
Abstract
Background
The commonality of Alzheimer's disease (AD) in the elderly suggests connections between aging and AD biology. APOE biology is also tied to AD.
Objective
We sought to link three aging hallmarks (loss of proteostasis, mitochondrial dysfunction, deregulated nutrient sensing) to APOE biology.
Methods
We altered SH-SY5Y cell proteostasis directly via heat shock, integrated stress response inhibition (ISRIB), or autophagy inhibition (chloroquine), and indirectly by perturbing mitochondria (mtDNA depletion; oligomycin). We also exposed induced pluripotent stem cell-derived neurons to ISRIB and chloroquine. Conversely, we mitigated protein stress with rapamycin. We assessed intervention impact on APOE expression.
Results
Increasing protein stress elevated and decreasing protein stress lowered APOE expression. In SH-SY5Y cells rapamycin blocked oligomycin-induced mTOR 2448 phosphorylation, Akt 473 phosphorylation, and APOE expression. In chloroquine-treated neurons rapamycin reduced mTOR phosphorylation and APOE expression.
Discussion
Protein stress initiates APOE expression and facilitates mitochondrial dysfunction's impact on APOE by engaging the mTOR pathway. Our findings link aging and AD biology.
Introduction
The incidence and prevalence of cognitive decline rises with advancing age and becomes increasingly common in elderly demographics. 1 Age-related cognitive decline syndromes often manifest as an inability to retain information, which predicts the presence of brain protein aggregations. Common aggregations include collections of intracellular tau and extracellular amyloid-β (Aβ) protein, although other proteins also accumulate. 2 The concomitant presence of cognitive decline, tau “tangles”, and Aβ “plaques” define a clinical entity, Alzheimer's disease (AD). 1 Multiple molecular parameters, though, change in AD, and an inability to place those changes within a comprehensive context, or fully understand cause and consequence relationships, ensures AD remains an uncurable, enigmatic diagnosis.
The sheer commonality of AD, along with the even higher prevalence of Aβ plaques in the elderly, who often have plaques despite the absence of measurable cognitive decline, 3 suggests connections between aging and AD biology. Proteostasis is clearly altered in AD, and a “loss of proteostasis,” defined as changes in protein balance and integrity, is a recognized “hallmark” of aging. 4 Age-related changes in proteostasis trigger cell adaptations that counter protein stress, which is a consequence of perturbed proteostasis. Age-related protein stress may present through changes in transcriptional activation, mRNA splicing, or translation; changes to post-translational modifications and oxidative stress; or inadequate protein degradation that occurs due to failing proteosome or autophagy activity. 4 Adaptive responses to acute protein stress, which may become maladaptive under chronic conditions, include the integrated stress response (ISR) and various unfolded protein responses (UPRs).5,6 ISR activation generally occurs with increasing age, while UPR activity declines.
“Mitochondrial dysfunction” is also a recognized aging biology hallmark. 4 Our understanding of how mitochondrial dysfunction drives aging is incomplete, as mitochondrial dysfunction includes a range of physiologic events that include, among others, oxidative and energy stress. Mitochondrial DNA (mtDNA) may play a critical role, as mtDNA can serve as a biological clock that accumulates changes over a lifetime and subsequently mediates a spectrum of age-related changes. 7 The mitochondrial dysfunction hallmark links to the loss of proteostasis hallmark, as mitochondria play a role in protein homeostasis,8,9 and cell events that affect protein homeostasis, such as autophagy, also influence mitochondrial integrity. 10 Changes to mitochondria that occur with advancing age are exaggerated in AD.11–14
Another aging hallmark includes “deregulated nutrient sensing.” 4 Nutrient sensing determines the anabolic versus catabolic state of a cell. In the setting of adequate nutrients, intact energy-generating infrastructures, and cell energy levels, nutrient sensing pathways promote cell growth and initiate protein, lipid, DNA, and carbohydrate synthesis. 15 During conditions of nutrient or energy scarcity nutrient sensing pathways mediate stress responses. 16 Nutrient sensing biology intertwines with mitochondrial and proteostasis biology. Prominent nutrient sensing pathways include the PI3-AKT and mTOR-related pathways, which link to each other. 17 In AD there is evidence of PI3-AKT and mTOR pathway activation. 18
