
Abstract
Background
Converging evidence supports a bidirectional link between epilepsy and Alzheimer's disease (AD). Limited data are available regarding the association of epilepsy with frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB).
Objective
To estimate the bidirectional association between epilepsy and AD, FTD, and DLB in the UK Biobank, a large, multi-source healthcare dataset.
Methods
Epilepsy, AD, FTD, and DLB cohorts were extracted. Cox proportional-hazard models were used to compare the AD risk in the epilepsy cohort against matched controls, and the epilepsy risk in the AD cohort against matched controls. Prevalent association of epilepsy with FTD and DLB was assessed using Chi-squared/Fisher test.
Results
Hazard-ratio (HR) for AD in the epilepsy population versus controls was 2.5 (1.8–3.4, p < 0.001); HR increased when epilepsy was diagnosed in the sixth-seventh decade and in individuals with epilepsy carrying the APOE ε4 allele. Conversely, HR for epilepsy in AD versus controls was 14.8 (9.7–22.5, p < 0.001). Epilepsy prevalence was higher in the FTD population compared to controls, with prevalence ratio (PR) of 5.3 (2.4–11.8, p = 0.001). Epilepsy PR in DLB versus controls approached but did not achieve statistical significance (2.4, 1.0–5.7, p = 0.068).
Conclusions
Our findings reinforce the notion of a bidirectional association between epilepsy and AD, providing proof-of-concept that epilepsy, especially late-onset, may be successfully integrated into AD risk frameworks. Furthermore, we found a high prevalence of epilepsy in the FTD population. Careful stratification of individuals with epilepsy could offer an opportunity to identify those at higher risk of future or covert AD and neurodegeneration.
Keywords
Introduction
Epilepsy and dementia are among the most prevalent neurological conditions worldwide, carrying a substantial burden in terms of death and disability. 1
Incidence of dementia increases sharply after the sixth decade, 2 while epilepsy incidence peaks at younger ages and rises again after the fifth decade,3,4 so that both conditions are prevalent in older adults.
Alzheimer's disease (AD) is the world leading cause of dementia. 5 At the pathophysiological level, AD is characterized by abnormal aggregation of amyloid-β (Aβ) and hyperphosphorylated tau. 5
Late-onset epilepsy (LOE) is a multifaceted clinical entity due to several mainly structural causes, the most common being cerebrovascular disease. 3 A notable proportion of LOE is reportedly attributable to underlying neurodegenerative processes, with AD playing a leading role. 3 However, it has been estimated that up to 20% or more LOE remain with unknown etiology.4,6
Longstanding epidemiological evidence supports a bidirectional association between epilepsy and dementia.7–15 Studies conducted on AD have repeatedly reported higher risk of seizures compared to controls or general population.7–10 Consistently, APOE ε4 genotype and dementia are risk factors for epilepsy at older ages. 16 On the other hand, epilepsy is associated with increased risk of incident dementia,11–15 with fewer data being available for the specific dementia subtypes.
A close link seemingly exists between the AD pathophysiologic process and epileptogenesis, especially at the level of temporal lobes, possibly due to network disturbance and hyperexcitability triggered by amyloid deposition.17–21 Aligning with these data, EEG epileptiform discharges are frequently observed in individuals with AD.22–24
Previous findings depicted high rates of positive AD biomarkers in individuals with LOE without overt cognitive impairment, 25 with increased risk of conversion to dementia compared to LOE without such profiles. 26 These observations are paralleled by the evidence that epilepsy can precede overt cognitive decline in ep-AD. 27 Last, a recent study elegantly found raised risk of incident LOE in individuals showing longitudinal reduction of plasma Aβ42/40 ratio in mid-to-late life. 28
In this evolving landscape, LOE of unknown cause has become a critical focus of interest, as it has been hypothesized that it might represent the first clinical manifestation of AD in some individuals, possibly at pre-cognitive stages.18,20,29
Intriguingly, on the other hand, pre-clinical evidence suggests that neuronal hyperactivity could contribute to the progression of amyloid pathology, 17 so that seizures and AD could form a positive loop. 18 In this regard, clinical AD progression seems faster in individuals with seizures 30 and in those with subclinical epileptiform discharges22,24; individuals with epileptic-AD (ep-AD) have more advanced AD pathology at death than AD without seizures, 30 and they could therefore represent a biological subgroup associated with worse prognosis.
In line with the above arguments, it has been argued that tackling epileptogenesis may improve cognitive outcomes in AD,19,31 so that ep-AD identification might be non-trivial.
Fewer data are available about the relation between epilepsy and other prevalent neurodegenerative dementias, namely frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB). Previous observations suggested lower risk of epilepsy in FTD compared to AD, 32 while a recent study reported high absolute lifetime prevalence of seizures in these individuals. 33 Data concerning DLB are even sparser, with variable risk estimates.
In an era of emerging disease-modifying therapies, identification of epilepsy cases due to potential covert neurodegeneration might allow timely treatment, prior to irreversible brain damage, at the same time offering a priceless spotlight into the earliest and presymptomatic stages of disease.
With this background in mind, we aimed to quantify the bidirectional association between the diagnoses of AD and epilepsy, both in terms of incidence and prevalence, using the UK Biobank, a large, population-based cohort of approximately 500,000 participants enrolled between 2006 and 2010, with retrospective and prospective linkage to multi-source health records, including linked primary care and hospital admissions data from the UK National Health Service (NHS).34,35
As a secondary study aim, we quantified the prevalent association of epilepsy diagnosis with FTD and DLB diagnoses, based on the same population dataset.
Methods
Study populations, epilepsy-AD analysis
For the study primary analysis, the full UK Biobank dataset (n = 502,199) was initially filtered to remove conditions with confounding power on the AD and/or epilepsy diagnoses and their association. Specifically, we excluded participants with any of the following diagnoses, irrespective of the timing of occurrence relative to the conditions of interest: congenital abnormalities of central nervous system (CNS), neurodevelopmental disorders, cerebral palsy, acute or chronic infection of the CNS, multiple sclerosis, CNS tumor, cerebrovascular disease (including ischemic/hemorrhagic stroke and vascular leukoencephalopathy), hydrocephalus, and other neurodegenerative conditions or dementias, including vascular dementia. International statistical classification of diseases and related health problems 10th revision (ICD-10) codes were used to identify diagnoses, based on the individual diagnostic entry provided by the UK Biobank (for the complete list, please see Supplemental Table 1).
The remaining dataset was further filtered to extract an AD cohort and an epilepsy cohort (Figure 1). The ICD-10 codes G30 or F00 were used to identify AD while G40 was used for epilepsy; these approaches have demonstrated good diagnostic accuracy in prior validation studies.36,37 For the principal analysis, only individuals with diagnoses made in hospital settings were deemed eligible, and all cases meeting these criteria were included.
