
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by memory loss and cognitive impairment. Despite its rapidly increasing global prevalence, effective disease-modifying therapies remain limited. Neuronal loss is a central pathological hallmark of AD, yet classical proteinopathy frameworks centered on amyloid-β (Aβ) deposition and tau hyperphosphorylation do not fully explain the extent and dynamics of neurodegeneration. Convergent upstream pressures—including Aβ/tau-associated proteotoxicity, mitochondrial dysfunction and oxidative stress, glucose hypometabolism/brain insulin resistance, and chronic neuroinflammation—lower the threshold for regulated neuronal death programs. Evidence from human postmortem brains and experimental AD models implicates multiple death modalities, including apoptosis, inflammasome-associated pyroptosis, cellular senescence with a senescence-associated secretory phenotype (SASP), and ferroptosis driven by iron-dependent lipid peroxidation. These signatures are often mixed and show region- and stage-dependent patterns, reflecting context- and model-specific drivers rather than mutually exclusive pathways. A crosstalk-and-convergence view highlights shared hubs—oxidative stress, mitochondrial failure, inflammasome/cytokine signaling, and SASP-mediated chronic inflammation—that connect these modalities through feed-forward loops, helping to explain the limited durability of single-pathway interventions. This review summarizes recent advances across these four pathways, discusses their mechanistic interplay, and outlines translational considerations (blood–brain barrier delivery, target specificity, and limited clinical evidence). We also highlight priorities for future work, including single-cell/spatial profiling, multi-omics integration, and biomarker-guided stratification to enable rational combination strategies.
Keywords
Introduction
Alzheimer's disease (AD), the most common chronic neurodegenerative disorder, poses a serious threat to the physical and mental health of the elderly population worldwide and has become a major global public health concern. Clinically, AD is characterized by progressive cognitive impairment and memory loss. Pathologically, it is marked by significant neuronal loss in brain tissue, extensive deposition of amyloid-β (Aβ) plaques, and the formation of neurofibrillary tangles (NFTs) resulting from hyperphosphorylation of tau protein.1–3 Epidemiological data indicate that, as of 2023, over 55 million individuals worldwide are living with AD. With the aging population, the number of new cases is projected to rise sharply, and the global prevalence of AD is expected to reach approximately 140 million by 2050. 4 However, current therapeutic strategies for AD are extremely limited. Most available drugs can only partially alleviate clinical symptoms, and no effective treatments have been developed to reverse or significantly delay the pathological progression of the disease. Therefore, an in-depth exploration of the pathogenic mechanisms of AD and the development of effective intervention strategies have become critical issues in the field of medical research. Notably, the persistent gap between prominent pathological hallmarks and limited disease-modifying efficacy highlights the need to re-examine neuronal loss through an integrated mechanistic framework, particularly from the perspective of regulated neuronal death programs and their convergence.
In the past, research on AD has mainly concentrated on two classical pathological processes: the aggregation of Aβ plaques and the abnormal hyperphosphorylation of tau protein, which leads to the formation of NFTs. 5 However, increasing evidence in recent years suggests that the traditional “amyloid cascade hypothesis” and “tau hypothesis” alone are insufficient to fully explain the complex pathological processes of AD. In particular, the diversity and complexity of neuronal cell death mechanisms have attracted growing attention. Studies have shown that the significant neuronal loss observed in AD patients is closely associated with multiple forms of cell death. These mechanisms include not only classical apoptosis but also emerging forms of programmed cell death that have been increasingly identified and studied in recent years, such as pyroptosis, cellular senescence, and ferroptosis.6–8 To contextualize these regulated neuronal death programs, we briefly summarize the core AD pathogenic mechanisms that impose shared upstream stress on vulnerable neural circuits.
Core pathogenic mechanisms of AD relevant to neuronal death
Amyloid cascade hypothesis
The “amyloid cascade hypothesis” posits that abnormal Aβ production/aggregation initiates a sequence of pathological events, including synaptic dysfunction, glial activation, oxidative injury, and ultimately neuronal loss. 9 Beyond plaque deposition, soluble Aβ species can impose proteotoxic stress and disrupt cellular homeostasis, thereby lowering the threshold for regulated cell death activation. 10
Tau phosphorylation/NFTs
In parallel, abnormal tau hyperphosphorylation and the formation of NFTs represent another central axis of AD pathogenesis. 11 Pathological tau disrupts microtubule stability and axonal transport, contributes to synaptic failure, and can propagate in a prion-like manner across neural circuits, thereby amplifying cellular stress and vulnerability to neuronal death. 12
Mitochondrial dysfunction and oxidative stress
Mitochondrial injury and bioenergetic failure are increasingly recognized as key contributors to AD progression. Impaired mitochondrial respiration, excessive reactive oxygen species (ROS) generation, and redox imbalance exacerbate oxidative damage to proteins, lipids, and DNA, creating permissive conditions for multiple death programs, including mitochondrial apoptosis and iron-dependent lipid peroxidation processes.13,14
Glucose hypometabolism/brain insulin resistance and neuroinflammation
Metabolic abnormalities, such as cerebral glucose hypometabolism and brain insulin resistance, can impair neuronal energy supply and stress-adaptive capacity, thereby sensitizing neurons to downstream injury. 15 Meanwhile, chronic neuroinflammation, driven by sustained microglial and astrocytic activation and accompanied by inflammatory mediators, forms a long-term pathological milieu that interacts with Aβ/tau pathology and reinforces oxidative and mitochondrial stress, further accelerating neurodegenerative cascades. 16
Importantly, original transcriptomic and experimental studies provide direct evidence that these pathogenic mechanisms are mechanistically intertwined rather than independent. Neuronal transcriptomic profiling in AD postmortem brains has repeatedly shown coordinated downregulation of energy-metabolism and mitochondrial programs in vulnerable cortical circuits, supporting a tight coupling between bioenergetic failure and oxidative-stress–related signatures.17,18 Consistently, experimental work indicates that Aβ can precipitate brain hypometabolism, network dysfunction, and behavioral abnormalities via ROS-generating pathways such as NOX2-linked oxidative stress, providing a causal bridge between Aβ proteotoxicity, redox imbalance, and metabolic suppression. 19 In parallel, Aβ has been reported to reprogram astrocytic glucose metabolism and insulin signaling in experimental AD settings, further reinforcing a glia-mediated metabolic axis that converges with neuroinflammation and neuronal vulnerability. 20 Metabolic stressors that mimic carbonyl/glycation burden (e.g., glyceraldehyde exposure) can also induce AD-like alterations in neuronal systems, supporting the convergence of metabolic dysregulation with classical AD pathology. 21 Moreover, direct perturbation of glucose uptake (e.g., the GLUT inhibitor WZB117) has been shown to induce cytotoxicity accompanied by increased Aβ production in neuronal cells, an effect preventable by β-hydroxybutyrate, further linking glucose restriction to amyloidogenic stress and neuronal injury. 22 Finally, modulation of cellular stress-response pathways that integrate metabolism with proteostasis, such as the integrated stress response, has been proposed as a mechanistic entry point to restore glucose metabolism and enhance neuronal resilience. 23 Together, these lines of evidence support viewing regulated neuronal death in AD as downstream of a coupled “proteotoxicity–metabolic failure–oxidative stress–inflammation” axis, which may manifest with region- and model-dependent dominance across death modalities.
Collectively, these core pathogenic mechanisms are not only hallmarks of AD but also represent shared upstream stressors that can initiate or amplify regulated neuronal death programs. 24 Aβ/tau-associated proteotoxicity, mitochondrial dysfunction/ROS, metabolic impairment, and chronic neuroinflammation jointly create a convergent pressure landscape that promotes apoptosis, inflammasome-associated pyroptosis, senescence/SASP-driven chronic damage, and ferroptosis.7,25,26 This perspective provides a mechanistic rationale for focusing on apoptosis, pyroptosis, cellular senescence, and ferroptosis in the sections below and for emphasizing their interactions rather than viewing them as isolated pathways.
Accordingly, the following sections summarize these four regulated neuronal death modalities in AD, apoptosis, pyroptosis, cellular senescence, and ferroptosis, highlighting their key molecular features and emerging interactions. As summarized in Figure 1, AD-associated stressors can engage multiple regulated neuronal death programs, including apoptosis (Figure 1A), pyroptosis (Figure 1B), cellular senescence (Figure 1C), and ferroptosis (Figure 1D).

Integrated overview of regulated neuronal death programs implicated in AD. Panels A–D summarize four regulated neuronal death modalities discussed in this review: (A) apoptosis (extrinsic and intrinsic pathways converging on executioner caspases), (B) pyroptosis driven by NLRP3 inflammasome activation and gasdermin-dependent pore formation, (C) cellular senescence with SASP-mediated inflammatory amplification, and (D) ferroptosis driven by iron-dependent lipid peroxidation and failure of antioxidant defenses.
To ground these mechanistic links in human disease, we next summarize postmortem evidence for regulated neuronal death signatures in AD brains and then compare these findings with evidence from experimental AD models and their limitations.
Postmortem evidence of regulated neuronal death in AD brains
Postmortem analyses of human AD brains provide an indispensable window into neuronal vulnerability and end-stage neuropathology, yet they also highlight a central challenge: signals of regulated neuronal death are often mixed, region- and stage-dependent, and influenced by comorbid pathology and perimortem factors. Accordingly, evidence supporting apoptosis, inflammasome-associated pyroptosis, senescence-like phenotypes, and ferroptosis in AD should be interpreted as overlapping signatures rather than mutually exclusive labels.27–29 These signatures can co-occur within the same brain region or even within the same cellular neighborhood, reflecting dynamic transitions across disease progression.
