mice. However, the expression of PSD95 and synaptophysin was significantly higher in 5XFAD mice administrated MPEP than in 5XFAD mice (Fig. 3B, C). Additionally, to verify whether the neurons of 5XFAD mice were improved after MPEP administration, we measured the expression of NeuN (neuronal nucleoprotein) and MAP2 (microtubule associated protein 2) (Fig. 3D, E), but in 5XFAD mice treated with MPEP, no statistical differences were observed. Then, we used electron microscopy to observe synaptic structure (Fig. 3F). In WT mice, we observed a clear basic synaptic structure and a large number of synapses per unit field of view. The dense regions of the presynaptic membrane and postsynaptic membrane were symmetrically distributed with uniform thickness. In 5XFAD mice, the synapse had severe oedema, the synaptic structure was severely damaged, a large area of the cell membrane had low electron density, microfilaments and microtubules in the cells disappeared, and the dense area of the presynaptic membrane and postsynaptic membrane became short, narrow, and sparse. In 5XFAD mice treated with MPEP, the synaptic structure and the synaptic gap recovered to some extent. These data certainly suggested that antagonizing mGluR5 with MPEP treatment in 5XFAD mice was efficient in rescuing synaptic deficits.

MPEP rescues synaptic loss and structure abnormalities in 5XFAD mice. A) Representative western blot showing PSD95, Synaptophysin, MAP2, NeuN, and GAPDH. B-E) Densitometric quantification of these proteins after normalization to GAPDH. 5XFAD mice showed marked decreases in the levels of PSD95 and synaptophysin, while MPEP prevented PSD95 and synaptophysin deficits, no significant change in NeuN or MAP2 was found. PSD95: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 7.587, p = 0.0014. WT versus 5XFAD, *p = 0.0160; 5XFAD versus 5XFAD + MPEP, *p = 0.0041. Synaptophysin: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 9.833, p = 0.0003. WT versus 5XFAD, *p = 0.0022; 5XFAD versus 5XFAD + MPEP, *p = 0.0009. NeuN: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 0.9488, p = 0.4359. WT versus 5XFAD, p = 0.4826; 5XFAD versus 5XFAD + MPEP, p = 0.8243. MAP2: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 1.550, p = 0.2325. WT versus 5XFAD, p = 0.6976; 5XFAD versus 5XFAD + MPEP, p = 0.4470. F) Representative transmission electron microscopy images showing the synaptic structure in each group of mice. The red arrowheads indicate the postsynaptic membrane (PD) and synaptic cleft (SC). Other remarks: Mitochondria (M), presynaptic membrane (PM), synaptic vesicles (SV), and synaptic corpuscles (SJ). Photomicrographs were taken at×15,000. Scale bar, 500 nm. Data were analyzed by one-way ANOVA and were shown as Mean±SEM. n = 6 for WT; n = 6 for WT + MPEP; n = 6 for 5XFAD; n = 6 for 5XFAD + MPEP. *p < 0.05, **p < 0.01, ***p < 0.001.
Antagonizing mGluR5 alleviated the pathological feature in 5XFAD mice
To further confirm whether MPEP ameliorates AD pathology, some relevant indicators were examined. Plaques and tangles share a common beta-plate-rich structure and can be stained in brain tissue with beta plate-binding dyes, such as thioflavin S (ThS) [14]; 6-month-old 5XFAD and 5XFAD+MPEP mouse brain sections were stained with ThS and the anti-Aβ monoclonal antibody 4G8. ThS-positive and 4G8-positive Aβ aggregates were observed in 5XFAD mice; however, the areas of ThS-positive plaques and Aβ plaques in 5XFAD+MPEP mice were markedly smaller than those in 5XFAD mice (Fig. 4A, B). Moreover, the phosphorylation level of APP (p-APP) at Thr668 in 5XFAD+MPEP mice was significantly lower than that in 5XFAD mice (Fig. 4C, D). To determine the effect of the mGluR5 antagonist MPEP on tau pathology, we performed western blot to test the expression of total and hyperphosphorylated tau. Treatment with MPEP decreased the level of tau hyperphosphorylation at serine 199/202 and threonine 231 in 5XFAD mice (Fig. 4E, F). These data demonstrated that 5XFAD mice that received MPEP showed improvement in some typical pathological changes of AD.