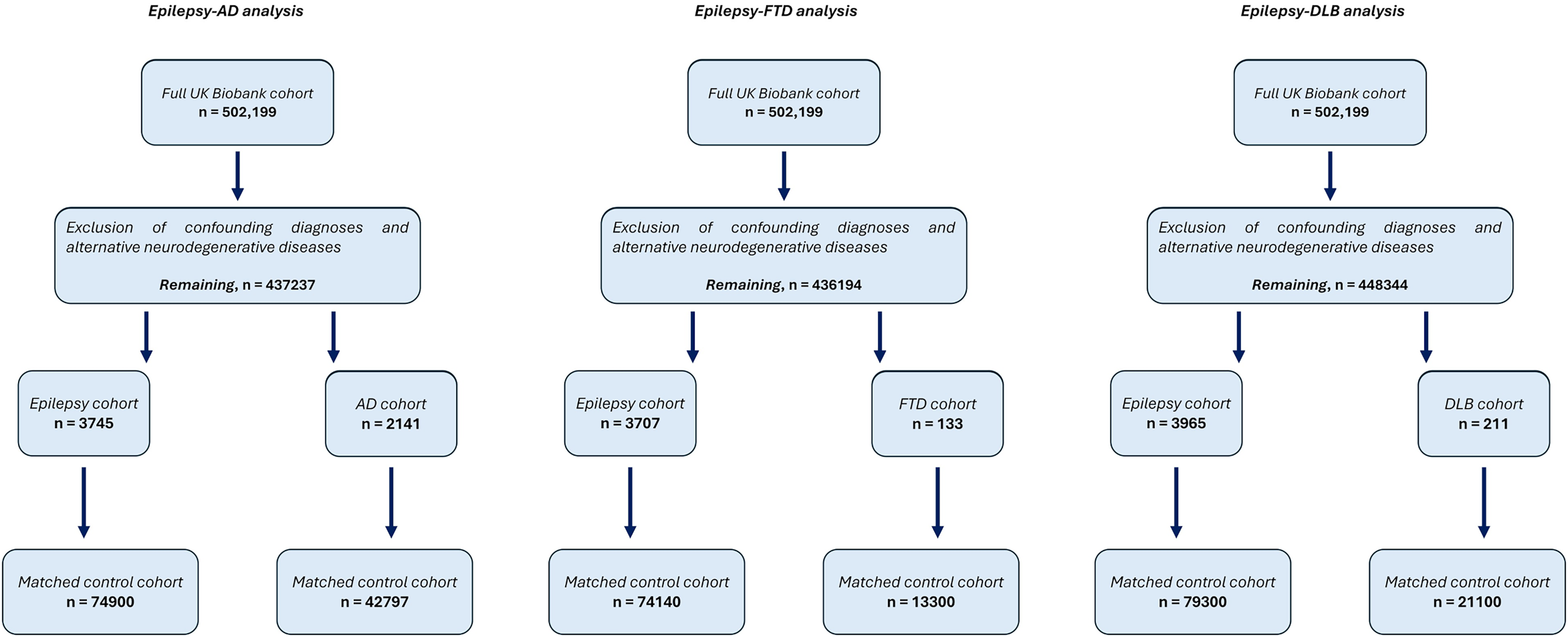
Study inclusion flowchart. The original UK Biobank dataset underwent parallel filtering, with slightly different criteria for each of the studied neurodegenerative condition (e.g. FTD and DLB were excluded from the epilepsy-AD analysis; FTD and AD were excluded from the epilepsy-DLB analysis; and so on. For further details, please refer to supplemental material). Abbreviations: Alzheimer's disease (AD), frontotemporal dementia (FTD), dementia with Lewy bodies (DLB).
Next, for every individual with epilepsy, controls without epilepsy matched by biological sex and year of birth were randomly chosen in a 1 to 20 ratio from the filtered dataset; the same was done for the AD group (Figure 1). With this approach, controls were allowed to have the same range of comorbidities as in the disease groups. To test consistency in the results, two supplemental seeds of matched controls were extracted and analyzed separately for both the AD and epilepsy groups.
Study populations, epilepsy-FTD, and epilepsy-DLB analysis
The full UK Biobank dataset was filtered with same logic for the epilepsy–FTD and epilepsy–DLB analyses, with identification of an FTD and a DLB cohort, respectively, along with their corresponding epilepsy cohorts (Figure 1; for details, please refer to Supplemental Tables 2 and 3). Matched controls were chosen with the same 1:20 ratio as above for the epilepsy cohorts; on the other hand, due to the smaller number of individuals, the chosen ratio was 1:100 for the FTD and LBD cohorts.
Primary and secondary outcomes
The primary study outcomes were incident and prevalent AD in individuals with epilepsy (IWE), and incident and prevalent epilepsy in individuals with AD. The primary objective was to assess the bidirectional epidemiological association between epilepsy and AD in the UK Biobank cohort.
To better characterize such associations along the natural history of the diseases, we further measured the outcomes of interest in population subgroups stratified by age at epilepsy or AD diagnosis.
To assess the consistency of our findings across different population settings, we conducted a sensitivity analysis using a broader cohort definition. The aim of the latter was to allow a more “natural” distribution of comorbid diagnoses in the disease cohorts and their controls, compared to the stricter clinical filtering used in the principal analysis. To do this, we retained most of the previously excluded diagnoses, and we only removed conditions with the highest confounding potential over the association studied, namely alternative neurodegenerative conditions and other causes of dementia. Furthermore, to reduce concern about under-ascertainment of individuals who primarily receive care in outpatient settings, we allowed epilepsy and dementia cases to be identified through primary care and death certificate data, in addition to hospital admission records.
In addition, as a separate sensitivity analysis, we estimated incident AD risk in the subpopulation of IWE with evidence of at least one anti-seizure medication in their treatment records. For details regarding sensitivity analyses, please see Supplemental Tables 4 and 5.
Secondary outcomes included the prevalence of epilepsy in individuals with FTD and DLB and, conversely, the prevalence of the latter conditions in IWE. Sensitivity analyses were performed to estimate epilepsy prevalence in FTD and DLB groups after loosening exclusion criteria and diagnostic requirements, as detailed above for the epilepsy-AD analysis (Supplemental Tables 6 and 7).
Follow-up, events, time at risk, and prevalence measures.
For AD incidence estimates, IWE were considered at risk from the age their epilepsy diagnosis was first reported; on the other hand, follow up for age and sex matched controls was started at a “surrogate epilepsy age”, corresponding to the age when diagnosis of epilepsy was first reported in their index patient.
Participants recruited in Wales, Scotland, and England were considered at risk until 31 May 2022, 31 August 2022, and 31 October 2022, respectively, corresponding to the UK Biobank censoring dates for linked hospital admission data from the three nations. Follow up persisted until diagnosis of AD, death, loss to follow up as declared by the UK Biobank, or censoring, whichever came first. An identical and opposite design was applied for incident epilepsy in the AD population, using the age when AD was first diagnosed as the origin of follow-up in the AD group, and the surrogate AD age in the control group (for more details, please refer to the Supplemental Material).
For both primary and secondary outcomes, prevalence was defined as the proportion of individuals with a recorded diagnosis in the study groups, at any time up to the above censoring dates for linked hospital-admission data.
Statistical analysis
Data were analyzed in Rstudio. 38 Chi squared test or Fisher's exact test were used to assess differences in categorical variables depending on the expected cell counts, while Wilcoxon test and Kruskal-Wallis test with post-hoc Dunn's correction were used for continuous variables. To account for the matched data structure, significant differences in epilepsy and dementia prevalence between the study groups and controls were confirmed using stratified conditional logistic regression. Prevalence measures were expressed as prevalence ratios (PRs) relative to controls.