Apoptosis-related postmortem signatures
Human studies have reported increased apoptotic markers in AD-vulnerable regions (e.g., hippocampus and association cortex), including TUNEL positivity, DNA fragmentation, elevated cleaved caspase-3, and dysregulation of Bcl-2 family proteins, together with evidence consistent with mitochondrial apoptotic engagement (e.g., cytochrome c–related signaling and mitochondrial stress markers).30,31 Importantly, the abundance of classical apoptotic markers in postmortem tissue can vary substantially across cohorts and Braak/Thal stages, which may reflect both biological heterogeneity and limitations of detecting transient caspase activation in end-stage samples.32,33
Pyroptosis/inflammasome-related postmortem signatures
Inflammasome activation has been repeatedly implicated in AD brain tissue, particularly within microglia-enriched compartments, with reported increases in components such as NLRP3, ASC, and cleaved caspase-1, alongside elevated mature IL-1β/IL-18 in affected regions.26,34,35 Evidence for gasdermin-dependent execution (e.g., GSDMD cleavage products) has also been described in some studies, although detection may depend on tissue preservation, antibodies, and the timing of pathway activation relative to death.36,37 Overall, postmortem findings support a prominent role for inflammasome-associated neuroinflammation in AD, while the degree to which full pyroptotic membrane rupture occurs broadly in human neurons versus glial populations remains an area of ongoing investigation.
Senescence-like phenotypes and SASP-related postmortem signatures
Cellular senescence (or senescence-like states in post-mitotic neurons) has been increasingly reported in AD brains. Human studies have documented elevated expression of cell-cycle inhibitors (e.g., p16^INK4a and p21), DNA damage response markers (e.g., γH2AX), altered nuclear integrity markers (e.g., Lamin B1 reduction), and increased inflammatory and matrix-remodeling factors consistent with senescence-associated secretory phenotype (SASP) activity.38–40 Importantly, these features often appear in both neurons and glial cells, and their distribution can differ by brain region and disease stage. 41 Because many senescence markers are not uniquely specific and can overlap with stress responses, multi-marker panels and spatially resolved validation are essential when interpreting postmortem senescence-like signatures in AD.
Ferroptosis-related postmortem signatures
Evidence consistent with ferroptotic vulnerability in AD includes iron accumulation (e.g., histochemical iron deposition, altered ferritin/transferrin-related markers), elevated lipid peroxidation products (e.g., 4-HNE, MDA, and related oxidative adducts), and reported dysregulation of antioxidant defense components linked to ferroptosis susceptibility (e.g., reduced GPX4 activity/expression in some studies, altered System Xc− (SLC7A11/SLC3A2) components, and lipid remodeling-associated proteins).42–46 Notably, iron and lipid peroxidation signatures can be strongly influenced by regional myelination, vascular factors, and inflammatory milieu, which may help explain variability across human cohorts.43,45,47
Why postmortem signals are often “mixed”
Several factors can blur the assignment of a single death modality in human tissue: (i) late-stage sampling bias, where transient pathway markers may have peaked earlier; (ii) brain-region and cell-type specificity, with distinct neuronal subtypes and glial populations exhibiting different vulnerabilities; (iii) comorbid pathologies (vascular injury, Lewy pathology, metabolic disease) that shift inflammatory and oxidative contexts; and (iv) perimortem variables (agonal state, postmortem interval, fixation) that affect protein cleavage products and redox-sensitive readouts.48–54 Together, these considerations motivate a parallel evidence framework that integrates postmortem observations with mechanistic insights from experimental systems.
Evidence from experimental ad models and key limitations
Experimental AD models provide mechanistic tractability and temporal resolution, enabling causal testing of regulated death programs. However, model-specific construction can strongly bias the dominant death signature observed, contributing to discrepancies relative to human postmortem data.55–57 Critically, there is no ideal animal model that fully recapitulates sporadic AD, the most common clinical form. Most widely used transgenic lines are driven by familial AD–linked mutations (APP/PSEN1/PSEN2) and often rely on overexpression strategies, which may accelerate amyloidosis and inflammation in ways that do not mirror sporadic disease trajectories.55,56,58
Transgenic amyloidosis models (e.g., APP/PS1, 5xFAD, knock-in lines)
These models robustly develop Aβ pathology and neuroinflammatory activation, and they have been used to support multiple death programs. Apoptosis-related readouts (e.g., caspase activation, DNA fragmentation), inflammasome pathway activation (e.g., NLRP3/ASC/caspase-1 signaling with IL-1β/IL-18 maturation), senescence-like markers (e.g., p16/p21 elevation and SASP-related inflammatory outputs), and ferroptosis-related changes (e.g., iron dysregulation, lipid peroxidation, GPX4-related vulnerability) have all been reported depending on age, brain region, and assay.59,60 Importantly, neuronal loss is often modest or variable across amyloidosis models relative to humans, and inflammatory timing can be compressed, complicating direct translation of pathway dominance.
Aβ injection/oligomer exposure models
Acute Aβ administration can induce rapid microglial activation, oxidative stress, and cytokine release, often producing strong inflammasome-related phenotypes and inflammatory injury.61–64 While useful for identifying proximal triggers and testing pathway inhibitors, these models may overemphasize acute inflammatory death mechanisms and do not reproduce decades-long chronic progression or the full spectrum of tau pathology typical of AD.
Tauopathy models and tau seeding paradigms
Tau-focused models capture NFT-like pathology and neuronal dysfunction more directly and can reveal death signatures associated with proteotoxic stress, mitochondrial impairment, and chronic inflammation.65–68 Senescence-like phenotypes and SASP-associated inflammation may be prominent in some tauopathy contexts, 69 and interactions between tau pathology and oxidative/lipid stress can also support ferroptosis-relevant vulnerability.70,71 Nevertheless, tauopathy models may not fully reproduce the amyloid–tau interplay characteristic of typical AD trajectories.
Aging, metabolic, vascular, and mixed-etiology models
Because sporadic AD is strongly shaped by aging and systemic risk factors, models incorporating aging acceleration, metabolic dysregulation (e.g., insulin resistance), vascular injury, or combined genetic and environmental stressors may better approximate the sporadic context.72–75 These paradigms can shift the dominant death signature toward chronic oxidative stress, mitochondrial dysfunction, and inflammatory priming—conditions that plausibly favor convergence among apoptosis, pyroptosis/inflammasome activation, senescence/SASP, and ferroptosis.76–78 However, each still represents a partial reconstruction of the human disease landscape.
Why models and human postmortem findings may diverge
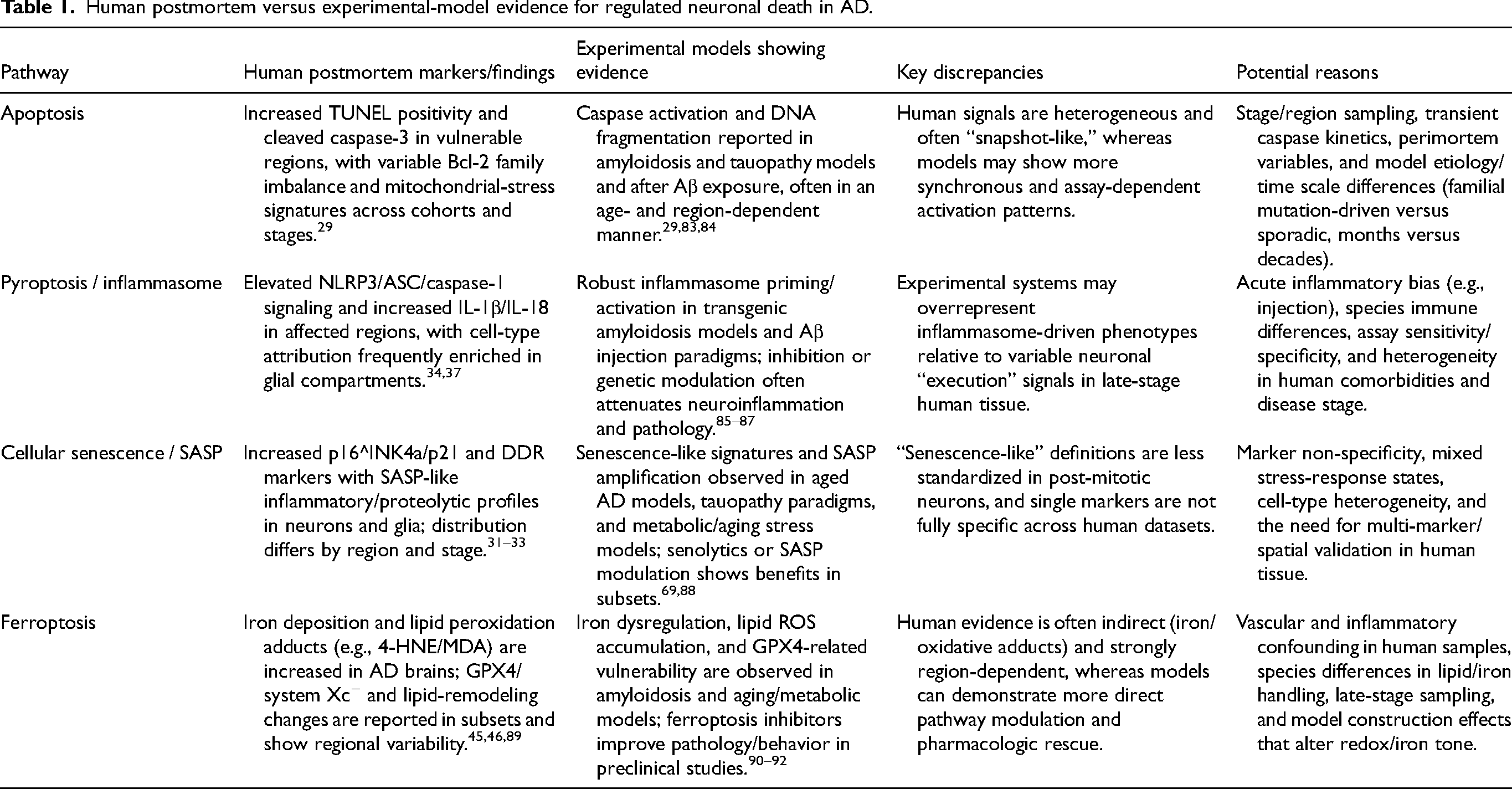
Discrepancies can arise because: (i) etiology differences (familial mutation-driven versus sporadic multifactorial disease); (ii) time scale (months in mice versus decades in humans); (iii) species-specific immune and lipid/iron metabolism; (iv) regional vulnerability mismatch and differences in neuronal subtype composition; and (v) model construction artifacts (overexpression, injection-induced injury, inflammatory skew).56,57,79–82 Consequently, pathway “dominance” observed in a given model should be viewed as context-dependent, and synthesis across model classes is required to infer clinically relevant convergence patterns. A comparative summary of human postmortem findings and experimental-model evidence across the major regulated neuronal death pathways is provided in Table 1.