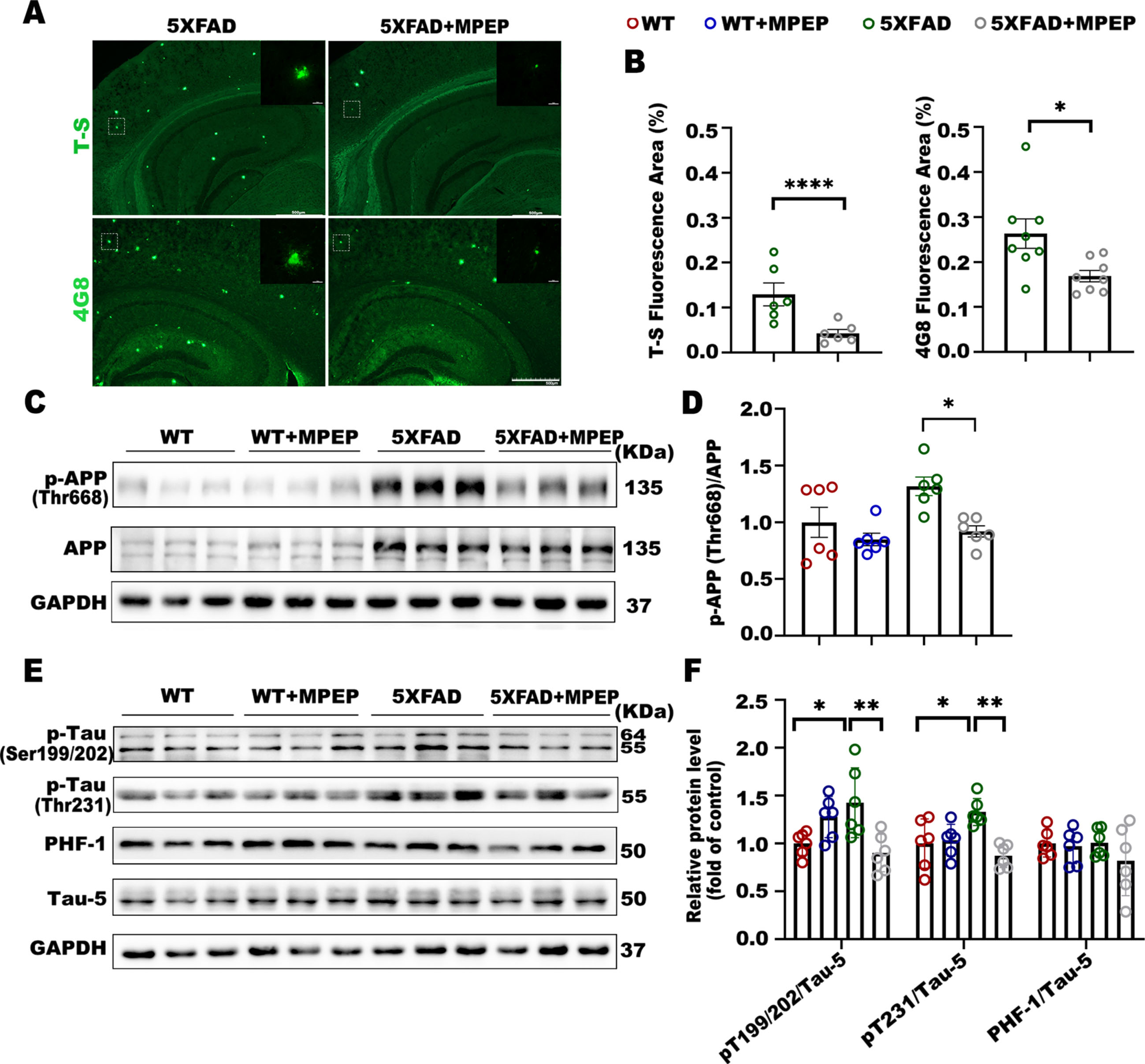
MPEP reverses Aβ and tau pathology in 6-month-old 5XFAD mice. A, B) Representative images of brain plaque deposition in 6-month-old 5XFAD and 5XFAD+MPEP mice. Scale bar, 500μm (x50); 10μm (x1000). Percentage of brain area covered in Thioflavin-S staining plaques in 5XFAD (n = 6 sections) and 5xFAD+MPEP (n = 6 sections), unpaired t test, *p < 0.0001, t = 7.398, df = 10. Percentage of brain area covered in amyloid plaques (4G8 staining) in 5XFAD (n = 8 sections) and 5xFAD+MPEP (n = 8 sections), unpaired t test, *p = 0.0176, t = 2.691, df = 14. C, D) Representative western blot showing p-APP, APP, and GAPDH. MPEP rescued the phosphorylation level of APP at Thr668 in 5XFAD mice. One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 5.760, p = 0.0052. WT versus 5XFAD, p = 0.0733; 5XFAD versus 5XFAD + MPEP, *p = 0.0188. E, F) Western blot analysis of tau pathology in WT, WT+MPEP, 5XFAD, and 5XFAD+MPEP mice. Densitometric quantification of the blots after normalization to total tau, Tau-5. Treatment of 5XFAD mice with MPEP decreased the level of phospho-Tau at Ser199/202 and Thr231. P-Tau (Ser199/202): One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 6.087, p = 0.0041. WT versus 5XFAD, *p = 0.0290; 5XFAD versus 5XFAD + MPEP, *p = 0.0061. P-Tau (Thr231): One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 6.977, p = 0.0021. WT versus 5XFAD, *p = 0.0226; 5XFAD versus 5XFAD + MPEP, *p = 0.0014. PHF-1: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 0.9141, p = 0.4519. WT versus 5XFAD, p > 0.9999; 5XFAD versus 5XFAD + MPEP, p = 0.4892. Data were shown as Mean±SEM. n = 6 for WT; n = 6 for WT + MPEP; n = 6 for 5XFAD; n = 6 for 5XFAD + MPEP. *p < 0.05, **p < 0.01, ****p < 0.0001.