Cox proportional hazards models were fitted to estimate hazard ratios (HRs) for incident diagnoses across the groups of interest (R survival package). For bivariate and multivariate Cox models, we used the following set of demographic and clinical covariates, also reported in Table 1: sex, age at censoring, diabetes, hypertension, smoke, alcohol, education, and APOE status (for further details regarding covariate quantification and attribution, please see the Supplemental Material). APOE information was unavailable for 7.7% and 7.8% of the AD and epilepsy cohort, respectively, and for 6.5% and 6.7 of their matched controls; In models including APOE status, individuals with missing APOE data were excluded. Potential synergistic effects between epilepsy (main exposure) and APOE status were evaluated using an interaction term. The proportional hazards assumption was assessed in every model using the scaled Schoenfeld residuals method. In the case of non-proportionality of covariate(s), stratified Cox models were used.
Demographic characteristics of the study groups and their matched controls, epilepsy-AD analysis. Age is reported as mean (standard deviation).
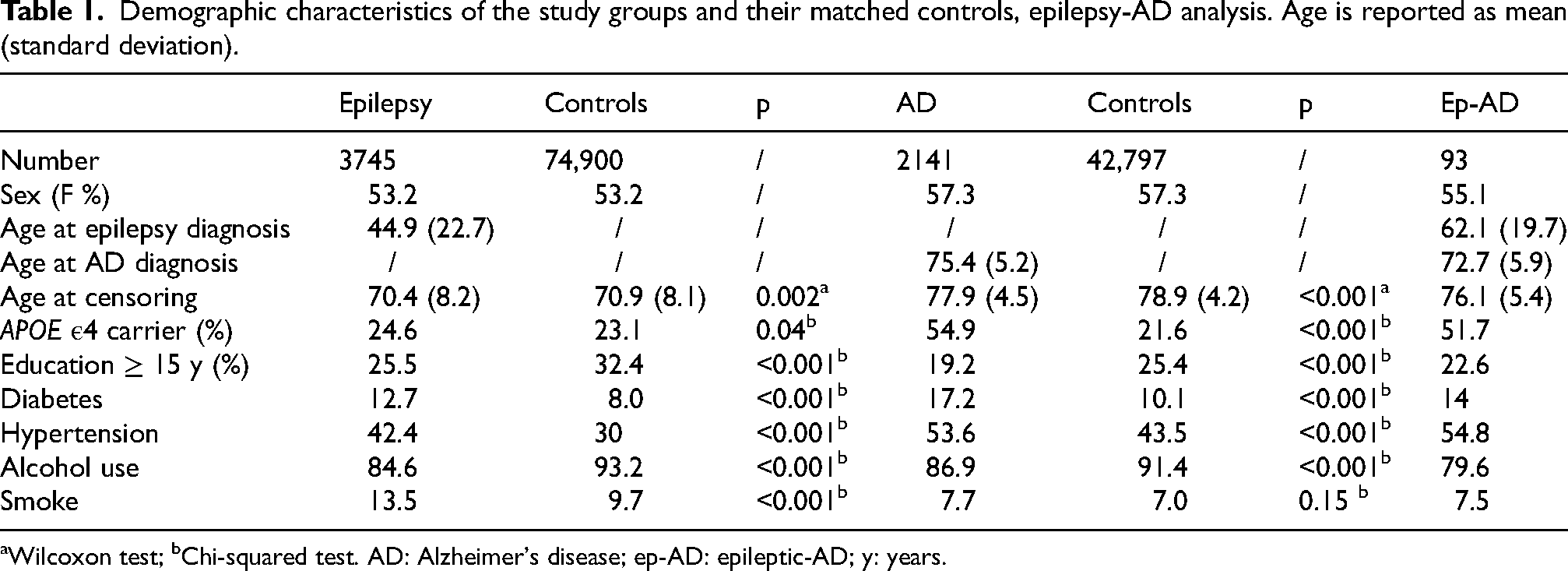
Wilcoxon test; bChi-squared test. AD: Alzheimer's disease; ep-AD: epileptic-AD; y: years.
We did not perform Cox proportional hazards models in the epilepsy-DLB and epilepsy-FTD analyses. This choice was based on the relatively small size of the DLB and FTD groups, with consequent limited number of incident events, making HR estimates potentially unstable and vulnerable to control sampling variability.
The Benjamini-Hochberg (BH) procedure was used to control for false discovery rate and adjust for multiple comparisons, with a significance threshold set at p < 0.05. To control for the family-wise alpha error, post-hoc pairwise comparisons in the Chi-squared/Fisher's and Kruskal-Wallis tests were adjusted with the stricter Bonferroni correction.
Results
Characteristics of the study cohorts
In the epilepsy-AD analysis, the AD and epilepsy cohorts were composed of 2141 and 3745 individuals, respectively (for details on the specific epilepsy diagnostic codes, please see Supplemental Table 8). The demographic characteristics of these cohorts are reported in Table 1. The epilepsy–FTD analysis included 133 individuals with FTD and 3707 with epilepsy, and the epilepsy–DLB analysis included 211 individuals with DLB and 3965 with epilepsy. Detailed information on these cohorts is available in Supplemental Tables 9 and 10.
Average age at dementia diagnosis was 75.4, 75.1, and 70.5 years for the AD, DLB, and FTD cohorts, respectively. Age differed across groups after Kruskal-Wallis test (p < 0.001). After pairwise comparisons, FTD individuals were younger at diagnosis than the other two groups (p < 0.001, Figure 2). On the other hand, female sex accounted for 57.3% of the AD population, with female prevalence being higher than FTD (42.1%, χ2 = 11.8, p = 0.002) and DLB (32.7%, χ2 = 47.0, p < 0.001). The APOE ε4 carrier rates were 54.9%, 51.6%, and 30.0% in the AD, DLB, and FTD cohorts, respectively. In addition, the carrier rate was 51.7% in the ep-AD group compared with 23.9% in IWE without AD. APOE ε4 distributions differed in the overall Chi-squared test (χ2 = 517.4, p < 0.001); post-hoc pairwise comparisons revealed significant differences between FTD and AD/DLB, as well as between epilepsy without AD and ep-AD (Figure 2).