Human postmortem versus experimental-model evidence for regulated neuronal death in AD.
Overview of major regulated neuronal death modalities in AD
Apoptosis is one of the earliest and most extensively studied forms of programmed cell death. Its typical morphological features include nuclear shrinkage (pyknosis), chromatin margination, chromatin condensation, and DNA fragmentation. 93 Recent studies have demonstrated that widespread neuronal apoptosis occurs in the brain tissue of individuals with AD, and its extent is strongly associated with disease severity. 94 In particular, the activation of the mitochondrial apoptotic pathway plays a crucial role in the pathogenesis of AD. Increased mitochondrial membrane permeability leads to the release of cytochrome c into the cytoplasm, which subsequently triggers the caspase cascade and ultimately results in neuronal apoptosis. 95 In addition, activation of the death receptor-mediated apoptotic pathway, including the aberrant expression of tumor necrosis factor (TNF) receptor family members, has been shown to significantly contribute to the progression of neuronal apoptosis in the brains of patients with AD. 96 Thus, intervention in apoptotic signaling pathways has been regarded as a potential and important strategy for the treatment of AD.
However, apoptosis alone is insufficient to fully explain the complex pathological changes observed in AD. In recent years, another form of inflammation-dependent programmed cell death, pyroptosis, has gradually attracted increasing research attention. Unlike traditional apoptosis, pyroptosis is characterized by its pronounced pro-inflammatory nature and membrane pore formation. A growing body of evidence indicates that the activation of the NLRP3 inflammasome during pyroptosis is closely associated with neuroinflammation in AD. 97 The massive release of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and interleukin-18 (IL-18), during pyroptosis not only directly exacerbates neuronal death but may also amplify neuroinflammatory responses, thereby accelerating the progression of AD. 98 Targeting the inhibition of NLRP3 inflammasome activation or the downstream effector gasdermin D (GSDMD) has recently been identified as a promising therapeutic strategy for the treatment of AD. 99
Meanwhile, cellular senescence, characterized by a state of permanent cell cycle arrest, has received increasing attention for its role in neuronal injury in AD. Studies have shown that large numbers of senescent neurons and glial cells are present in the brain tissue of AD patients. These senescent cells secrete a variety of SASP factors, including interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and matrix metalloproteinases (MMPs), which exacerbate neuroinflammation and neuronal damage in the AD brain. 39 Recently, senolytic agents, which specifically target and eliminate senescent cells, have demonstrated significant therapeutic potential in preclinical models of neurodegenerative diseases, providing new opportunities for the development of AD treatments. 100
Meanwhile, ferroptosis, a newly identified form of regulated cell death, has been increasingly implicated in the pathogenesis of AD. Ferroptosis is an iron-dependent form of programmed cell death characterized by excessive lipid peroxidation catalyzed by iron ions. Studies have demonstrated that abnormal iron homeostasis is evident in the brain tissue of AD patients, with excessive intracellular Fe2+ accumulation. This accumulation promotes the generation of large amounts of ROS through the Fenton reaction, leading to lipid peroxidation of cellular membranes and ultimately resulting in neuronal ferroptosis. 101 Furthermore, disturbances in iron homeostasis exhibit significant crosstalk with other key pathological processes in AD, including tau aggregation and Aβ plaque formation, collectively exacerbating disease progression. Consequently, targeting iron homeostasis or blocking ferroptosis represents a promising therapeutic approach for the treatment of AD.
Although the aforementioned forms of cell death each play important roles in the pathology of AD, they do not exist in isolation. Instead, they exhibit complex interactions and regulatory crosstalk, forming an intricate pathological network that drives the progression of AD. Therefore, a comprehensive investigation of the interconnections and underlying mechanisms among these different forms of cell death may provide novel theoretical foundations and potential targets for the early diagnosis, disease assessment, and targeted therapy of AD.
This review focuses on four major types of cell death—apoptosis, pyroptosis, cellular senescence, and ferroptosis—providing a detailed discussion of their roles, underlying mechanisms, and interactions in the context of AD. Based on current research findings, we also propose potential therapeutic targets and future research directions, aiming to offer more precise and effective strategies for the diagnosis and treatment of AD.
To provide pathway-specific mechanistic detail, we next describe each regulated neuronal death modality in dedicated sections, beginning with apoptosis.
Mechanisms and research progress of apoptosis in AD
Apoptosis, as a key mechanism of neuronal injury in AD, has long been a focus of research. According to traditional views, the major pathological changes in AD are characterized by the accumulation of Aβ plaques and the hyperphosphorylation of tau protein, resulting in the formation of NFTs. These two hallmark pathologies synergistically contribute to the progressive loss of neurons, ultimately leading to irreversible cognitive decline. 102 Nevertheless, accumulating evidence suggests that the aggregation and deposition of pathogenic proteins alone are insufficient to account for the extensive and rapid neuronal degeneration observed in AD. Among the various mechanisms of programmed cell death, neuronal apoptosis is thought to play a pivotal role in mediating this process. 103
Apoptosis is an active form of cell death regulated by highly conserved genes. It is characterized by distinct morphological features, including chromatin condensation, nuclear pyknosis, DNA fragmentation, cytoplasmic densification, and maintenance of plasma membrane integrity. 104 Unlike necrosis, apoptosis does not trigger a significant inflammatory response in the surrounding tissues. However, excessive activation of apoptosis leading to neuronal loss has been confirmed to be closely associated with cognitive impairment in AD. Recent studies have further highlighted the prominence of apoptosis in the AD brain, particularly in regions associated with memory and cognition, such as the cortex and hippocampus. In these areas, the number of apoptotic neurons is significantly increased and shows a strong positive correlation with disease severity. 105
The initiation of apoptosis is primarily mediated through two distinct pathways: the extrinsic and intrinsic pathways. The extrinsic pathway, also known as the death receptor pathway, is typically activated by members of the TNF superfamily, such as Fas ligand (FasL) or TNF-α. Studies have demonstrated significantly elevated levels of pro-inflammatory factors, including TNF-α and FasL, in the brain tissue of patients with AD. These ligands bind to death receptors expressed on the neuronal cell membrane, such as TNF receptor (TNFR) and Fas, leading to the recruitment of adaptor proteins, including TNFR-associated death domain protein (TRADD) and Fas-associated death domain protein (FADD). This recruitment subsequently results in the formation of the death-inducing signaling complex (DISC). 106 The assembly of the death-inducing signaling complex (DISC) leads to the rapid activation of caspase-8, which initiates a caspase cascade by subsequently activating the downstream executioner caspase-3, culminating in apoptosis. Moreover, the NF-κB signaling pathway has been found to be broadly activated in AD, where it facilitates neuronal apoptosis by enhancing the transcription of key molecules involved in the extrinsic apoptotic pathway. 107 Consequently, targeting the expression of pro-inflammatory cytokines or blocking the activation of death receptors represents a promising therapeutic approach for the treatment of AD. As shown in Figure 2, the extrinsic death receptor pathway and the intrinsic mitochondrial pathway converge on caspase-3 activation, thereby executing neuronal apoptosis in AD. These two pathways converge on the activation of caspase-3, ultimately resulting in programmed neuronal death.

Apoptosis mechanisms contributing to neuronal injury in AD. Schematic overview of extrinsic (death receptor–mediated) and intrinsic (mitochondria-mediated) apoptotic pathways in neurons, highlighting their convergence on executioner caspase activation.
In addition, the intrinsic apoptotic pathway, also known as the mitochondrial pathway, is a key contributor to the neuropathology of AD. This pathway is primarily characterized by increased mitochondrial membrane permeability and decreased mitochondrial membrane potential, leading to the opening of the mitochondrial permeability transition pore (MPTP) and the subsequent release of cytochrome c (Cyt-c) into the cytosol.108,109 The released Cyt-c binds to apoptotic protease-activating factor-1 (Apaf-1) in the cytoplasm, forming the apoptosome. This complex subsequently activates the initiator caspase-9, which in turn triggers the caspase cascade by activating the effector caspase-3, ultimately leading to neuronal apoptosis. Notably, significant mitochondrial dysfunction has been observed in the brain tissue of AD patients, including impaired ATP production and excessive generation of ROS. These dysfunctions can induce endoplasmic reticulum (ER) stress and DNA damage, activating the p53 signaling pathway, which further promotes the expression and oligomerization of pro-apoptotic proteins such as Bax and Bak.110,111 Together, these alterations contribute to increased mitochondrial membrane permeability, enhanced Cyt-c release, and an amplification of neuronal apoptosis. Moreover, the marked downregulation of mitochondrial anti-apoptotic proteins, such as Bcl-2, shifts the balance toward pro-apoptotic signals, thereby accelerating neuronal apoptotic processes in AD.
It is noteworthy that the classical pathological changes in AD, such as Aβ accumulation and tau hyperphosphorylation, can facilitate the induction of apoptosis through various mechanisms. Aβ oligomers markedly elevate intracellular calcium concentrations, and the resulting calcium homeostasis disruption promotes the opening of the MPTP, further accelerating the activation of the intrinsic mitochondrial apoptotic pathway in neurons, 10 In addition, tau protein hyperphosphorylation compromises the stability of axonal microtubules, triggering ER stress and mitochondrial dysfunction, thereby exacerbating neuronal apoptosis. 112 Consequently, attenuating Aβ aggregation and tau hyperphosphorylation may substantially mitigate neuronal apoptosis in AD.