Antagonizing mGluR5 with MPEP reduced neuroinflammation in 5XFAD mice
Neuroinflammation caused by abnormal glial activation drives the pathogenesis and progression of disorders such as AD [15]. To explore whether mGluR5 antagonism was associated with neuroinflammation, we measured the markers of astrocytes and microglia by western blot (Fig. 5A). The expression levels of glial fibrillary acidic protein (GFAP) (Fig. 5B) and Ionized calcium binding adaptor molecule 1 (Iba1) (Fig. 5C) were significantly increased in 5XFAD mice compared to wild-type mice, and these levels decreased in the 5XFAD+MPEP. Tumor necrosis factor alpha (TNF-α) and Interleukin 1 beta (IL-1β) are two key inflammatory factors; therefore, we also examined whether these factors were affected by antagonizing mGluR5. The protein expression of TNF-α (Fig. 5D) and IL-1β (Fig. 5E) in MPEP-treated 5XFAD mice was significantly lower than that in 5XFAD\enlargethispage 2pt mice. Then, we used immunostaining to examine the distribution and reactivity of GFAP in brain tissue (Fig. 5F). The expression and reactivity of GFAP in 5XFAD+MPEP mice was lower than that in 5XFAD mice (Fig. 5G). In conclusion, MPEP effectively reduced the activation of glial cells and the occurrence of neuroinflammation, thus downregulating the expression of inflammatory factors in 5XFAD mice.