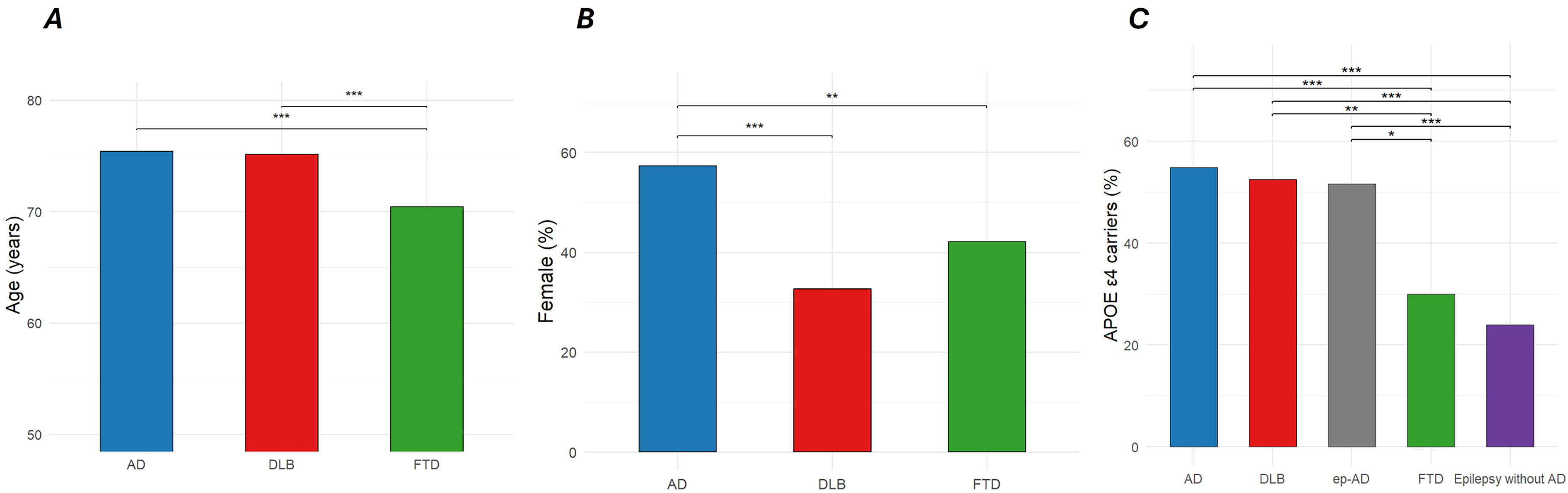
Age at diagnosis, sex and APOE-ε4 carrier status in the studied cohorts. A) individuals with FTD were significantly younger than AD and DLB at diagnosis; B) female sex is significantly underrepresented in DLB and FTD in comparison with AD; C) APOE-ε4 allele prevalence is significantly lower in FTD compared to AD and DLB. Ep-AD individuals have significantly higher APOE-ε4 rate than epilepsy without AD; Figure legend: * = p < 0.05, ** = p < 0.01, *** = p< 0.001. Abbreviations: Azheimer’s disease (AD), Dementia with Lewy Bodies (DLB), epileptic-AD (ep-AD), frontotemporal dementia (FTD).
The AD risk in the epilepsy population
Of the total epilepsy population, 3689 individuals with available follow-up were free of AD at the time of epilepsy diagnosis, and they were enrolled to fit Cox proportional hazards models. When compared with matched controls without epilepsy, unadjusted HR for incident AD was 2.5 (95% CI 1.8–3.4, p < 0.001). HR remained significantly elevated both after adjustment for individual covariates and in the multivariable Cox model encompassing all the covariates (Figure 3). Average duration of follow-up was 25.9 years in the epilepsy cohort and 26.0 years in matched controls, while AD incidence rate (IR) after age 70 was 400 per 100,000 person-years and 152 per 100,000 person-years, respectively. When stratified by age at epilepsy diagnosis, HR for incident AD appeared to be highest in the subgroup diagnosed between age 50 and 60 (4.7, 95%, CI 2.3–9.2, p < 0.001), followed by diagnosis between age 60 and 70 (3.4, 95% CI 1.7–6.6, p < 0.001) (Figure 4). HR appeared to decline in individuals with diagnosis of epilepsy after age 70 (1.8, 95% CI 0.9–3.7, p = 0.12). Although marginally non-significant, the younger age subgroups also displayed higher risk estimates (Figure 4); furthermore, when considered as a whole, IWE diagnosed before age 50 showed a higher risk compared with matched controls (HR 2.2, 95% CI 1.3–3.8, p = 0.003).
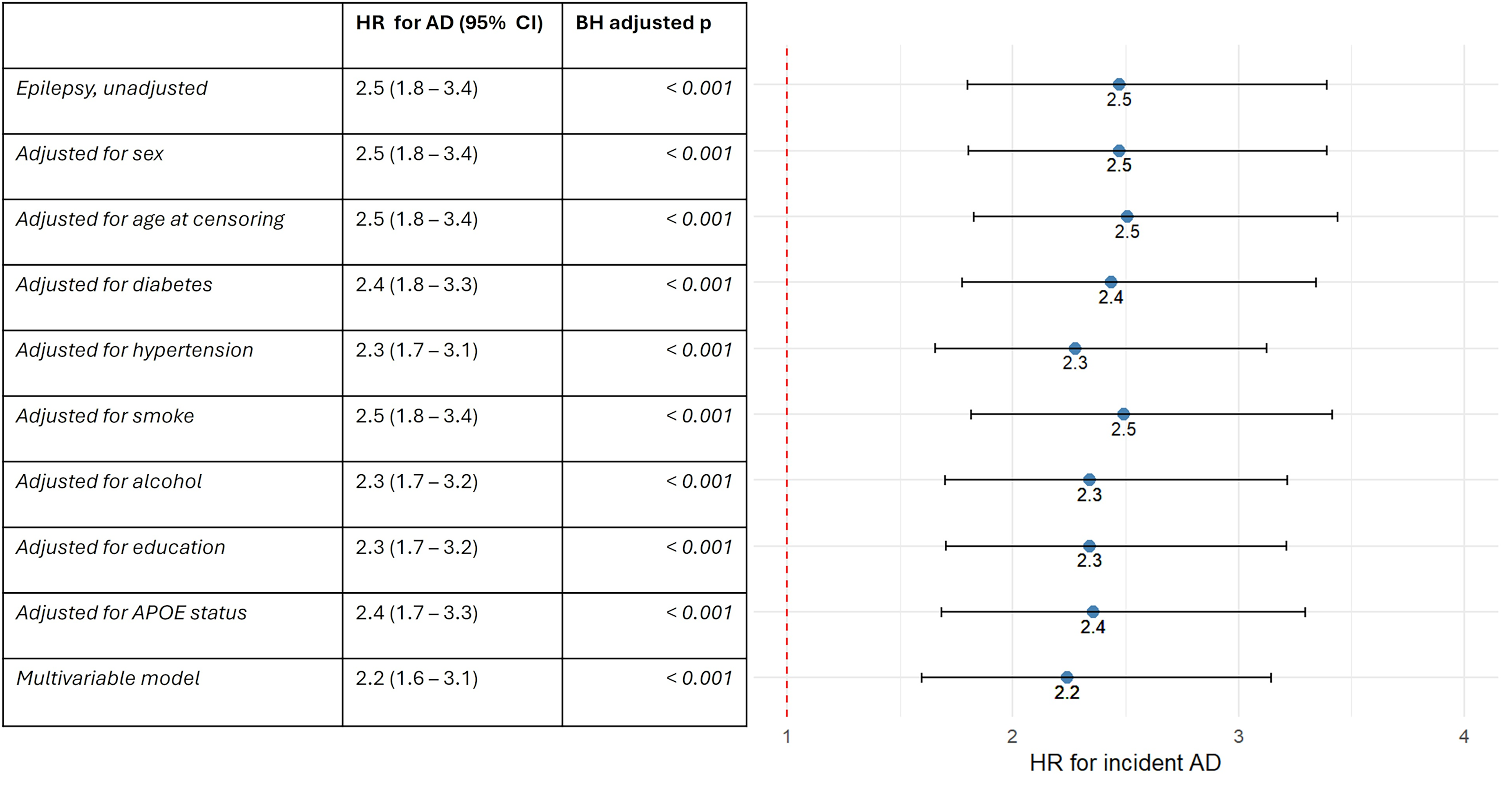
Forest plot of HR for incident AD in the epilepsy population. More details are available in the supplemental material. Abbreviations: hazard ratio (HR), Alzheimer’s disease (AD), confidence interval (CI), Benjamini-Hochberg (BH).