At present, therapeutic strategies aimed at modulating apoptosis in AD are primarily concentrated in two areas. The first involves the direct inhibition of apoptosis by targeting critical molecules in both the intrinsic and extrinsic pathways, including TNF-α, Fas receptors, caspase family proteins, and the formation of mitochondrial membrane pores. The second approach focuses on improving mitochondrial function and attenuating oxidative stress to indirectly prevent apoptosis. Mitochondria-targeted antioxidants, such as MitoQ, and mitochondrial membrane stabilizers, such as cyclosporin A, have shown significant neuroprotective effects in preclinical animal models and are considered promising candidates for clinical trials. 113 In addition, TNF-α neutralizing antibodies and caspase inhibitors have shown promising efficacy in reducing neuronal apoptosis in animal models, offering valuable preclinical evidence for the development of novel therapeutic strategies for AD.
Despite the preliminary progress achieved in current research, the complete mechanisms underlying the relationship between apoptosis and AD remain to be fully elucidated. In particular, the interactions between apoptosis and other forms of regulated cell death, such as pyroptosis, cellular senescence, and ferroptosis, are not yet clearly understood. Elucidating these complex interactions is crucial for the development of precise therapeutic strategies. Moreover, given the pronounced heterogeneity of neuronal populations, future studies aimed at uncovering cell type-specific apoptotic mechanisms will provide a stronger theoretical foundation and technological support for the precision and personalization of AD diagnosis and treatment.
In summary, apoptosis, as one of the major forms of neuronal death in AD, plays a central role in the disease's onset and progression. In-depth research into the apoptotic pathways and their targeted modulation may represent a key strategy for alleviating neuronal injury and slowing the progression of AD. Moving forward, it will be necessary to integrate multi-omics approaches to explore the regulatory networks connecting apoptosis with other modes of cell death and to develop precise therapeutic agents targeting apoptosis, thereby opening new avenues for the clinical management of AD.
Mechanisms and research progress of pyroptosis in AD
Pyroptosis, an inflammation-related form of programmed cell death, has garnered increasing attention in recent years. Its hallmark features include the formation of membrane pores, rapid cellular swelling and lysis, and the massive release of pro-inflammatory cytokines. 114 Distinct from classical apoptosis, pyroptosis exhibits rapid loss of plasma membrane integrity, leading to the release of intracellular contents and inducing robust inflammatory responses. In the central nervous system (CNS), especially in the brains of AD patients, pyroptotic cell death in both neurons and glial cells has been increasingly reported, showing a strong correlation with disease progression and representing a prominent focus in recent AD pathological studies.
Originally, pyroptosis was characterized as a form of inflammatory programmed cell death mediated by caspase-1. In recent years, accumulating evidence has revealed that its mechanisms predominantly involve the activation of the NLRP3 inflammasome and the subsequent cleavage of GSDMD. In the context of AD, pathologically aggregated Aβ has been shown to directly stimulate both microglia and neurons, resulting in elevated oxidative stress, as well as the activation of the nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways. 115 These pathways further enhance the expression and activation of NLRP3. Upon activation, NLRP3 interacts with the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), facilitating the recruitment of pro-caspase-1. Pro-caspase-1 is then cleaved into its mature form, caspase-1, which possesses proteolytic activity. 116 Activated caspase-1 facilitates the processing of pro-inflammatory cytokines, such as pro-IL-1β and pro-IL-18, into their mature forms, and induces the cleavage of GSDMD, releasing its pore-forming N-terminal fragment (GSDMD-N). The GSDMD-N fragment embeds into the plasma membrane to form numerous nanopores, resulting in rapid osmotic imbalance, cellular swelling, and the release of intracellular contents. This cascade triggers pyroptosis and the subsequent release of pro-inflammatory cytokines, further amplifying neuroinflammation and exacerbating neuronal injury in AD. As shown in Figure 3, upstream inflammatory signaling and oxidative stress promote NLRP3 inflammasome assembly and caspase-1 activation, culminating in GSDMD-N-mediated membrane pore formation, cytokine release (IL-1β/IL-18), and inflammatory cell death. The figure depicts the activation of NLRP3 by Aβ and tau, the formation of the inflammasome complex with ASC and caspase-1, and the GSDMD-N-mediated membrane pore formation leading to inflammatory cell death.
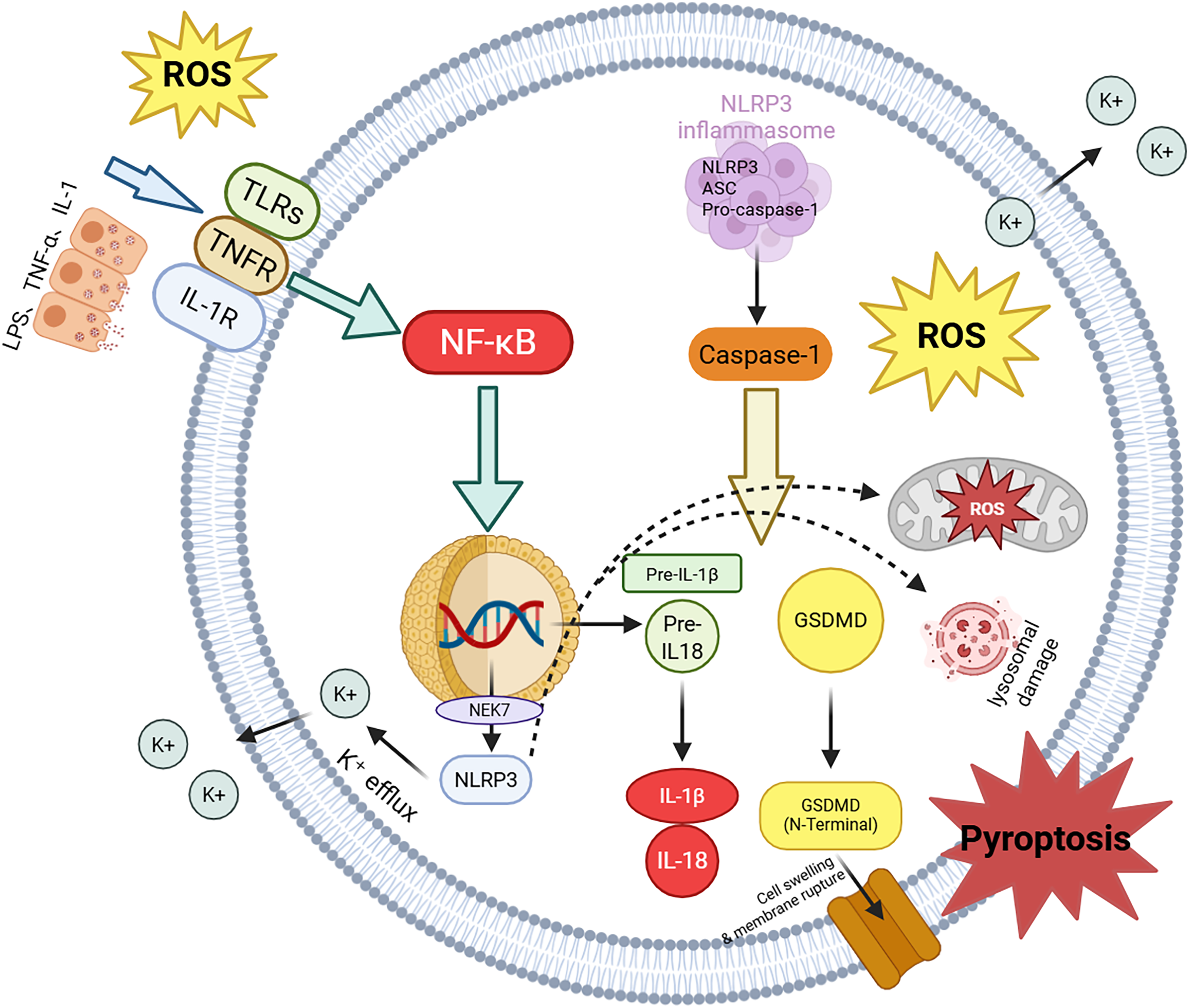
NLRP3 inflammasome–mediated pyroptosis in AD. Schematic overview showing how Aβ/tau-associated stress promotes inflammasome activation, leading to caspase-1–dependent gasdermin cleavage, cytokine maturation (IL-1β/IL-18), and inflammatory cell death.
An increasing body of evidence has established the critical involvement of the NLRP3 inflammasome in the pathogenesis of AD. Notably, significantly elevated expression of NLRP3, ASC, and caspase-1 has been detected in the brains of AD patients, particularly within cognition-related regions such as the hippocampus and cerebral cortex. These elevated levels show a positive correlation with the degree of cognitive decline. Furthermore, in transgenic AD animal models, including APP/PS1 mice, genetic ablation of NLRP3 has been shown to enhance neuronal survival, attenuate neuroinflammatory responses by reducing the release of pro-inflammatory cytokines, and markedly improve cognitive performance. 117 These results provide further evidence supporting the pivotal role of the NLRP3 inflammasome-centered pyroptosis pathway in the pathogenesis of AD.