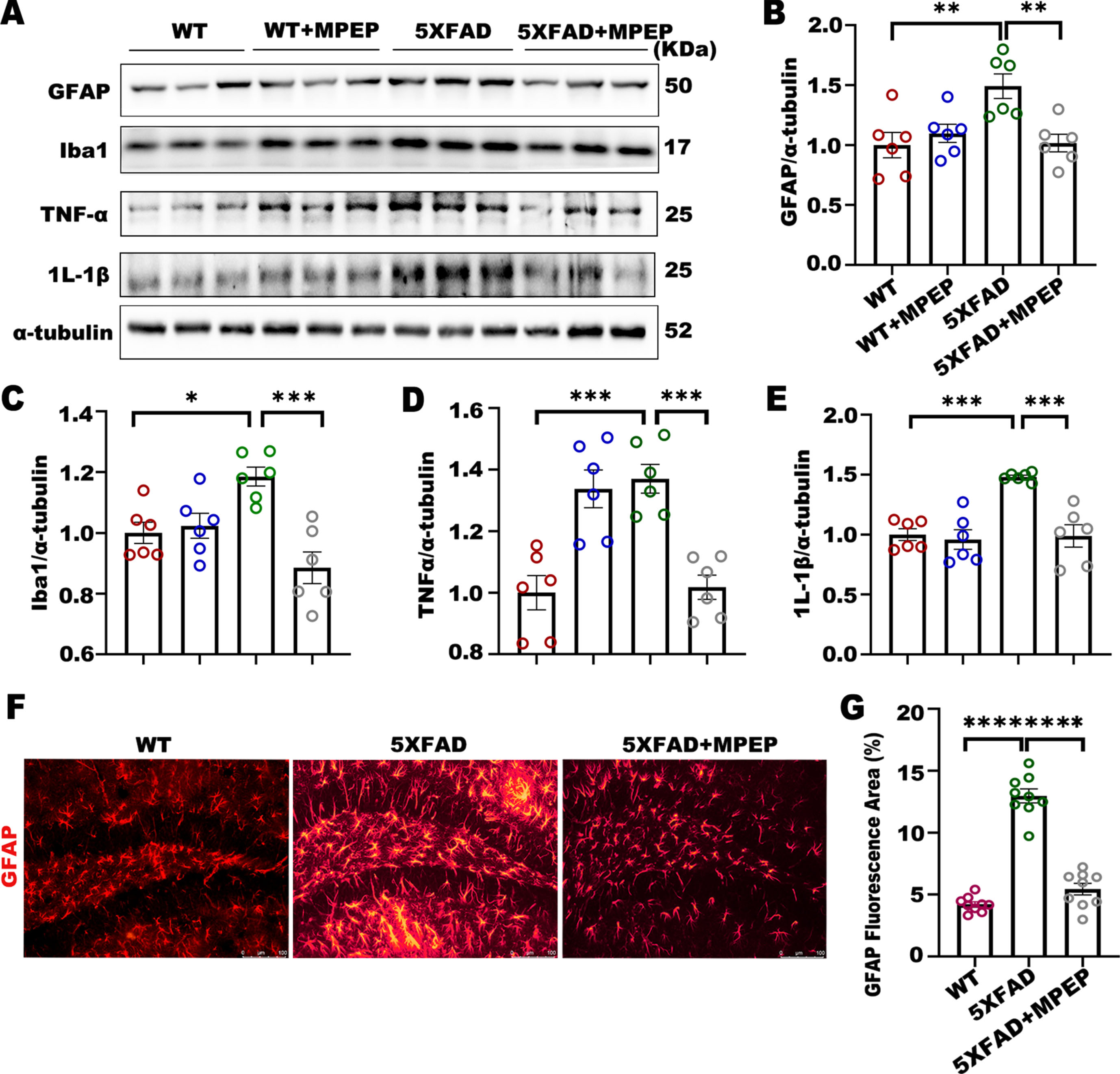
MPEP prevents neuroinflammation at 6 months of age in 5XFAD mice. A) Representative western blot showing GFAP, Iba1, TNF-α, and IL-1β and their quantification after normalization to α-tubulin. B-E) 5XFAD mice showed marked increases in GFAP, Iba1, TNF-α, and IL-1β compared to WT mice, and treatment with MPEP significantly decreased these levels. GFAP: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 6.510, p = 0.0030. WT versus 5XFAD, *p = 0.0051; 5XFAD versus 5XFAD + MPEP, *p = 0.0070. Iba1: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 9.317, p = 0.0005. WT versus 5XFAD, *p = 0.0200; 5XFAD versus 5XFAD + MPEP, *p = 0.0002. TNF-α: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 15.12, p < 0.0001. WT versus 5XFAD, *p = 0.0003; 5XFAD versus 5XFAD + MPEP, *p = 0.0005. IL-1β: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 14.02, p < 0.0001. WT versus 5XFAD, *p = 0.0003; 5XFAD versus 5XFAD + MPEP, *p = 0.0002. F, G) Representative image showing GFAP immunostaining in the hippocampi of 6-month-old WT (n = 9 sections), 5XFAD (n = 9 sections), and 5XFAD+MPEP mice (n = 9 sections); marked GFAP staining was observed in 5XFAD mice compared to the WT mice, while MPEP attenuated this change. Scale bar, 100μm (x100). One-way ANOVA with Tukey’s post hoc test, F (2, 24) = 117.2, p < 0.0001. WT versus 5XFAD, *p < 0.0001; 5XFAD versus 5XFAD + MPEP, *p < 0.0001. Data were analyzed by one-way ANOVA and were shown as Mean±SEM. n = 6 for WT; n = 6 for WT + MPEP; n = 6 for 5XFAD; n = 6 for 5XFAD + MPEP. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
mGluR5 knockdown alleviated pathological features in 5XFAD mice
To verify the effect of antagonizing mGluR5 in 5XFAD mice, we further evaluated by crossing 5XFAD transgenic mice with mGluR5-null mice. First, we examined p-APP levels, and the findings were consistent with our previous experiments, p-APP at Thr668 in 5XFAD/mGluR5(+/-) mice was markedly lower than that in 5XFAD mice (Fig. 6A, B). Similarly, Aβ plaque distribution was also significantly reduced (Fig. 6C, D). Next, we examined the expression of phospho-Tau (p-Tau) and confirmed that the expression of pT199/202 in 5XFAD/mGluR5(+/-) mice was markedly lower than that in 5XFAD mice, but pT231 and PHF-1 expression did not change significantly (Fig. 6E, F). Inhibition of neuroinflammation was also validated in 5XFAD/mGluR5(+/-) mice. The protein expression of GFAP and Iba1 was significantly lower in 5XFAD/mGluR5(+/-) mice than in 5XFAD mice, as was the expression of TNF-α and IL-1β (Fig. 6G, H).