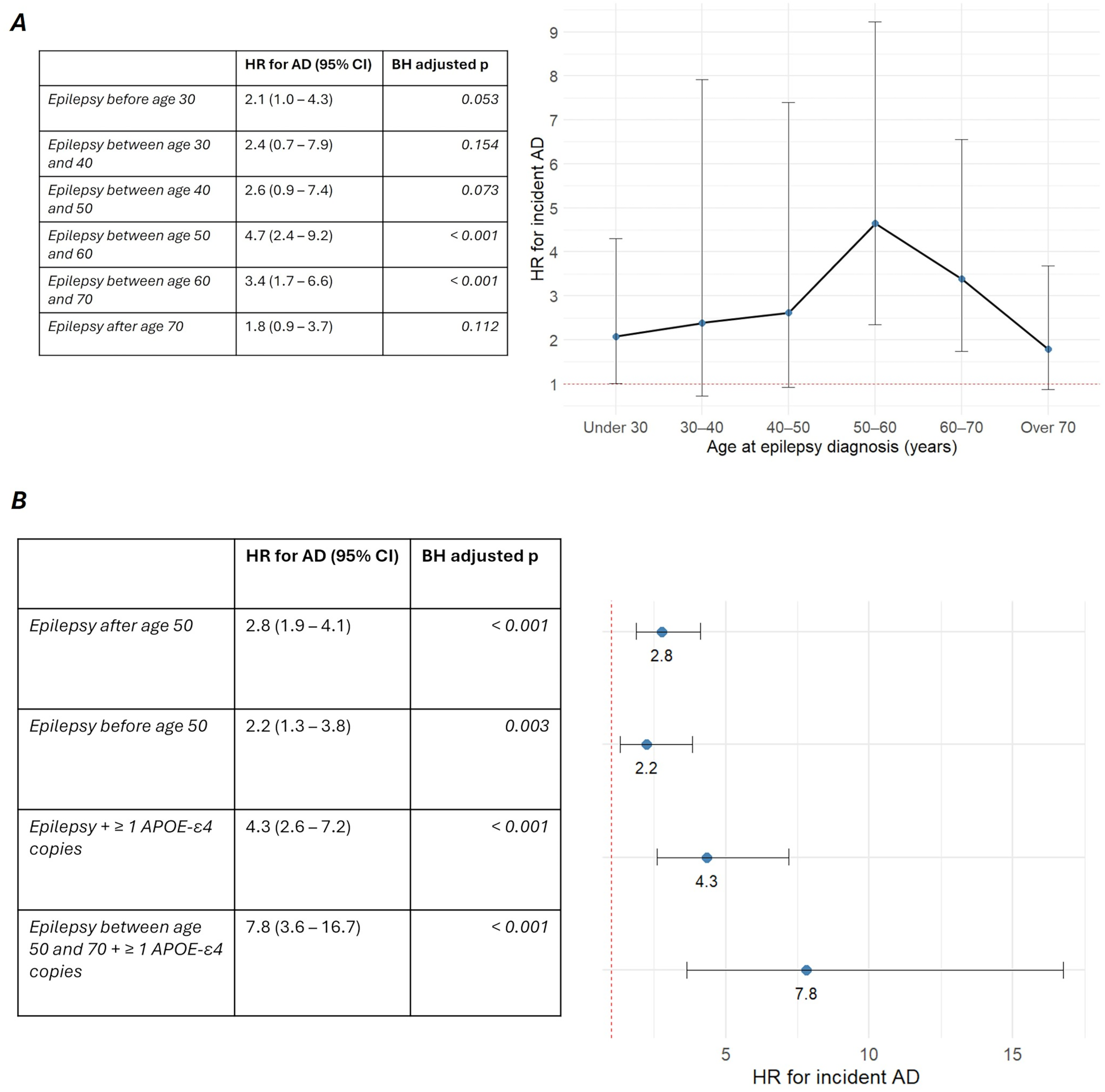
Stratified measures of incident AD risk in the epilepsy population. A) HR for incident AD stratified by age at epilepsy diagnosis; B) Forest plot of HR for incident in individuals with diagnosis of epilepsy before and after age 50 and in APOE-ε4 carriers. Abbreviations: hazard ratio (HR), Alzheimer’s disease (AD), confidence interval (CI), Benjamini-Hochberg (BH).
We then fitted Cox models to estimate AD risk in the population of IWE carrying at least an APOE ε4 copy (n = 820) versus matched controls; HR appeared higher in this group (4.3, 95% CI 2.6–7.2, p < 0.001), particularly so in the subgroup with diagnosis between age 50 and 70 (7.8, 95% CI 3.6–16.7, p < 0.001, Figure 4). Epilepsy and APOE ε4 carrier status seemingly exerted an additive more than synergistic effect on the risk of incident AD, as the multivariable model did not reveal significant interaction between the two variables. For more details regarding the analyses and covariates, please refer to Supplemental Tables 11–14.
Consistent with incidence data, prevalence of AD was higher in epilepsy as compared to controls (PR 4.7, 95% CI 3.8–5.9, p < 0.001).
In the sensitivity analyses, relaxing the exclusion criteria and diagnosis requirements did not change significantly the risk estimates, with HR for incident AD of 2.2 (95% CI 1.8–2.7, p < 0.001). To adjust for the confounding effect of incident stroke, we fitted a Cox model with the latter modelled as a time-varying covariate. Even in this case, HR was mostly unaffected (2.3, 95% CI 1.8–2.7, p < 0.001). Moreover, in the separate analysis only considering IWE with at least an anti-seizure medication in their treatment record, we consistently observed an HR of 2.3 (95% CI 1.4–3.7, p = 0.001).
Last, comparison against two additional seeds of matched controls revealed largely consistent result; particularly, unadjusted HR for incident AD in epilepsy versus seed 2 was 2.5 (95% CI 1.8–3.4, p < 0.001), while versus seed 3 it was 2.4 (95% CI 1.8–3.3, p < 0.001), with overlapping results observed also in the sub-analyses stratified by age band and APOE ε4 carrier status (Supplemental Tables 15 and 16).
The epilepsy risk in the AD population
Of the total AD population, 2069 individuals were free of epilepsy at the time of AD diagnosis, and they were enrolled to fit Cox proportional hazards models. When compared with matched controls, unadjusted HR for incident epilepsy was 14.8 (95% CI 9.7–22.5, p < 0.001). HR remained significantly elevated both after adjustment for individual covariates and in the multivariable Cox model encompassing all the covariates (Figure 5, for more details please refer to Supplemental Tables 17 and 18). Average duration of follow-up was 2.5 years in the AD cohort and 3.5 years in matched controls, while epilepsy IR was 640 per 100,000 person-years 43 per 100,000 person-years, respectively. Risk appeared higher in the subgroups with diagnosis at younger ages, with IR of 1147 and 1267 per 100,000 person-years respectively for the subgroup with diagnosis of AD before age 65 and before age 70; on the other hand, IR was lower in individuals with diagnosis after age 75, at 245 per 100,000 person-years.