It is noteworthy that the relationship between pyroptosis and the hallmark pathological features of AD, including Aβ deposition and tau hyperphosphorylation, has become increasingly evident in recent years. Studies have shown that Aβ deposition can not only directly activate the NLRP3 inflammasome in microglia and neurons, leading to the release of inflammatory cytokines, but also that these cytokines further promote the excessive phosphorylation and aggregation of tau protein, exacerbating neuronal injury. This pyroptosis-induced inflammatory cascade not only intensifies neuroinflammation but is also closely associated with the rapid progression of neurodegeneration in AD. In addition, the massive release of pro-inflammatory cytokines, such as IL-1β and IL-18, during pyroptosis activates inflammatory signaling pathways in surrounding unaffected cells, further promoting their pyroptosis or even other forms of cell death, such as apoptosis and cellular senescence. This vicious cycle accelerates disease progression and contributes to the rapid deterioration observed in AD. 118
In light of the critical involvement of pyroptosis in AD pathology, therapeutic strategies targeting this cell death pathway have gained increasing attention in recent years. Current pharmacological interventions primarily focus on two key targets: the inhibition of NLRP3 inflammasome activation and the blockade of GSDMD-mediated membrane pore formation. Notably, studies have shown that MCC950, a well-characterized NLRP3 inflammasome inhibitor, significantly attenuates neuronal pyroptosis in AD animal models and leads to marked improvements in spatial learning and cognitive performance in mice. 119 Furthermore, GSDMD gene knockout using CRISPR-Cas9 technology has been shown to confer significant neuroprotection in mouse models, further validating the pyroptosis pathway as an important therapeutic target for mitigating neuronal injury in AD. In addition, other inflammasome-associated inhibitors, including IL-1β monoclonal antibodies and IL-18 antagonists, have exhibited prominent anti-inflammatory and neuroprotective effects in preclinical animal studies, offering promising new avenues for the development of clinical therapeutics. 120
Although remarkable progress has been made in the study of pyroptosis, numerous challenges remain unresolved. Notably, the mechanisms underlying the differential expression and regulation of pyroptosis in distinct neuronal populations and across different brain regions remain poorly understood. Additionally, the intricate crosstalk between pyroptosis and other forms of regulated cell death, including apoptosis, ferroptosis, and cellular senescence, has not been fully characterized. To address these gaps, future research should leverage advanced methodologies, such as integrated multi-omics technologies, single-cell sequencing, and spatial transcriptomics, to achieve a more holistic and nuanced understanding of pyroptosis and its interplay with other cell death modalities in the context of AD pathology.
In summary, pyroptosis serves as a critical inflammation-mediated neuronal death pathway in the pathogenesis of AD. Comprehensive elucidation of its molecular mechanisms and interaction networks will offer valuable insights for the discovery of early diagnostic biomarkers and the development of precision therapeutics aimed at mitigating disease progression in AD.
Building on this inflammation-linked death program, the next section summarizes cellular senescence and SASP-driven chronic tissue remodeling as an additional contributor to neuronal vulnerability in AD.
Mechanisms and research progress of cellular senescence in AD
Cellular senescence is defined as a physiological and pathological condition marked by irreversible cell cycle arrest, loss of proliferative capacity, dysregulated metabolic activity, and the acquisition of a SASP. While initially considered a hallmark of natural aging, accumulating evidence indicates that premature cellular senescence under pathological conditions plays a critical role in the progression of numerous chronic diseases. Of particular interest is the senescence of neurons and glial cells, which has emerged as a key feature in the pathogenesis of AD. 100
In recent years, accumulating evidence has demonstrated the widespread presence of prematurely senescent neurons and glial cells in the brains of patients with AD. Notably, the burden of these senescent cells shows a strong correlation with disease severity and the degree of cognitive dysfunction.121,122 Neuronal senescence is characterized by distinct molecular features, including telomere attrition, heightened DNA damage responses, significant upregulation of cell cycle inhibitors such as p16^INK4a and p21, and increased activity of senescence-associated β-galactosidase (SA-β-Gal). Telomere shortening, in particular, plays a pivotal role in this process. Telomeres, which cap the ends of chromosomes, are essential for preserving genomic stability and regulating cellular replication. In the brains of AD patients—especially within hippocampal neurons—telomere length is markedly reduced. This telomere attrition promotes chromosomal instability, induces a sustained DNA damage response, and activates both the p53-p21 and p16^INK4a signaling pathways, resulting in irreversible cell cycle arrest and accelerating neuronal senescence. 8 In addition, premature telomere shortening is significantly associated with the pathological accumulation of Aβ and hyperphosphorylated tau, indicating that telomere attrition may serve as a critical mechanism underlying neuronal senescence and facilitating the progression of AD. 100
The SASP represents one of the hallmark pathological characteristics of cellular senescence. 123 Extensive research has demonstrated that senescent neurons and glial cells are capable of secreting a wide range of pro-inflammatory cytokines, chemokines, and proteolytic enzymes—such as IL-1β, IL-6, IL-8, TNF-α, and MMPs. These components of the SASP induce chronic neuroinflammatory responses within the local brain tissue, leading to damage of neighboring healthy neurons and further enhancing glial cell overactivation. This persistent inflammatory microenvironment contributes to sustained neuronal degeneration and exacerbates cognitive impairment. As shown in Figure 4, senescent neurons and glial cells exhibit characteristic molecular hallmarks and produce SASP factors that exert paracrine effects on surrounding healthy cells, thereby amplifying local inflammation and accelerating neurodegeneration through a self-reinforcing vicious cycle. The figure illustrates the morphological and molecular features of senescent neurons and glial cells, the secretion of SASP factors, and their paracrine effects on surrounding healthy cells, which collectively accelerate neurodegeneration. Moreover, findings from animal models have confirmed that SASP factors significantly accelerate the pathological progression of AD, in part by promoting tau hyperphosphorylation and Aβ aggregation. 124 In addition, SASP components are capable of exerting paracrine effects, promoting the senescence of adjacent healthy cells and establishing a vicious cycle that accelerates the pathological progression of AD. As a result, the targeted elimination of senescent cells and the suppression of SASP factor secretion have emerged as promising therapeutic strategies to delay AD progression.

Cellular senescence and SASP-driven bystander effects in AD. Schematic overview of senescence hallmarks in neurons and glia and the paracrine actions of SASP factors that sustain neuroinflammation and accelerate neurodegeneration; potential intervention points include senolytic and SASP-modulating strategies.
In recent years, a novel therapeutic strategy termed “senolytics” has gained increasing attention in the field of cellular senescence research. 125 The core concept of senolytic therapy is the selective clearance or inhibition of senescent cells to mitigate their deleterious effects and consequently slow disease progression. Currently, a variety of senolytic compounds with well-established anti-senescent activity have been developed, with the combination of dasatinib and quercetin representing one of the most well-studied and effective examples. 126 Preclinical animal studies have demonstrated that the combined administration of dasatinib and quercetin significantly decreases the accumulation of senescent cells in the brains of AD models, including APP/PS1 transgenic mice. This treatment also attenuates the release of SASP factors and leads to notable improvements in neuronal function and cognitive abilities.8,127,128 These research findings provide the first direct evidence confirming the therapeutic potential of senescent cell clearance strategies in AD. Moreover, other senolytic agents, including Navitoclax, Fisetin, and Piperlongumine, have exhibited significant neuroprotective effects in animal models of neurodegenerative diseases. Collectively, these findings highlight the clearance of senescent cells as a promising strategy for the future development of precision therapies targeting AD. 129
Although substantial advances have been made in elucidating the role of cellular senescence in AD, several critical challenges remain unresolved. First, the current lack of senescent cell-specific biomarkers hampers the precise identification and efficient targeting of these cells for clearance. Second, the functional heterogeneity of senescent cells across different brain regions and distinct cell types has yet to be fully characterized. Furthermore, the intricate crosstalk between cellular senescence and other regulated cell death mechanisms in AD, including apoptosis, pyroptosis, and ferroptosis, remains poorly understood. Future research should focus on leveraging cutting-edge technologies such as single-cell RNA sequencing, spatial transcriptomics, and proteomics to gain deeper insights into the specific roles of senescent cells within the AD pathological microenvironment. Particular emphasis should be placed on deciphering the regulatory mechanisms of SASP secretion and elucidating its interaction networks with other forms of neuronal cell death.
In conclusion, cellular senescence significantly contributes to the progression of AD pathology. Elucidating the mechanisms of neuronal and glial cell senescence, deciphering the regulatory networks governing SASP secretion, and clarifying their complex interactions with AD pathological processes will provide a robust theoretical basis for the development of early intervention strategies. Moreover, these advancements are anticipated to accelerate the clinical translation of anti-senescence therapies, particularly senolytics, ultimately offering novel avenues for the prevention, intervention, and treatment of AD.
Mechanisms and research progress of ferroptosis in AD
Ferroptosis is a newly recognized form of programmed cell death distinguished by iron-dependent lipid peroxidation and oxidative damage to cellular membranes, primarily due to impaired activity of glutathione peroxidase 4 (GPX4). 130 Distinct from apoptosis, pyroptosis, and cellular senescence, ferroptosis is a caspase-independent form of cell death that lacks the classical morphological hallmarks of apoptosis. Instead, its defining ultrastructural features include mitochondrial shrinkage, reduced mitochondrial cristae, or complete cristae loss. In recent years, an increasing number of studies have revealed the critical involvement of ferroptosis in neuronal damage and cognitive dysfunction, particularly in the context of AD. Consequently, ferroptosis has gained recognition as an emerging hotspot in AD pathogenesis research. 131
Mechanistically, ferroptosis is typically initiated when cellular antioxidant defenses that detoxify phospholipid hydroperoxides are overwhelmed, most prominently involving the System Xc−–GSH–GPX4 axis and lipid peroxidation-prone membrane substrates. 132
Iron is an essential element that participates in critical biological processes such as oxygen transport, electron transfer, and energy metabolism under physiological conditions. However, under pathological circumstances, dysregulation of iron homeostasis has been identified as a key initiator of ferroptosis. In AD, abnormal iron accumulation represents a hallmark pathological alteration, predominantly observed in neurons within the hippocampus, cortex, and basal ganglia. The excessive buildup of iron ions facilitates the generation of ROS via the Fenton reaction, which subsequently promotes lipid peroxidation, particularly in membrane phospholipids enriched with polyunsaturated fatty acids (PUFAs). This lipid peroxidation drives ferroptotic cell death in neuronal populations.133,134 A growing body of evidence indicates that in the brains of patients with AD, particularly during the early stages, there is a marked imbalance in iron metabolism, accompanied by increased levels of lipid peroxidation products, such as 4-hydroxynonenal (4-HNE). These observations implicate ferroptosis as a potential contributor to the early pathogenesis of AD.