mGluR5 knockdown alleviated pathological features in 5XFAD mice. mGluR5 knockout alleviates AD-related pathology and ameliorates neuroinflammation in 5XFAD mice. A, B) Representative western blot showing mGluR5, p-APP, and APP. Densitometric quantification of these proteins after normalization to GAPDH and APP. 5XFAD mice showed a marked increase in p-APP at Thr 668 compared with WT mice, while mGluR5 knockout decreased these levels. p-APP/APP: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 7.583, p = 0.0014. WT versus 5XFAD, p = 0.0013; 5XFAD versus 5XFAD/mGluR5(+/-), *p = 0.0070. C, D) Immunofluorescence analysis showed decreases in the Aβ plaque areas in coronal brain sections from 5XFAD/mGluR5(+/-) mice at 6 months of age (×50. Scale bar, 500μm; magnification:x1000. Scale bar, 10μm). Percentage of brain area covered in amyloid plaques (4G8 staining) in 5XFAD (n = 8 sections) and 5xFAD+MPEP (n = 8 sections), unpaired t test, *p = 0.0042, t = 3.409, df = 14. E, F) Western blot analysis of tau pathology in WT, WT/mGluR5(+/-), 5XFAD, and 5XFAD/mGluR5(+/-) mice. Densitometric quantification of the blots after normalization to total tau (Tau 5). mGluR5 knockout reduced tau phosphorylation at sites 202 and 231; there was no decrease in PHF-1 in 5XFAD/mGluR5(+/-) mice compared to 5XFAD mice. G, H) Representative western blot showing GFAP, IBA1, TNF-α, and IL-1β and their quantification after normalization to α-tubulin in the four groups. 5XFAD mice showed marked increases in GFAP, IBA1, TNF-α, and IL-1β, whereas 5XFAD/mGluR5(+/-) mice showed significantly decreased levels of these proteins. Data were analyzed by one-way ANOVA and were shown as Mean±SEM. n = 6 for WT; n = 6 for WT/mGluR5(+/-); n = 6 for 5XFAD; n = 6 for 5XFAD/mGluR5(+/-). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Antagonizing mGluR5 activated autophagy and anti-inflammatory by inhibiting PI3K/AKT/mTOR pathway
mGluR5 exerted its neuroprotective effects via the Phosphatidylinositol-3-kinase (PI3K)/AKT pathway [16]. Therefore, to examine whether the expression of PI3K and AKT in brain tissues was affected by MPEP, we measured the expression of the p85 regulatory subunit and p110 catalytic subunit of PI3K and p-AKT respectively (Fig. 7A). We found no significant difference in the expression of p110, but the protein expression of p85 were significantly lower than those in 5XFAD mice. Mammalian target of rapamycin (mTOR) as a serine/threonine protein kinase, is distributed downstream of AKT. Western blot analysis revealed that there were no significant differences in the total amount AKT and mTOR between the 5XFAD group and the 5XFAD+MPEP group, while protein phosphorylation levels decreased sharply and significantly in MPEP-treated 5XFAD mice (Fig. 7B). We also examined autophagy marker proteins (Fig. 7C), and it turned out that the protein level of LC3-II/I and beclin1 was increased significantly in the 5XFAD+MPEP group. Simultaneously, treatment with MPEP decreased p62 in compared to that in 5XFAD mice. In addition, the protein level of B-cell lymphoma-2 (Bcl-2) was increased in the 5XFAD+MPEP group (Fig. 