This study sought to contextualize links between aging and AD biology by connecting three aging hallmarks that additionally associate with AD (protein stress, mitochondrial dysfunction, and nutrient sensing) with APOE biology. Polymorphisms in the APOE gene affect both longevity and AD risk, neuron APOE expression increases during stress states, and apolipoprotein E (APOE) protein reportedly influences protein aggregation.19–22 We also explored the recently reported connection between mitochondrial function and APOE expression,23–25 a process that appears to utilize the nutrient sensing RAS-MEK-ERK pathway. 23 Finally, we leveraged this study to explore the biological implications of manipulating AD-associated aging hallmarks with rapamycin, an mTOR antagonist that potentially mitigates aging,26–29 and impacts Aβ and tau biology. 30
Methods
SH-SY5Y ρ+ and ρ0 cell culture
SH-SY5Y cells (APOE ε3/ε3; ATCC CRL-2266) with (ρ+) or without (ρ0) mtDNA were maintained in high-glucose, L-glutamine Dulbecco's Modified Eagle Medium (DMEM; Gibco 11965) supplemented with 10% fetal bovine serum (FBS), 100 μg/mL sodium pyruvate, 50 μg/mL uridine, and 1% penicillin/streptomycin. The SH-SY5Y ρ0 cells were generated through chronic ethidium bromide exposure, with no reversion to ρ+ status occurring after the removal of ethidium bromide from the culture medium. 31 Both SH-SY5Y ρ+ and ρ0 cell lines were seeded at 25–50% density and maintained side-by-side in a humidified incubator at 37°C and 5% CO2. Cells were harvested at ∼80% density for subsequent analyses. While glial cells typically generate most of the APOE in the brain, 21 our previous observation that neuronal SH-SY5Y cells express APOE under stress conditions, 23 in conjunction with our possession of a ρ0 version of the line, led us to select this model for these studies.
Maintenance of human induced-pluripotent stem cells
An APOE ε3/ε3 human induced pluripotent stem cell (iPSC) line was obtained from Jackson labs (JIPSC001268). The iPSCs were cultured on Matrigel-coated cultureware using a feeder free system. Passaging was performed using ReLeSR (STEMCELL Technologies) to select for non-differentiated colonies.
Induction, differentiation, and maintenance of iPSC-derived forebrain neurons
Cultureware was precoated with Corning™ Matrigel™ hESC-Qualified Matrix (Fisher Scientific 08-774-552) dissolved in DMEM/F12 with GlutaMAX (ThermoFisher 10565018). iPSCs were induced into neural progenitor cells (NPCs) using the embryoid body protocol method, as outlined by STEMCELL Technologies. NPCs were grown in STEMdiff™ Neural Progenitor Medium (STEMCELL Technologies 05833) supplemented with 0.5% penicillin/streptomycin by volume (ThermoFisher 15140122). NPC media was changed daily. NPCs were grown and maintained at 12 passages or fewer. Upon reaching ∼80% confluency, NPCs were differentiated into forebrain neurons with the STEMdiff™ Forebrain Neuron Differentiation Kit (STEMCELL Technologies 08600). Forebrain neuron differentiation media was replaced every other day. After one week, forebrain neurons were replated and matured in BrainPhys™ Neuronal Medium (STEMCELL Technologies 05790) supplemented with NeuroCult™ SM1 Neuronal Supplement (STEMCELL Technologies 05711), N2 Supplement-A (STEMCELL Technologies 07152), ascorbic acid, Human Recombinant BDNF (STEMCELL Technologies 78005.1), Human Recombinant GDNF (STEMCELL Technologies 78058.1), dibutyryl cAMP (STEMCELL Technologies 100-0244), and penicillin/streptomycin. Neurons matured for 2 weeks with half-media changes every other day. All cell culture was maintained in a humidified incubator at 37°C and 5% CO2. NPCs and neurons were replated with ACCUTASE™ (STEMCELL Technologies 07920).
Heat shock
SH-SY5Y ρ+ cells were grown in a T75 flask. The flask, which contained DMEM with FBS, pyruvate and uridine, and penicillin/streptomycin was placed in a floating position in a hot water bath set to 42°C for one hour as previously described. 32 Following heat shock, the cells were immediately placed in fresh media and returned to the standard humidified CO2 and temperature-controlled conditions. Cells were harvested for downstream analyses 24 h after completing the heat shock intervention.