Beyond iron overload per se, emerging studies highlight that iron availability can be further amplified by iron-handling pathways such as ferritin turnover and ferritinophagy, in which NCOA4-mediated delivery of ferritin to lysosomes releases labile Fe2+ and thereby fuels Fenton chemistry and lipid peroxidation. 135
A hallmark of ferroptosis is the diminished activity of GPX4, an essential membrane-associated antioxidant enzyme. GPX4 plays a pivotal role in catalyzing the reduction of lipid hydroperoxides, thereby protecting cells from ferroptotic death. In AD, substantial evidence indicates that both GPX4 protein expression and its enzymatic activity are significantly reduced in brain tissues. This impairment compromises the clearance of lipid peroxides, exacerbates membrane lipid peroxidation, and ultimately facilitates the induction of neuronal ferroptosis. 136 Further evidence from animal models has demonstrated that GPX4 gene deficiency or functional impairment leads to aggravated neuronal ferroptosis and is associated with pronounced cognitive impairment. Conversely, strategies aimed at enhancing GPX4 activity or administering antioxidant supplements, such as vitamin E or selenium, effectively attenuate neuronal ferroptosis and significantly ameliorate cognitive deficits in AD animal models.137,138 Consequently, dysregulation of GPX4 expression and activity occupies a central position in the ferroptosis pathway involved in the pathogenesis of AD.
Upstream of GPX4, the cystine/glutamate antiporter System Xc− imports cystine for glutathione (GSH) biosynthesis; impairment of System Xc− function or depletion of GSH reduces GPX4-dependent lipid peroxide detoxification, thereby lowering the threshold for neuronal ferroptosis.101,139,140
In parallel, ferroptosis susceptibility is strongly shaped by lipid remodeling pathways that enrich membranes with peroxidation-prone PUFA-phospholipids (PUFA-PLs), including ACSL4- and LPCAT3-dependent incorporation of PUFAs into phospholipids, which provides key substrates for iron-driven lipid peroxidation. 141
In addition to the canonical GPX4-centered defense, alternative anti-ferroptotic systems have been described, including the FSP1–CoQ10 axis, the GCH1–BH4 pathway, and NRF2-regulated antioxidant networks; dysfunction of these compensatory systems may further exacerbate lipid redox imbalance and ferroptotic vulnerability in AD-relevant settings.142–144
In recent years, increasing attention has been directed toward the interaction between ferroptosis and the core pathological features of AD, including Aβ accumulation and tau protein hyperphosphorylation. Substantial evidence indicates that aberrant Aβ aggregation in the brain facilitates the activation of divalent metal transporter 1 (DMT1), leading to increased iron uptake and aggravating disturbances in iron homeostasis. Additionally, hyperphosphorylated tau impairs the normal axonal transport of iron within neurons, resulting in intracellular iron overload. This excess iron further intensifies oxidative stress in an iron-dependent manner, thereby accelerating the ferroptotic process. 145 Moreover, ROS produced during ferroptosis have been shown to aggravate tau hyperphosphorylation and Aβ accumulation, establishing a self-perpetuating vicious cycle that accelerates the progression of AD. Emerging evidence further indicates that therapeutic strategies aimed at restoring iron homeostasis or inhibiting lipid peroxidation can effectively attenuate Aβ deposition and tau pathology, leading to significant improvements in both neuropathological alterations and cognitive deficits in AD animal models. 146 These findings further clarify the intricate and reciprocal relationship between ferroptosis and the core pathological features of AD. As shown in Figure 5, dysregulated iron uptake and Fe2+-dependent oxidative reactions drive lipid peroxidation (“lipid ROS”), while compromised antioxidant defense (e.g., the GSH/GPX4 axis) lowers the threshold for neuronal ferroptosis and reinforces a feed-forward loop with tau hyperphosphorylation and Aβ deposition.

Ferroptosis mechanisms contributing to neuronal injury in AD. Schematic overview illustrating iron dyshomeostasis–driven lipid peroxidation and compromised antioxidant defenses (e.g., GPX4-related pathways) that culminate in neuronal ferroptosis, with bidirectional interactions with Aβ and tau pathology.
At the mechanistic level, this reciprocity can be conceptualized as a feed-forward chain: Aβ- and tau-associated disturbances promote iron dyshomeostasis (e.g., via DMT1 upregulation and impaired axonal iron trafficking), which increases the labile iron pool and lipid ROS; lipid peroxidation products and oxidative stress, in turn, aggravate tau phosphorylation and Aβ pathology, thereby sustaining a vicious cycle that progressively amplifies neuronal vulnerability to ferroptosis. 147
At present, therapeutic interventions targeting ferroptosis are primarily centered around several key approaches. The first strategy focuses on the regulation of iron homeostasis through pharmacological agents, particularly iron chelators such as deferoxamine (DFO) and deferasirox, which are designed to reduce excess iron accumulation and mitigate iron-induced oxidative damage. 131 The second strategy focuses on enhancing the activity of GPX4, through the administration of selenium-containing compounds or lipid-soluble antioxidants, such as vitamin E, 148 Furthermore, direct inhibitors of lipid peroxidation have been developed, including ferroptosis-specific inhibitors such as Ferrostatin-1 (Fer-1) and Liproxstatin-1 (Lip-1). 149 These pharmacological interventions have exhibited significant neuroprotective effects in AD animal models. For example, long-term administration of the iron chelator DFO has been shown to effectively attenuate abnormal brain iron accumulation, reduce oxidative stress, and improve cognitive performance in AD mouse models. Similarly, GPX4 agonists markedly suppress neuronal ferroptosis and delay disease progression in experimental models. Collectively, these findings offer preliminary evidence supporting the potential clinical value of ferroptosis-targeted therapeutic strategies in the treatment of AD.
In addition, strategies that modulate cystine import/GSH availability (thereby stabilizing the System Xc−–GSH–GPX4 axis), inhibit PUFA-PL remodeling (e.g., ACSL4/LPCAT3-related pathways), suppress ferritinophagy-mediated iron release (e.g., NCOA4-linked mechanisms), or activate compensatory anti-ferroptotic defenses (FSP1–CoQ10, GCH1–BH4, NRF2) have been proposed as complementary directions for ferroptosis-oriented neuroprotection.139,150,151
Although substantial advances have been made in elucidating the role of ferroptosis in AD, several critical challenges remain unresolved. First, the differential regulation of iron homeostasis across distinct brain regions and neuronal subtypes is not yet clearly defined. Second, the intricate interplay between ferroptosis and other forms of neuronal cell death, including apoptosis, pyroptosis, and cellular senescence, warrants further comprehensive investigation. Moreover, the clinical translation of ferroptosis-targeted therapeutic strategies remains at an early stage. Comprehensive assessments of optimal drug dosing, safety, and long-term therapeutic efficacy are still lacking and require validation through extensive preclinical and clinical studies.
Furthermore, the relative contributions of distinct ferroptosis control modules (System Xc−/GPX4, lipid remodeling, ferritinophagy-driven iron release, and GPX4-independent defense axes) may vary across disease stages and cell types, which highlights the need for cell-type- and region-resolved analyses to identify the dominant actionable nodes in AD.
In conclusion, ferroptosis represents a novel and unique mechanism of regulated neuronal death that plays a pivotal role in the neuropathology of AD. Future research should aim to delineate the core regulatory pathways governing ferroptosis and its interaction networks with other cell death modalities. Additionally, further exploration of ferroptosis-targeted therapeutic interventions holds great promise for advancing AD prevention and precision therapy, with the ultimate goal of improving disease outcomes and enhancing patient quality of life.
In particular, clarifying how ferroptosis-relevant modules (System Xc−–GSH–GPX4, PUFA-PL remodeling, ferritinophagy/iron release, and GPX4-independent defense systems) intersect with Aβ/tau pathology will strengthen mechanistic evidence and facilitate more rational target selection for therapeutic development.
Given the increasing evidence that ferroptosis intersects with other regulated cell death modalities in AD, it is important to further consider how apoptosis, pyroptosis, cellular senescence, and ferroptosis interact and converge to drive neuroinflammation and neuronal loss.
Crosstalk and convergence of regulated cell death pathways in AD
Accumulating evidence indicates that apoptosis, pyroptosis, cellular senescence, and ferroptosis do not operate as isolated processes in AD. Instead, these regulated cell death (RCD) modalities are dynamically interconnected and converge on shared upstream pathological stressors and downstream neuroinflammatory amplification, collectively accelerating neuronal dysfunction and loss. Common AD-related drivers, including Aβ accumulation, tau hyperphosphorylation, mitochondrial injury, excessive ROS generation and lipid peroxidation, dysregulated iron homeostasis, Ca2+ overload, and chronic glial-driven neuroinflammation, may activate multiple RCD programs in parallel or sequentially, thereby shaping a self-reinforcing network rather than a single linear pathway.7,14,24,27,29,152 Notably, Aβ/tau pathology and oxidative stress frequently function as shared triggers, whereas sustained neuroinflammation and progressive neuronal loss represent convergent outcomes of this network (Figure 6A,C).