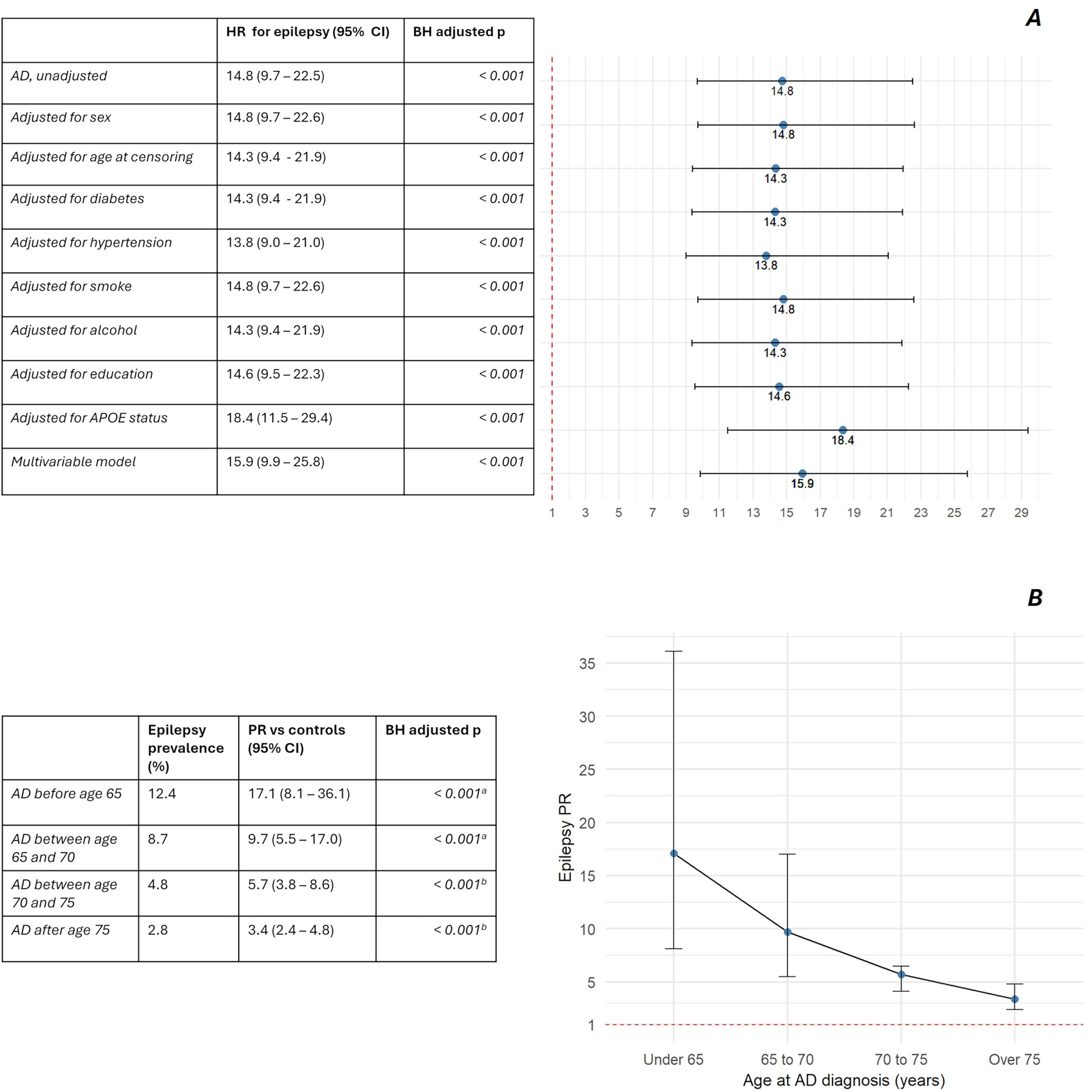
Measures of incident and prevalent epilepsy risk in the AD population. A) Forest plot of HR for incident epilepsy after adjustment for confounders in bivariate and multivariate Cox models. Further information is available in the supplemental material; B) PR of epilepsy stratified by age at AD diagnosis. Abbreviations: hazard ratio (HR), Prevalence ratio (PR), Alzheimer’s disease (AD), confidence interval (CI), Benjamini-Hochberg (BH). Figure legend: a) Fisher exact test; b) Chi-squared test.
Epilepsy prevalence was higher in AD compared to matched controls (PR 5.2, 95% CI 4.2–6.5, p < 0.001). Coherently with incident risk, PRs of epilepsy were higher in the AD subgroups with earlier diagnosis, declining with older ages (Figure 5).
Sensitivity analysis, with relaxed exclusion criteria and diagnostic requirements, estimated a similar HR for epilepsy of 15.2 (95% CI 11.7–19.9, p < 0.001). After adjustment for incident stroke as described above, HR remained elevated at 11.8 (9–15.5, p < 0.001). Expectedly, absolute epilepsy prevalence was higher in this more loosely filtered cohort, at 6.3%, as compared to 4.3% in the principal analysis.
Last, comparison against two additional seeds of matched controls revealed largely consistent results; particularly, unadjusted HR for incident epilepsy in AD versus seed 2 was 15.9 (95% CI 10.4–24.4, p < 0.001), while versus seed 3 it was 16.8 (10.9–26.0, p < 0.001) (Supplemental Table 19).
The time relation between epilepsy and AD
Of the 93 ep-AD individuals, epilepsy was earlier in 46.2%, AD was earlier in 35.5% and they were concurrent in 18.3%; In 53% of the cases, the two diagnoses were separated by an interval shorter than three years (Figure 6). Among AD-first individuals, no diagnosis of epilepsy was observed beyond six years from the time of AD onset. Age at AD diagnosis was lower in the ep-AD group than in AD without epilepsy (72.7 versus 75.5, p < 0.001).
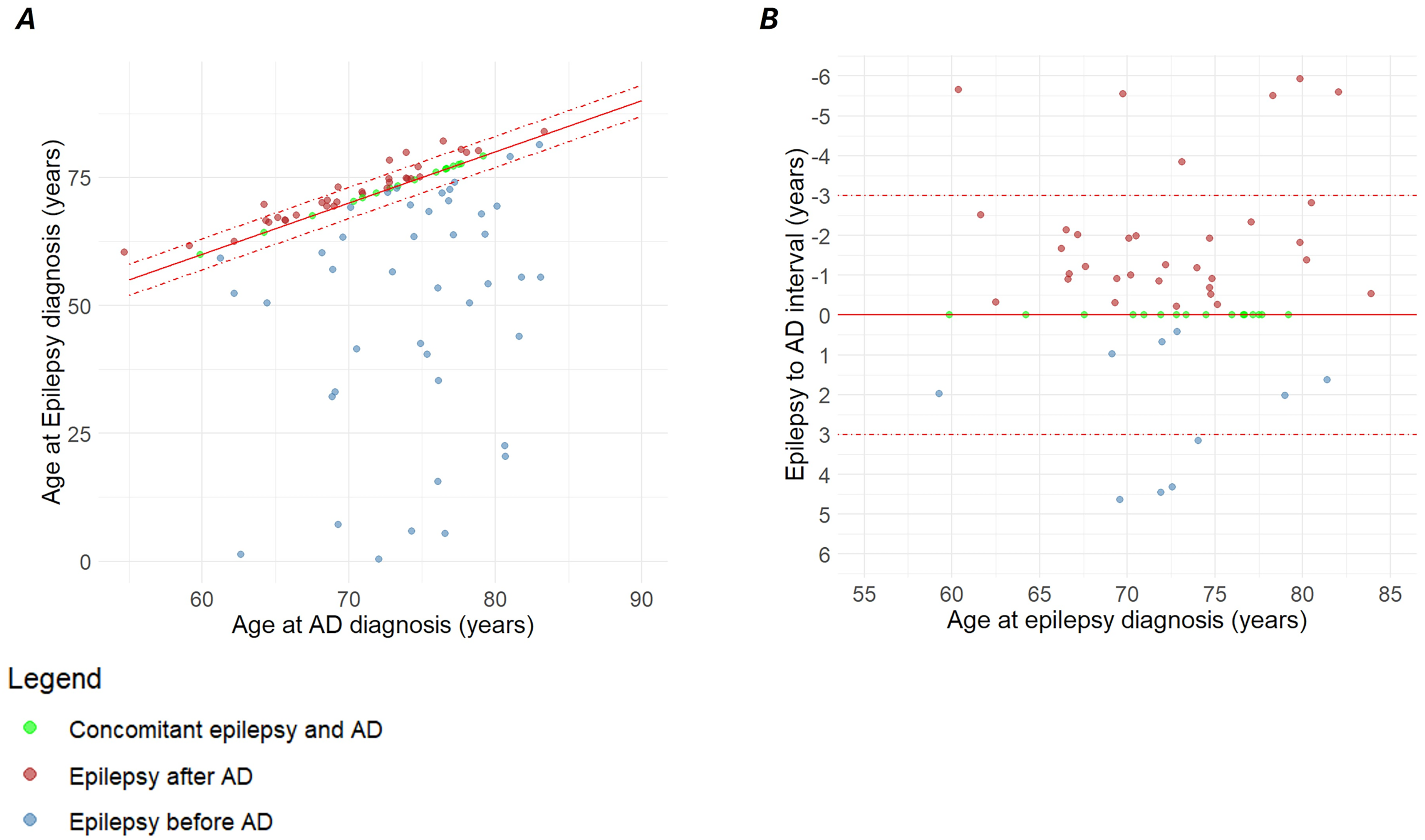
Temporal relation between AD and epilepsy diagnoses in individuals with ep-AD. A) Scatter plot of the whole ep-AD population. Concurrent diagnoses lie along the solid red line, while diagnoses separated by less than three years are located between the dashed red lines; B) Magnified view shows the detail of the latter. Abbreviations: Alzheimer’s disease (AD), epileptic-AD (ep-AD).