ISRIB treatment
SH-SY5Y ρ+ cells were incubated for 5 days in DMEM containing FBS, supplemented with pyruvate and uridine, penicillin/streptomycin, and 1 μM integrated stress response inhibitor (ISRIB) dissolved in DMSO (Sigma Aldrich SML0843). The concentration of ISRIB was guided by prior literature. 33 Neurons were incubated for 5 days in BrainPhys™ Neuronal Medium and supplements plus 1 μM ISRIB. Fresh ISRIB was added after each media change. For the neurons, half-media changes were performed every other day. SH-SY5Y ρ+ cells and iPSC-derived neurons were harvested after completing their 5-day ISRIB exposures.
Chloroquine treatment
SH-SY5Y ρ+ cells were incubated for 24 h in DMEM containing FBS, supplemented with pyruvate and uridine, penicillin/streptomycin, and 40 μM chloroquine (Sigma Aldrich C6628-25G). The 40 μM chloroquine concentration was selected based on prior literature. 34 iPSC-derived neurons in BrainPhys™ Neuronal Medium and supplements were also treated with 40 μM chloroquine for 24 h. SY5Y ρ+ cells and iPSC-derived neurons were harvested after completing their 24-h chloroquine exposures.
Oligomycin treatment
SH-SY5Y ρ+ cells were incubated for 24 h in DMEM containing FBS, pyruvate and uridine, penicillin/streptomycin, and 5 μM oligomycin. 23 The oligomycin was initially dissolved in DMSO. The SY5Y ρ+ cells were harvested at the conclusion of their 24-h oligomycin exposure.
Rapamycin treatment
SH-SY5Y ρ+ and ρ0 cells were treated with rapamycin for 1 to 7 days, with full-media changes every other day. The media consisted of DMEM with FBS, pyruvate and uridine, penicillin/streptomycin, and 20 nM rapamycin (CST9904). The rapamycin was initially dissolved in ethanol. Concentration and treatment duration was guided by prior studies. 35 The iPSC-derived neurons were maintained in BrainPhys™ Neuronal Medium and supplements, plus 20 nM rapamycin, for 7 days. Fresh rapamycin was added after each media change. For the neurons, half-media changes were performed every other day. SH-SY5Y ρ+ cells, SH-SY5Y ρ0 cells, and iPSC-derived neurons were harvested upon completing their specified period of rapamycin exposure.
Combination exposures
We performed combination exposures in which SH-SY5Y ρ+ cells were pretreated with rapamycin and then placed in medium containing oligomycin or chloroquine. These combination exposures were guided by the literature, 35 with minor modifications. The SH-SY5Y ρ+ cells were pretreated with 20 nM rapamycin (in DMEM containing FBS, pyruvate and uridine, and penicillin/streptomycin) for 24 h prior to adding 5 μM oligomycin or 40 μM chloroquine. The rapamycin-pretreated SH-SY5Y ρ+ cells were then maintained in rapamycin plus oligomycin, or rapamycin plus chloroquine, for an additional 24 h. At the end of the 24-h co-exposure period the cells were collected for assays. We also pretreated iPSC-derived neurons with 20 nM rapamycin (in BrainPhys™ Neuronal Medium and supplements) for 24 h prior to adding 40 μM chloroquine. The rapamycin-pretreated iPSC-derived neurons were then maintained in rapamycin plus chloroquine for the next 24 h. At the end of the 24-h co-exposure period the neurons were collected for assays.
MTOR knockdown
MTOR knockdown was achieved through small interfering RNA (siRNA) transfection. MTOR- and non-targeting siRNA were purchased from Dharmacon (ON-TARGETplus, SMARTPool). Transfection was performed according to the manufacturer's instructions and were guided by a previous study 23 with minor modifications. Lipofectamine RNAiMAX (ThermoFisher 13778150) was selected as a transfection reagent. Transfection components were incubated in Opti-MEM I Reduced Serum Media before mixing into penicillin/streptomycin-free DMEM, supplemented with pyruvate, uridine, and 10% FBS. Total siRNA concentration in the transfection mixtures was 50 nM. Cells were incubated in transfection media for two days, at which point they were transitioned into media containing DMEM, FBS, pyruvate and uridine, and penicillin/streptomycin. Cells were harvested after two days (four days post-transfection).