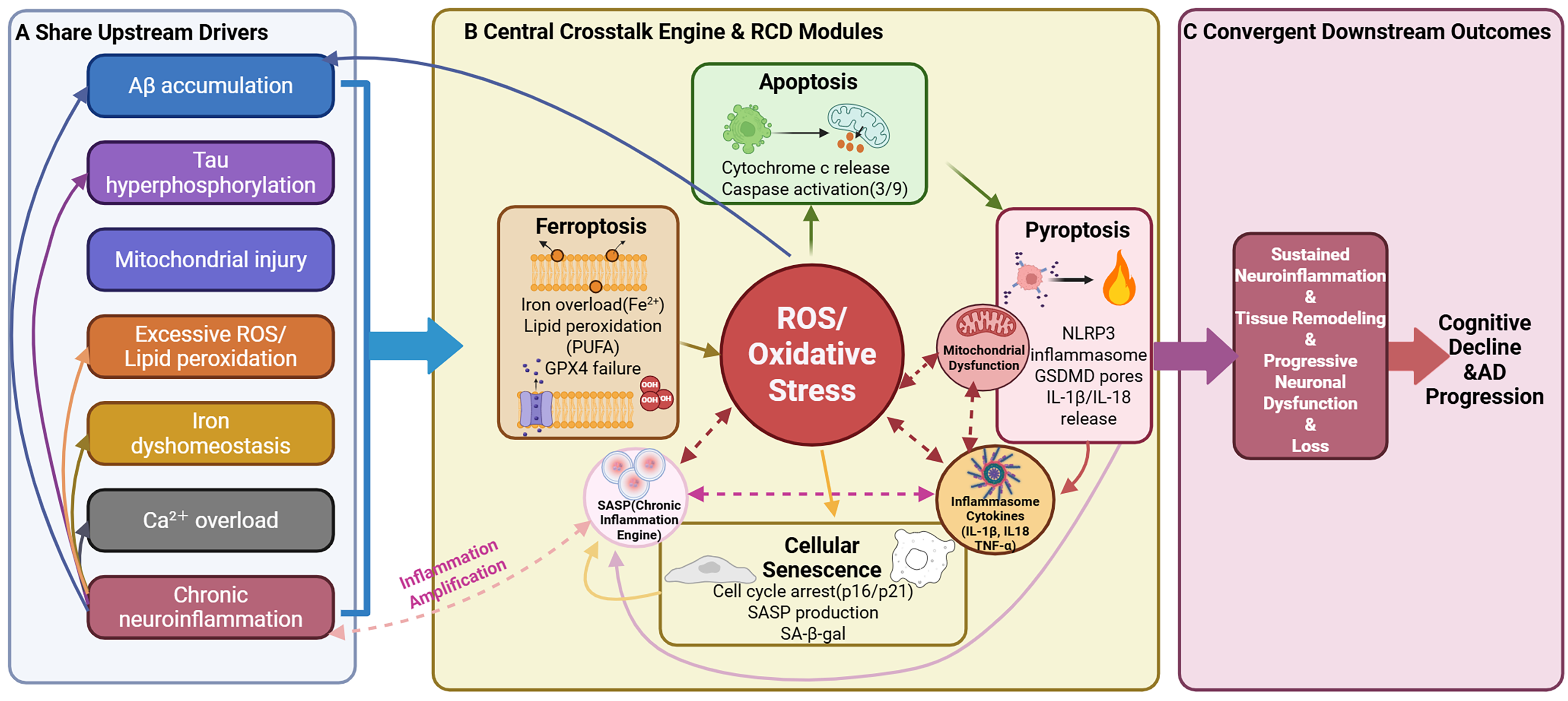
Crosstalk and convergence of regulated cell death pathways in AD. Panels A–C summarize shared upstream stressors, central crosstalk hubs (oxidative stress/mitochondrial dysfunction, inflammasome-cytokine signaling, and senescence/SASP), and feed-forward loops linking apoptosis, pyroptosis, cellular senescence, and ferroptosis to sustained neuroinflammation and progressive neuronal loss.
Several “bridging nodes” mechanistically link these RCD modalities, with oxidative stress and mitochondrial dysfunction serving as central crosstalk engines (Figure 6B). 14 Oxidative stress acts as a central integrator: ROS promotes intrinsic apoptosis via mitochondrial membrane damage and cytochrome c release, enhances inflammasome priming/activation to strengthen pyroptosis with IL-1β/IL-18 release, and drives ferroptosis by initiating PUFA-enriched lipid peroxidation when antioxidant defenses (e.g., GPX4 activity) are compromised. 153 Inflammatory cytokines released during pyroptosis (together with elevated TNF-α and related mediators) intensify glial activation and secondary neuronal injury, lower apoptotic thresholds, and promote SASP, thereby sustaining a pro-inflammatory microenvironment.154,155 Mitochondrial dysfunction further amplifies these cascades by increasing ROS output, impairing ATP generation, and weakening redox buffering, which sensitizes neurons to apoptosis and ferroptotic collapse. Meanwhile, SASP functions as a “chronic inflammation engine,” maintaining inflammatory priming and oxidative stress and progressively lowering the threshold for multiple RCD programs. 156
These interconnections generate positive feedback loops that drive neuroinflammation and neuronal loss, as highlighted by the circular feed-forward circuitry (Figure 6B). Aβ/tau-associated stress promotes ROS production and mitochondrial dysfunction, which accelerates apoptosis and lipid peroxidation toward ferroptosis; oxidative and mitochondrial stress can also prime inflammasome pathways, increasing pyroptosis and cytokine release that further amplifies neuroinflammation and oxidative stress. Chronic inflammation and tissue remodeling facilitate senescence and SASP, sustaining an environment that reinforces pyroptosis while potentiating apoptotic and ferroptotic susceptibility. Collectively, this convergent, self-perpetuating network may help explain why targeting a single RCD pathway in isolation often yields limited benefit, whereas strategies that disrupt shared hubs (e.g., oxidative stress, mitochondrial failure, and inflammatory amplification) or employ rational multi-target interventions may offer more durable disease-modifying effects.
This crosstalk perspective provides an integrative framework for interpreting the pathway-specific evidence summarized above and sets the stage for the following Discussion on key insights and translational implications.
Discussion
AD, one of the most prevalent neurodegenerative disorders globally, is predominantly characterized by the progressive damage and loss of neurons, which ultimately results in irreversible cognitive decline and memory loss. This condition poses a heavy burden on both society and families. Historically, it has been believed that the onset and progression of AD are primarily linked to the accumulation of Aβ plaques and the abnormal hyperphosphorylation of tau protein, leading to the formation of NFTs. 157 Recent research, however, highlights that a singular focus on mechanisms such as protein aggregation or inflammation is inadequate to comprehensively explain the extensive and rapid neuronal damage observed in AD. Growing evidence suggests that multiple forms of neuronal cell death contribute significantly to the pathogenesis of AD. This review offers a systematic overview of several key neuronal death mechanisms that have attracted considerable attention in AD research, including apoptosis, pyroptosis, cellular senescence, and ferroptosis. The review also delves into the underlying mechanisms of these cell death pathways, their interrelationships, and potential therapeutic strategies, with the goal of providing a more precise and effective theoretical foundation for future diagnostic and therapeutic approaches in AD.
Integrated interpretation of pathway-specific evidence
Apoptosis, recognized as one of the most classic and extensively studied forms of programmed cell death, plays a pivotal role in the neuronal damage observed in AD. This process encompasses both the extrinsic death receptor pathway (such as TNF-α and FasL) and the intrinsic mitochondrial pathway (including increased mitochondrial membrane permeability and cytochrome c release). Studies have shown that apoptosis levels are significantly elevated in the brain tissue of AD patients and are strongly associated with disease severity. 103 However, targeting the apoptotic pathway alone is inadequate to completely mitigate neuronal damage in AD, suggesting the involvement of additional cell death mechanisms.
Pyroptosis, an emerging form of inflammatory programmed cell death, has been increasingly recognized for its significant role in the neuropathological processes of AD. The key characteristics of pyroptosis include the activation of the NLRP3 inflammasome, cleavage of GSDMD, and the subsequent release of pro-inflammatory cytokines such as IL-1β and IL-18. 158 Extensive research has demonstrated that pyroptosis is significantly elevated in the brains of AD patients. The inflammatory cytokines released during pyroptosis contribute to neuroinflammation, which further exacerbates tau protein pathology and Aβ accumulation, establishing a self-perpetuating vicious cycle that accelerates the progression of the disease. 119 These findings indicate that traditional anti-inflammatory therapies alone may not be sufficient to yield substantial clinical outcomes. In contrast, the development of therapeutics specifically targeting the pyroptosis pathway may offer enhanced therapeutic potential.
Cellular senescence, an irreversible state of cell cycle arrest, is accompanied by the release of numerous SASP factors. Recent studies have revealed that senescent cells are prevalent in the brain tissues of AD patients. Through SASP, these cells secrete inflammatory cytokines, chemokines, and matrix-degrading enzymes, which significantly amplify the local neuroinflammatory microenvironment, damaging adjacent healthy neurons and further promoting neuronal apoptosis, pyroptosis, or ferroptosis. The rise of Senolytics (senescent cell clearance) therapies has demonstrated considerable anti-AD potential in animal models, indicating that senescent cell-targeted precision therapies may represent a promising avenue in future AD treatment strategies.
Ferroptosis, an iron-dependent form of programmed cell death, has gained increasing attention in recent years. In the brain tissues of AD patients, disruptions in iron homeostasis and a reduction in GPX4 enzyme activity exacerbate iron ion-dependent lipid peroxidation, thereby promoting neuronal death. Of particular interest is the clear interaction between ferroptosis and the classic pathological hallmarks of AD, including Aβ accumulation and tau protein pathology. The oxidative damage induced by ferroptosis further accelerates tau hyperphosphorylation and Aβ aggregation, establishing a crucial link in the pathogenesis of AD. 43 Consequently, modulating iron homeostasis or enhancing GPX4 enzyme activity may serve as novel and promising therapeutic approaches for the prevention and treatment of AD.