In the sensitivity analysis, the relative timing of epilepsy and AD remained largely unchanged, with 50% of the cases having diagnoses separated by less than 3 years. By design, the latter analysis allowed epilepsy and AD to be diagnosed in primary care and not solely in hospital settings, yet only a minority of epilepsy and AD individuals exclusively relied on this source. This prevented us to draw conclusions about the ep-AD relative timing in the primary care subgroup, due to paucity of events.
Epilepsy-FTD analysis
Prevalence of epilepsy was higher in the FTD population than matched controls, with PR of 5.3 (2.4–11.8, p = 0.001); the reverse analysis similarly showed higher FTD prevalence in the epilepsy cohort compared to controls, with PR of 5.7 (95% CI 2.3–14.1, p = 0.002). After relaxing exclusion criteria in the sensitivity analysis, prevalence of epilepsy in the FTD cohort rose to 7.3% as compared to 4.5% in the primary analysis (Supplemental Figure 1). Prevalence estimates were largely similar when comparing epilepsy and FTD populations to two additional seeds of matched controls; particularly, PR of epilepsy in the FTD group versus seed 2 was 5.4 (95% CI 2.4–12.0, p = 0.001), while versus seed 3 it was 5.0 (95% CI 2.2–11.0, p = 0.002). The reverse analysis confirmed higher prevalence of FTD in the epilepsy population (PR against seed 2: 4.00, 95% CI 1.67, 9.60, p = 0.007; PR against seed 3: 7.5, 95% CI 2.9–19.1, p < 0.001) (Supplemental Tables 20 and 21).
Epilepsy-DLB analysis
Albeit close to the significance threshold, the epilepsy prevalence in the DLB population was not different from controls, with PR of 2.4 (95% CI 1.0–5.7, p = 0.068). A similar finding was observed in the reverse analysis for the difference in DLB prevalence between IWE and controls (PR 2.6, 95% CI 1.0–6.5, p = 0.062). After relaxing exclusion criteria in the sensitivity analysis, prevalence of epilepsy in the DLB cohort rose to 3.6% as compared to 2.4% in the primary analysis (Supplemental Figure 1). Prevalence estimates were similar when comparing epilepsy and DLB populations to two additional seeds of matched controls; particularly the difference in epilepsy prevalence between the DLB cohort and seed 2 was marginally significant (PR 2.8, 95% CI 1.1–7.1, p = 0.044), and again close to the significance threshold versus seed 3 (PR 2.3, 95% CI 0.9–5.9). Consistently, in the reverse analysis, DLB prevalence was close to the significance threshold versus seed 2 (PR 2.4, 95% 1.0–5.7, p = 0.063), while it was marginally different versus seed 3 (PR 2.7, 95% CI 1.1–6.4, p = 0.049) (Supplemental Tables 22 and 23).
Discussion
Epilepsy and AD
The risk of conversion to AD was more-than-double in the UK Biobank epilepsy cohort compared to controls, aligning with previous HR estimates of 2.0–4.0 in epilepsy or LOE.11–15 These results were essentially unchanged using two additional seeds of matched controls, confirming their robustness to sampling variability.
We provided higher resolution risk estimates for incident AD stratified by age at epilepsy onset, expanding previous literature which mostly relied on binary cutoffs. With this approach, risk appeared higher in the sixth and seventh decade, despite being higher than controls even with diagnosis earlier in life.
We were able to depict these findings despite an incidence of AD in the UK Biobank that is lower than reported in other settings, likely reflecting the cohort's overall healthier and highly educated profile39,40 or earlier discontinuation of follow-up.
In our view, the consistently elevated AD risk in epilepsy populations, across different study designs and baseline characteristics, further supports the reliability of this epidemiological association, aligning with converging evidence of a robust link between AD and epileptogenesis, at the molecular, genetic and neurophysiological level.17,18,20,22–24,41
Intriguingly, a 2019 study found that cognitively unimpaired Individuals with LOE had higher prevalence of AD CSF biomarkers than controls, showing a high rate of conversion to dementia. 26 Consistent with this, a 2023 study found that mid-to-late life longitudinal reduction of plasma Aβ42/40 ratio significantly increased the risk of later LOE. 28
In this scenario, we believe our findings might support the hypothesis of LOE of unknown origin as the presenting manifestation of AD in some individuals,18,20,29 warranting close clinical monitoring in this population.
Incident AD risk seemingly declined in individuals with LOE diagnosed after age 70. Although this may partly reflect the shorter follow-up in this group, we cannot exclude that the association is weaker at this age, and further studies are needed to specifically address this question.
The HR for AD appeared higher in IWE carrying at least one APOE ε4 allele.
We believe this finding holds substantial clinical relevance, as it suggests that epilepsy could be integrated into AD risk frameworks alongside established risk factors. Furthermore, it provides proof of concept that multimodal measures of AD risk may represent a feasible approach to identify LOE cases at higher likelihood of conversion.
The latter point is especially critical, because not all populations of LOE with unknown origin exhibit abnormal AD biomarker profiles, 42 so that deep, biomarker-assisted phenotyping will be key to accurate risk stratification.
When considered as a whole, our epilepsy subgroup with onset before age 50 showed a more-than-twofold risk of incident AD compared to controls. Fewer data exist about dementia in individuals with younger-onset epilepsy, depicting an increased risk for these populations, similarly to their older counterparts.13,14
We argue that our findings reinforce the concept of seizure and epilepsy as a risk factor for AD later in life, and not solely as a possible manifestation of underlying AD.