7D). Our results showed that the inflammatory factors IL-1β and TNF-α were downregulated after the administration of MPEP in 5XFAD mice. IκB kinase (Ikappa B kinase or IKK) is a downstream signaling molecule of AKT. NF-κB is a major regulator of inflammation and is involved in a variety of inflammatory pathways [17]. To explore how MPEP regulated inflammation, we examined the IKK/NF-κB signaling system (Fig. 7E). We confirmed that the phosphorylation levels of IKK (p-IKK) and NF-κB (p-P65) in MPEP-treated 5XFAD mice were significantly lower than those in 5XFAD mice (Fig. 7F). Based on these results, we speculated that the antagonizing mGluR5 could activate anti-inflammatory pathways in 5XFAD mice, which may protect neuronal synapses and ameliorate cognitive dysfunction.

MPEP activated autophagy and anti-inflammatory by inhibiting PI3K/AKT/mTOR pathway. A, B) Representative western blot showing p110, p85, PI3K, p-AKT, AKT, and GAPDH. Densitometric quantification of these proteins after normalization to GAPDH, PI3K, and AKT. There was no decrease in p110 in 5XFAD+MPEP mice, but p85 was reduced in the 5XFAD+MPEP group compared to 5XFAD control group. p110/PI3K: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 0.2685, p = 0.8473. WT versus 5XFAD, p > 0.9999; 5XFAD versus 5XFAD + MPEP, p > 0.9999. p85/PI3K: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 9.616, p = 0.0004. WT versus 5XFAD, p = 0.0767; 5XFAD versus 5XFAD + MPEP, *p = 0.0015. Treatment with MPEP significantly decreased the levels of p-AKT. p-AKT/AKT: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 11.38, p = 0.0001. WT versus 5XFAD, p = 0.9163; 5XFAD versus 5XFAD + MPEP, *p = 0.0008. C, D) Representative western blot showing p-mTOR, mTOR, LC3II/I, p62, beclin1, Bcl-2, and GAPDH. Densitometric quantification of these proteins after normalization to GAPDH and mTOR. MPEP significantly decreased the levels of p-mTOR in 5XFAD mice, and elevated the level of LC3II/I, beclin1 and Bcl-2 in 5XFAD mice and suppressed the expression of p62 protein. p-mTOR/mTOR: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 8.474, p = 0.0008. WT versus 5XFAD, p = 0.2451; 5XFAD versus 5XFAD + MPEP, *p = 0.0007. LC3II/I: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 12.73, p < 0.0001. WT versus 5XFAD, p = 0.9578; 5XFAD versus 5XFAD + MPEP, *p = 0.0006. p62: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 16.44, p < 0.0001. WT versus 5XFAD, p > 0.9999; 5XFAD versus 5XFAD + MPEP, *p = 0.0003. beclin1: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 60.48, p < 0.0001. WT versus 5XFAD, *p = 0.0161; 5XFAD versus 5XFAD + MPEP, *p < 0.0001. Bcl-2: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 13.15, p < 0.0001. WT versus 5XFAD, p = 0.9745; 5XFAD versus 5XFAD + MPEP, *p < 0.0001. E, F) Representative western blot showing p-IKK, IKK, p-P65, P65 (NF-KB), and GAPDH. Densitometric quantification of these proteins after normalization to GAPDH, IKK, or P65. MPEP treatment significantly decreased the levels of p-IKK, and p-P65. P-IKK/IKK: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 9.916, p = 0.0003. WT versus 5XFAD, p = 0.1088; 5XFAD versus 5XFAD + MPEP, *p = 0.0001. p-P65/P65: One-way ANOVA with Tukey’s post hoc test, F (3, 20) = 26.83, p < 0.0001. WT versus 5XFAD, p = 0.4630; 5XFAD versus 5XFAD + MPEP, *p < 0.0001. Data were analyzed by one-way ANOVA and were shown as Mean±SEM. n = 6 for WT; n = 6 for WT + MPEP; n = 6 for 5XFAD; n = 6 for 5XFAD + MPEP. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DISCUSSION
Previous studies have shown that mGluR5 was age-dependent in 5xFAD mice as measured by PET [18]. In this study, we provided experimental evidence that mGluR5 expression increased in 5XFAD mice as Aβ levels increased. Moreover, the expression of mGluR5 in 6-month-old and 12-month-old 5XFAD mice was higher than that in WT mice of the same age. Aβo-PrPC-mGluR5 complexes activate mGluR5 signaling and downstream pathways. Aβ oligomers were previously reported to activate mGluR5 and promote mGluR5 clustering at synapses [19]. The mGluR5 pathway may alter Aβ in a feedback manner to exacerbate AD and enhance Aβ production [20]. Blocking the binding of Aβ to PrP reverses memory deficits in transgenic AD mice [21]. In this study, with the increase of Aβ in 5XFAD mice, we observed that specificity of mGluR5 interactions with PrP were higher than those in WT mice of the same age.
Cognitive dysfunction in 5XFAD mice is predicted to be an age-dependent effect, and there is a progressive loss of learning and memory from 4 to 12 months of age as Aβ plaques develop [22]. The noncompetitive selective mGluR5 antagonist 2-methyl-6-(phenylethynly)-pyridine (MPEP) is a negative allosteric regulator of mGluR1 and mGluR5 that binds to a site other than glutamate within the seven-transmembrane region. A previous study showed that MPEP was centrally active following peripheral administration and penetrates the blood-brain barrier, antagonizing excitations mediated by agonists of Group I mGlu receptors in vivo [23]. In this study, MPEP was selected to antagonize the mGluR5 signaling. We found that MPEP reversed cognitive deficits and improved long-term and short-term memory in 6-month-old 5XFAD mice. Our results confirm that mGluR5 may be a target for AD intervention. Synaptic loss is a major hallmark and a significant feature of cognitive decline [24]. We proved that the protein expression of synaptophysin and PSD95 in 5XFAD mice that received MPEP was markedly increased, whereas NeuN (neuronal nucleoprotein) and MAP2 were not changed. These results suggested that mGluR5 antagonist MPEP may affect presynaptic and postsynaptic membranes but not neuronal cells.