RNA extraction, cDNA preparation, and real-time PCR
SH-SY5Y ρ+ neuronal cells, SH-SY5Y ρ0 neuronal cells, and iPSC-derived neurons were washed with ice-cold phosphate buffered saline (PBS), and RNA from cell cultures was isolated using TRIzol reagent, chloroform, and isopropanol extraction as was performed in other studies.23,25 cDNA was prepared from 1 μg input RNA with BIO-RAD iScript Reverse Transcription Supermix (1708841) for reverse transcription according to their protocol. All quantitative PCR reactions were prepared with ThermoFisher TaqMan Universal Master Mix II with UNG (4440038) and gene specific ThermoFisher Taqman primers (Table 1). Real-time PCR was performed using a QuantStudio 3 Real-Time PCR System (96 well) and software. Relative gene expression was determined using the 2−ΔΔCt method. ACTB served as the endogenous reference for all qPCR assays. Each sample was tested in duplicate or greater, and the reported data reflect the average of the replicates.
Taqman primers used for quantitative PCR.

Immunoblotting
Cells were washed with ice-cold PBS and scraped into radioimmunoprecipitation assay (RIPA) buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher 1861284). Cell/RIPA suspensions were vigorously vortexed, incubated on ice for 15-min, vortexed again, and centrifuged at 14,000 g for 15 min at 4°C. The supernatant was collected in a fresh tube for analysis and loading.
For western blot immunochemistry, protein concentrations were measured with a bicinchoninic acid (BCA) assay. Samples were diluted to allow for the addition of ∼80–100 μg of protein per well and boiled with 1X Laemmli-SDS buffer. Samples were run alongside a prestained protein ladder (10 to 180 kDa, ThermoFisher, 26616). Following gel electrophoresis, we performed wet transfers to polyvinylidene difluoride (PVDF) membranes, which were blocked for one hour with bovine serum albumin (BSA) and maintained overnight in primary antibody solution at 4°C. After imaging, membranes were stripped with Restore™ PLUS Western Blot Stripping Buffer (ThermoFisher 46430) and redeveloped in primary. Quantification of targets was performed through densitometry. All targets were normalized to β-tubulin. Densitometry was performed with Image Lab Software (BIO-RAD) using protocols applied in other studies.23,25 Table 2 lists the antibodies used in this study.
Antibodies used for western blot immunochemistry.

Statistics
Data analysis was performed using GraphPad Prism statistical software. For comparisons between two groups, we used unpaired, two-way Student's t-tests, with significance defined as p
Results
We exposed human neuronal SH-SY5Y ρ+ cells to interventions intended to increase protein stress (through protein denaturation, increased protein production, or decreased protein clearance) and determined their impact on APOE mRNA expression. First, we subjected SH-SY5Y ρ+ cells to heat shock for one hour, and 24 h later we observed a 150% increase in the HSPA5 mRNA level (Figure 1A). HSPA5 encodes the GRP78/BiP protein, which is induced by protein stress in the endoplasmic reticulum and mediates an endoplasmic reticulum-based UPR. 36 24 h after the heat shock event, the SH-SY5Y APOE mRNA level exceeded the baseline level by 225% (Figure 1B).