Potentially supporting this view, evidence exists that neuronal hyperexcitability could favor the AD pathophysiological process17,18 and surgical series revealed upregulated amyloid and tau pathways in brain tissue form IWE with drug-resistant epilepsy. 43 Furthermore, studies found higher PET amyloid levels in adult individuals with young-onset epilepsy compared with controls,44,45 and an association between epilepsy and AD at the genetic level has been suggested, 46 so that the mechanisms of the epilepsy-AD interplay could even be broader.
In the reverse analysis, HR for epilepsy was high in AD, positioning at the higher end of risk estimates.7–10
Individuals with dementia and recurrent seizures have notably higher mortality and poorer cognitive performance, 47 and paroxysmal network hyperexcitability could have a direct role in worsening cognitive outcomes in AD. 19
Accordingly, our observations prompt careful screening for seizures in this population, as their recognition has meaningful prognostic implications.
Prevalence of epilepsy appeared highest in the individuals with AD before age 65, progressively declining in the older subgroups; ep-AD were significantly younger at diagnosis than their epilepsy-free counterparts. These data align with several studies depicting higher epilepsy risk in younger AD individuals,8,9,27,30 as well as a higher rate of EEG abnormalities in these subjects. 48
AD and epilepsy were separated by less than three years in half the AD-ep individuals. Albeit limited by the lack of detailed clinical information, these findings support the possible early occurrence of seizures in the course of AD, in line with previous observations 27 ; Of note, seizures in AD are often non-convulsive and can more easily go unrecognized, making harder to timely detect their onset. 27
Epilepsy and the other neurodegenerative dementias
Limited data exist about the relation between FTD and seizures, with previous findings depicting a lower incidence in FTD than AD, albeit higher than control populations. 32
In our FTD cohort, absolute prevalence of epilepsy was 4.5%, with high PR compared to controls, to a similar extent as in AD; sensitivity analysis with less strict exclusion showed higher prevalence of 7.3%. These numbers align with recent findings on a Finnish population 33 and suggest an increased lifetime seizures risk in FTD.
Despite being higher than controls, APOE ε4 carrier rate in the FTD group was markedly different from the AD population, suggesting that the contribution of misdiagnosed atypical AD to our observations is limited. This is corroborated by a higher male prevalence, in line with epidemiological observations in behavioral-variant FTD. 49
The FTD neuropathological spectrum is wide, with frontotemporal lobar degeneration (FTLD)-tau and FTLD-TDP accounting for most cases. 50 Preclinical findings suggest that aberrant tau could be related to seizures and hyperexcitability independent of Aβ deposition, 51 but molecular evidence remains sparse. Further studies are needed to shed light on the possible link between epileptogenesis and the FTLD proteinopathies.
Overall, prevalence of epilepsy appeared higher in the DLB cohort compared to matched controls, despite exceeding the significance threshold only against one control seed. Furthermore, prevalence seemed lower than AD and FTD, even with relaxed exclusion in the sensitivity analysis. Male sex was notably overrepresented in DLB, aligning with the reportedly higher risk in males, 52 while APOE ε4 carrier rate overlapped with previous estimates, 53 together suggesting good diagnostic accuracy.
Our data are consistent with earlier findings of 2.6% epilepsy prevalence in DLB, 54 and contrast with a previous study, finding similar seizure risk for DLB and AD. 32
Of note, in the latter study seizure-incidence dramatically increased at older ages; conversely, our DLB population was relatively young, with average censoring around age 77, and we think this might contribute explaining the observed differences.
Seizure assessment in DLB is complicated by the possible confounding effect of cognitive fluctuations; furthermore, establishing the seizure risk specifically attributable to DLB is hindered by the high rate of AD copathology. 55 Molecular evidence regarding the relationship between alpha-synuclein and epileptogenesis is sparse; further studies will be necessary to clarify the mechanisms of the possible DLB-epilepsy association.
Strengths and limitations
The major strength of our work is the availability of data from a large cohort, enabling us to study separate conditions against matched controls from the same population, with high statistical power. Moreover, the robustness of our findings to varying baseline assumptions was tested through extensive sensitivity analyses.
Limitations of our study include use of retrospective data, its ICD-based design, and absence of imaging or biological information. In this context, differential diagnosis of neurodegenerative dementia is constrained by phenotypic overlap, and possible residual confounding from unaccounted brain lesions, such as white matter changes. To mitigate this issue, we implemented rigid inclusion criteria to label diseases, with exclusion of most known confounders, encompassing any clinical or subclinical vascular involvement at the resolution allowed by ICD diagnoses; this reasonably allowed increased diagnostic specificity and better estimation of the associations studied, as corroborated by the APOE ε4, age and sex distributions in the dementia groups. Another limitation is the possible diagnostic misattribution of non-neurodegenerative causes of cognitive decline, such as epilepsy- or ASM-related dysfunction. However, the APOE ε4 carrier rate in the ep-AD subgroup was notably greater than the general epilepsy cohort, being highly similar to the general AD population, strongly supporting diagnostic reliability.
A further limitation of our study is that the ICD codes used convey limited information about AD clinical stages, such that the diagnosis of mild cognitive impairment due to AD is not specifically captured, preventing more detailed inference about this clinically relevant subgroup.
Despite the notion that seizures in AD are often focal,23,27 epilepsy was considered as a whole in our analysis; this was done because most enrolled individuals had a code for unspecified epilepsy (G40.9), not allowing grouping based on the presumed syndrome.
By design, all individuals in the epilepsy and AD cohorts had received care in hospital settings, raising concern for potential surveillance bias compared with controls, who were not subject to the same constraint. Nevertheless, controls were allowed to have the same comorbidities as the disease groups, with the great majority having hospital-recorded codes for alternative conditions (88.0% and 92.6% of controls of the epilepsy and AD cohorts, respectively), strongly limiting this concern.
Finally, the UK Biobank cohort is healthier and more health-conscious than the general UK population, with potential implications for the epidemiology of the diseases studied, and this could possibly limit the generalizability of our findings.39,40
Conclusions
In an era of rapid diagnostic advancement, epilepsy could offer an opportunity to identify individuals at higher risk of future or covert AD and neurodegeneration, with relevance for present and emerging treatments.
Supplemental Material
sj-pdf-1-alz-10.1177_13872877261450962 - Supplemental material for The bidirectional association of epilepsy with Alzheimer's disease and other neurodegenerative dementias: A longitudinal observational study on the UK Biobank
Supplemental material, sj-pdf-1-alz-10.1177_13872877261450962 for The bidirectional association of epilepsy with Alzheimer's disease and other neurodegenerative dementias: A longitudinal observational study on the UK Biobank by Enrico Fratto, Jolanda Buonocore, Francesco Fortunato, Ilaria Sammarra, Camilla Calomino, Basilio Vescio, Andrea Quattrone, Antonio Gambardella and Aldo Quattrone in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 147093.
ORCID iDs
Ethical considerations
UK Biobank has ethical approval from the North West Multi-Centre Research Ethics Committee.
Consent to participate
All participants provided written informed consent at recruitment. The study was conducted in accordance with the principles of the Declaration of Helsinki and the UK Biobank Ethics and Governance Framework, ensuring participant confidentiality and adherence to relevant legislation.
Consent for Publication
Not applicable
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.