Amyloid plaques formed by the deposition of Aβ outside neurons result in neurotoxicity to adjacent cells [25], and the hyperphosphorylation of tau protein in neurons [26] is directly related to learning, memory, and cognitive dysfunction. Tau and Aβ pathology occurred before the expected onset of clinical symptoms [27, 28]. After the onset of disease, treatment of neuronal degeneration becomes more difficult, and fine-tuned neuronal circuits and cognitive ability do not easily recover in the later stage of the disease [29]. The use of drugs for the early prevention and treatment of AD is an important research goal. Imbalance in Aβ production and clearance lead to Aβ accumulation. Autophagy is considered to be one of the most important physiological mechanisms for clearing Aβ [30, 31]. However, the accumulation of Aβ induces autophagy dysfunction in transgenic AD model mice [32]. mTOR promotes anabolic metabolism and inhibits autophagy. Therefore, the regulation of autophagy through mTOR provides a new therapeutic strategy for neurodegenerative diseases [33]. Our study results demonstrated that MPEP induced autophagy by downregulating the PI3K/AKT/mTOR pathway. Beclin1 is a well-known key regulator of autophagy, and it suppresses the autophagic process when it becomes inactive, in AD transgenic mice, reduction in beclin1 have been associated with increased Aβ accumulation [34]. Bcl-2 proteins in the mitochondria and ER are important regulators of autophagy and apoptosis [35]. We found that autophagy activation could lead to the decrease of p62 level, and the increase of LC3-II/I, beclin1, and Bcl-2 were observed in 5XFAD+MPEP mice compared with 5XFAD mice, which indicates that antagonizing mGluR5 in 5XFAD mice activates autophagy, thereby clearing the Aβ plaque load and protecting neurons from damage.
Studies showed that autophagy played a key role in cleaning the extracellular “garbage” like Aβ. Disturbing autophagy process leads to gliosis and neuroinflammation, which contribute significantly to the development and progression of AD. Neuroinflammation directly affect the stability and maintenance of synaptic contacts, providing a suitable microenvironment around active synapses but also exhibit a form of excitability based on intracellular Ca2 + increases, which lead to glutamate release [36, 37]. In neuroinflammation, microglial and astrocyte hyperplasia may also be chronic causes of weakened neuronal integrity and lead to tau protein degeneration in neurodegenerative diseases [38]. In this study, the expression levels of the astrocyte marker GFAP and the microglial marker Iba1 in 5XFAD+MPEP mice were both lower than that in transgenic AD mice, which confirmed that pharmacological blockade of mGluR5 with specific antagonist MPEP could significantly prevent neuroinflammation in 6-month-old 5XFAD mice. NF-κB induces the expression of various pro-inflammatory genes, including those encoding cytokines and chemokines, and also participates in inflammasome regulation. When inflammatory stimuli induce IκB phosphorylation through the IKK complex, the NF-κB heterodimer (p50/p65) freely translocate into the nucleus and trigger a cascade reaction that releases abundant inflammatory factors [39]. Our results indicated that antagonizing mGluR5 in 5XFAD mice may alleviate neuroinflammation by reducing the activation levels of IKK and NF-kB.
The present study showed a significant reduction In Aβ plaque load and neuroinflammation due to autophagy activation via PI3K/AKT and mTOR-dependent pathways. This is the first study that mGluR5 antagonism may be effective in reducing neuroglial activation and promoting synaptic recovery in 5XFAD mice, thereby ameliorate the progression of AD-related cognitive deficits. Our study provides a new basis for the future treatment of AD in 5xFAD mice model and the development of new drugs. However, further pharmacological studies are necessary to validate the utility of mGluR5 antagonists in the treatment of AD patients.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by the National Natural Science Foundation of China (No. 81100944), Natural Science Foundation of Guangdong Province (2018A030313565, 2019A1515010903, 2022A1515010077), and Guangzhou municipal Science and Technology Project (201904010040). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.