In conclusion, the four cell death mechanisms detailed in this article do not operate independently within the pathological process of AD, but instead, exhibit significant interactions and mutual enhancement. This intricate network of interactions constitutes the comprehensive pathological framework of neuronal damage in AD. Specifically, the inflammatory cytokines released during pyroptosis collaborate with the SASP factors from senescent cells, synergistically exacerbating neuroinflammation. The ensuing amplification of inflammation and oxidative stress further aggravates mitochondrial dysfunction, thereby facilitating the onset of apoptosis and ferroptosis. 159 The intricate, multi-level interactions among different cell death pathways greatly enhance the complexity of the pathological processes in AD. This complexity suggests that therapies targeting a single cell death pathway may not produce substantial therapeutic benefits. Consequently, future research should prioritize the development of multi-target, integrated intervention strategies, particularly those that concurrently target the key pathways of apoptosis, pyroptosis, cellular senescence, and ferroptosis, in order to achieve early intervention and effective disease management.
Therapeutic targeting: translational barriers and safety considerations
Although targeting apoptosis, pyroptosis, cellular senescence, and ferroptosis holds clear mechanistic appeal, a balanced evaluation requires careful consideration of translational barriers and safety constraints. For each pathway, the theoretical advantages of intervention must be weighed against challenges in blood–brain barrier (BBB) delivery, target specificity, systemic off-target effects, and the current scarcity of robust clinical evidence in AD.
Apoptosis-focused strategies may theoretically mitigate neuronal loss by interrupting caspase-dependent execution pathways or stabilizing mitochondrial integrity. However, BBB penetration remains a practical limitation for many candidate agents, and CNS delivery may be further complicated by active efflux transporters and the need for sustained exposure in chronic disease.160–162 Moreover, because apoptotic signaling is fundamental for tissue homeostasis and immune regulation, insufficient target specificity may lead to systemic adverse effects, including impaired physiological cell turnover or unintended immunomodulation.163–165 Importantly, most apoptosis-targeting approaches in AD remain supported predominantly by preclinical evidence, and translating pathway inhibition into durable clinical benefit remains challenging given disease heterogeneity and pathway redundancy.
For pyroptosis, inhibiting upstream inflammasome signaling (e.g., NLRP3-related pathways) offers the advantage of simultaneously reducing inflammatory cytokine maturation and limiting inflammation-driven neuronal injury. Nevertheless, BBB penetration and appropriate CNS exposure remain key hurdles, particularly for small-molecule inhibitors whose brain distribution may be restricted. In addition, inflammasome pathways contribute to host defense; therefore, inadequate selectivity or excessive pathway suppression may increase susceptibility to infection or interfere with immune surveillance. Compounds frequently discussed in this context (including MCC950) have comparatively limited clinical trial evidence in AD, 166 and thus require rigorous evaluation of long-term safety, dosing windows, and on-target/off-target profiles before clinical translation can be realistically advanced.
Senescence-targeting approaches, including senolytics and SASP-modulating strategies, are conceptually attractive because they aim to dismantle a chronic pro-inflammatory microenvironment and reduce sustained bystander damage. However, BBB delivery and cell-type specificity remain substantial challenges, as senescent phenotypes may differ across neural and glial populations and may vary with disease stage. A major safety concern is that eliminating senescent cells or broadly suppressing SASP could disrupt tissue repair processes or alter immune regulation; moreover, long-term senolytic exposure has raised concerns regarding systemic toxicities in non-CNS tissues. At present, evidence for senescence-targeted interventions in AD is largely derived from animal studies and early-stage translational exploration, underscoring the need for more definitive clinical validation. 167
Ferroptosis-targeted interventions (e.g., iron chelation, reinforcement of antioxidant defenses such as GPX4-related pathways, or inhibition of lipid peroxidation with agents such as Fer-1) may directly counteract iron-dependent oxidative membrane damage and thereby protect vulnerable neurons.151,168–170 However, translating these strategies faces several obstacles, including BBB penetration, pharmacokinetic stability, and achieving sufficient target engagement within relevant brain regions. Because iron metabolism and lipid redox pathways are essential for systemic physiology, poor specificity may increase the risk of anemia, metabolic disturbance, or interference with normal cellular redox signaling. Moreover, many ferroptosis inhibitors, including Fer-1, are supported primarily by preclinical data, and comprehensive clinical assessments of dosing, 171 long-term safety, and therapeutic durability in AD are still lacking.
Overall, these considerations suggest that enthusiasm for pathway-specific therapeutics should be tempered by practical constraints. Future efforts may benefit from (i) improving BBB-permeable delivery platforms, (ii) identifying biomarkers that enable patient stratification and target engagement monitoring, and (iii) prioritizing rational multi-target strategies that disrupt shared hubs (oxidative stress, mitochondrial dysfunction, and inflammatory amplification) while minimizing systemic toxicity.
Why death signatures differ across models and brain regions
Notably, an important and still unresolved question is why distinct experimental AD models—and even different brain regions within the same disease context—often display divergent dominant neuronal death signatures. This heterogeneity likely reflects multiple, partially independent axes. First, regional microenvironments differ substantially across the hippocampus, association cortex, locus coeruleus, and basal forebrain, including baseline iron burden, myelin/lipid composition, vascular supply, and the intensity and phenotype of glial activation; such factors can bias local susceptibility toward oxidative lipid damage/ferroptosis, inflammasome-associated pyroptosis, or mitochondrial apoptosis.124,172,173 Second, disease stage may shift the prevailing death program: early phases may feature compensatory stress responses and inflammatory priming, whereas late phases are more prone to bioenergetic collapse, cumulative mitochondrial injury, and irreversible degeneration, potentially altering the balance among apoptosis, pyroptosis, senescence/SASP-driven chronic injury, and ferroptosis.172,174 Third, cell-type-specific vulnerability can further shape pathway choice, as excitatory versus inhibitory neuronal populations and surrounding astrocytes/microglia differ in redox buffering capacity, iron handling, cytokine responsiveness, and lipid metabolism, thereby creating cell-selective thresholds for each regulated death modality. 175 Finally, model construction introduces systematic bias: transgenic amyloidosis models driven by APP/Aβ overexpression tend to amplify inflammatory and oxidative cascades early and may preferentially highlight inflammasome signaling or lipid peroxidation, whereas tauopathy, injection-based, aging, or metabolic-stress paradigms can emphasize distinct upstream triggers and thus different downstream death programs.176,177 Collectively, these considerations support the view that regulated neuronal death in AD is not governed by a single universal mechanism, but rather by context-dependent pathway dominance shaped by region, stage, cell identity, and model-specific drivers—an insight that further underscores the need for spatially resolved, longitudinal, and multi-model validation when inferring clinically relevant death mechanisms.
Systems-level priorities for future progress
Given these translational constraints, future progress will likely depend on systems-level approaches that can resolve cell-type- and region-specific death programs and identify convergent regulatory hubs. In particular, integrating single-cell and spatially resolved profiling with multi-omics datasets may enable biomarker-guided patient stratification, clarify optimal intervention windows across disease stages, and provide a rational basis for mechanism-driven combination therapies targeting multiple regulated cell death pathways.
Currently, research on drugs targeting the aforementioned cell death mechanisms remains predominantly at the preclinical stage, and the clinical translation of these findings faces significant challenges. The mechanisms of neuronal death may exhibit substantial heterogeneity across different brain regions, disease stages, and patient subtypes, rendering traditional single-target treatment approaches inadequate for achieving precise intervention. Consequently, future AD treatment strategies should make full use of advanced technologies such as single-cell genomics, spatial transcriptomics, and systems biology to comprehensively reveal the interaction networks and core regulatory mechanisms underlying these cell death pathways, thereby facilitating the development of individualized, precision-based therapeutic approaches.
In summary, a comprehensive understanding of the specific mechanisms and interrelationships of various neuronal death modalities, such as apoptosis, pyroptosis, cellular senescence, and ferroptosis, in the context of AD will provide novel theoretical insights and technical support for early diagnosis, disease progression monitoring, and the development of precision-based therapeutic strategies. This approach will ultimately contribute to the effective prevention, treatment, and clinical intervention of AD.
Conclusion
In summary, neuronal loss in AD represents a complex pathological process driven by multiple mechanisms, with apoptosis, pyroptosis, cellular senescence, and ferroptosis serving as key contributors. Rather than acting as parallel and independent events, these regulated cell death pathways form an interconnected network with shared upstream drivers and self-reinforcing feedback loops, which collectively intensify neuroinflammation, oxidative stress, and the progression of AD hallmarks, including Aβ accumulation and tau abnormalities. Consequently, therapeutic strategies targeting a single pathway in isolation may yield limited disease-modifying effects, whereas integrated interventions that disrupt shared hubs may be more promising.
To advance this field, several research priorities should be emphasized:
Single-cell and spatially resolved analyses (e.g., single-cell transcriptomics and spatial transcriptomics) to map cell-type- and brain-region-specific preferences of death programs across disease stages, and to define when and where each pathway becomes dominant. Multi-omics integration (transcriptomics, proteomics, metabolomics/lipidomics, and metallomics/iron-related profiling) to identify convergent regulatory hubs and actionable nodes that couple oxidative stress, inflammation, mitochondrial dysfunction, and iron dyshomeostasis. Rational combinatorial strategies that jointly target complementary mechanisms, such as anti-inflammatory/anti-pyroptotic modulation, suppression of lipid peroxidation/ferroptosis, and clearance or reprogramming of senescent cells/SASP, with careful evaluation of synergy and safety. Biomarker and imaging development to translate pathway activity into measurable indicators (e.g., CSF/plasma markers and neuroimaging readouts), enabling pathway-guided patient stratification and monitoring of target engagement and treatment response.
From a translational perspective, future clinical development should prioritize stage- and subtype-informed therapeutic frameworks. Specifically, patient stratification based on disease stage, molecular phenotype, and biomarker-defined pathway activation may guide the selection of appropriate targets and dosing windows; subsequently, mechanism-driven combination regimens can be tested to determine whether multi-target intervention provides superior and durable clinical benefit compared with single-target approaches. Collectively, these efforts may facilitate precision prevention and treatment strategies and ultimately improve outcomes for patients with AD.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.