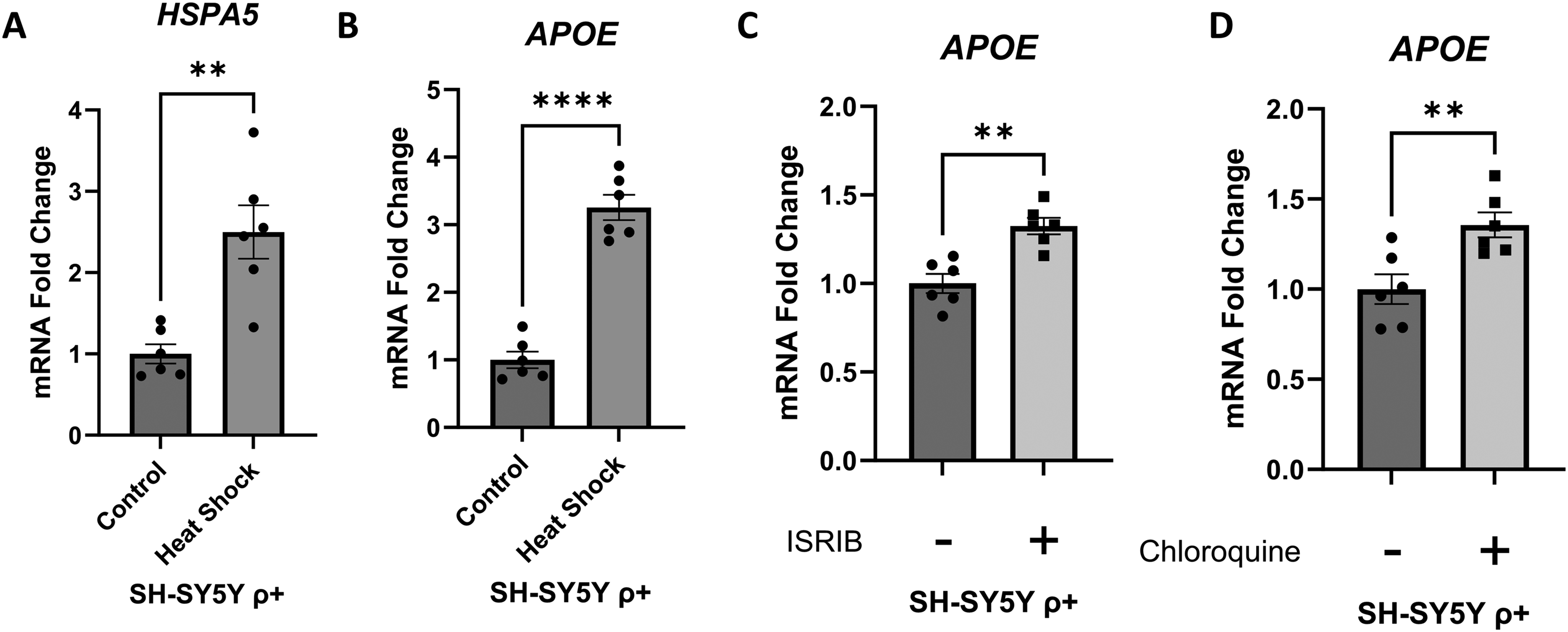
Effect of interventions that promote protein stress on SH-SY5Y ρ+ cell APOE mRNA. (A) 24 h after a heat shock intervention HSPA5 mRNA increased, which verifies activation of a heat shock response. (B) The heat shock intervention increased APOE mRNA. (C) A 5-day ISRIB exposure increased APOE mRNA. (D) A 24-h chloroquine exposure increased APOE mRNA. Error bars represent SEM. Statistics reflect unpaired, two-tailed Student's t-tests. **p ≤ 0.01, ****p ≤ 0.0001.
Next, we treated SH-SY5Y ρ+ cells with ISRIB, a chemical ISR inhibitor. 6 Under stress conditions, the ISR limits protein translation through a series of events that includes kinase-mediated phosphorylation of eIF2's α subunit at its serine 51 position (eIF2αP). 6 This phosphorylation causes eIF2B, whose physiologic role is to maintain eIF2 activity by supplying it with fresh GTP, to form an inactive eIF2αP-eIF2B complex. ISRIB prevents the formation of the inactive eIF2αP-eIF2B complex, which allows eIF2B to remain functional and protein production to proceed at less-restricted levels. 6 A 5-day exposure to ISRIB increased the SH-SY5Y ρ+ cell APOE mRNA level by ∼35% (Figure 1C).
As a third intervention we added chloroquine to the SH-SY5Y ρ+ cell medium. Chloroquine accesses lysosomes, where it buffers protons and through this action reduces lysosome acidification. 37 Reducing lysosome acidification prevents the activation of some proteolytic enzymes such as cathepsin L, as well as lysosome-autophagosome fusion.34,38 At adequate chloroquine concentrations, autophagy-mediated protein degradation cannot proceed, which disrupts proteostasis and increases protein stress. 39 A 24-h exposure to chloroquine increased the SH-SY5Y ρ+ cell APOE mRNA level by 35% (Figure 1D).
We previously demonstrated that mitochondrial dysfunction increases SH-SY5Y cell APOE mRNA expression. 23 In that earlier study, mtDNA depletion, which creates a state of respiratory chain incompetence as well as protein stress, 8 robustly increased APOE mRNA (an ∼65-fold increase) and APOE protein levels. 23 To now determine whether protein stress contributes to increased APOE expression in cells with impaired mitochondria, we leveraged SH-SY5Y ρ0 cells. We assessed the impact of reducing protein stress on SH-SY5Y ρ0 cell APOE mRNA expression by treating them with rapamycin, a drug that inhibits the mTORC1 complex. 28 mTORC1 inhibition activates autophagy and reduces translation, both of which alleviate protein stress. 40 A 72-h exposure to 20 nM rapamycin decreased mTOR phosphorylation at its serine 2448 position, when normalized to tubulin or referenced to total mTOR protein (Figure 2A). A change in the mTOR serine 2448 phosphorylation level does not by itself necessarily establish a state of mTOR activation or inhibition but does indicate the rapamycin concentration we used was sufficient to engage mTOR-related pathways, and suggests the rapamycin treatment reduced mTORC1-mediated phosphorylation of its p70 ribosomal S6 kinase (p70S6 K) target, as mTORC1-activated p70S6 K in turn phosphorylates mTOR at its serine 2448 position. 41

Effect of rapamycin on SH-SY5Y ρ0 cell mTOR activity, markers of protein homeostasis, and APOE mRNA. (A) A 72-h rapamycin exposure decreased mTOR serine 2448 phosphorylation and decreased the phosphorylated mTOR/total mTOR ratio. (B) The 72-h rapamycin exposure decreased 4E-BP1 threonine 70 phosphorylation and the phosphorylated 4E-BP1/total 4E-BP1 ratio. (C) Over the course of a 1-week exposure period rapamycin progressively lowered APOE mRNA levels. Statistics reflect unpaired, two-tailed Student's t-tests (A and B) and one-way ANOVA followed by a Tukey test (C). Error bars represent SEM. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
To further evaluate rapamycin's impact on SH-SY5Y ρ0 cell proteostasis, we assessed the phosphorylation status of 4E-BP1, a protein that modifies cell protein translation. 42 In its unphosphorylated form, 4E-BP1 forms a complex with eIF4E. In its free, unbound state, eIF4E initiates translation, but when complexed to unphosphorylated 4E-BP1 it is inactive. Unphosphorylated 4E-BP1, therefore, essentially acts as a “brake” on protein synthesis. mTORC1 phosphorylates 4E-BP1, which prevents it from binding to eIF4E. 42 Through this effect mTORC1 signaling promotes protein synthesis. Rapamycin (after 3 days) decreased 4E-BP1 phosphorylation, when normalized to tubulin or referenced to total 4E-BP1 protein, which indicates mTORC1 inhibition and is consistent with a decrease in translation that would predictably reduce protein stress (Figure 2B).
Over the course of a 1-week rapamycin treatment, SH-SY5Y ρ0 cell APOE mRNA levels progressively fell. After 1 day it was reduced by 25% relative to the baseline level, and by 7 days it was reduced by 75% from the baseline (Figure 2C).
In the SY5Y ρ+ cells, rapamycin effectively prevented the chloroquine-induced increase in APOE mRNA (Supplemental Figure 1A). To address the question of whether rapamycin's impact reflected mTOR inhibition versus an off-target effect, we used siRNA to knock down MTOR in SH-SY5Y ρ0 cells. Relative to SH-SY5Y ρ0 cells treated with a non-targeting siRNA, the ρ0 cells that received the MTOR-specific siRNA showed an ∼50% reduction in their APOE mRNA level (Supplemental Figure 1B).
We further explored the mitochondrial dysfunction-proteostasis-APOE expression nexus by exposing SH-SY5Y ρ+ cells to oligomycin, which inhibits ATP synthase (complex V) of the respiratory chain, 43 in the absence and presence of rapamycin. Oligomycin by itself (over the course of a 24-h treatment) increased mTOR serine 2448 phosphorylation, which indicates ATP synthase inhibition affected mTOR signaling (Figure 3A). Rapamycin (begun 24 h prior to the introduction of oligomycin and continued for the duration of the 24-h oligomycin treatment) prevented the oligomycin-induced mTOR phosphorylation, which is consistent with a role for mitochondria-triggered protein stress in driving that interaction (Figure 3A).
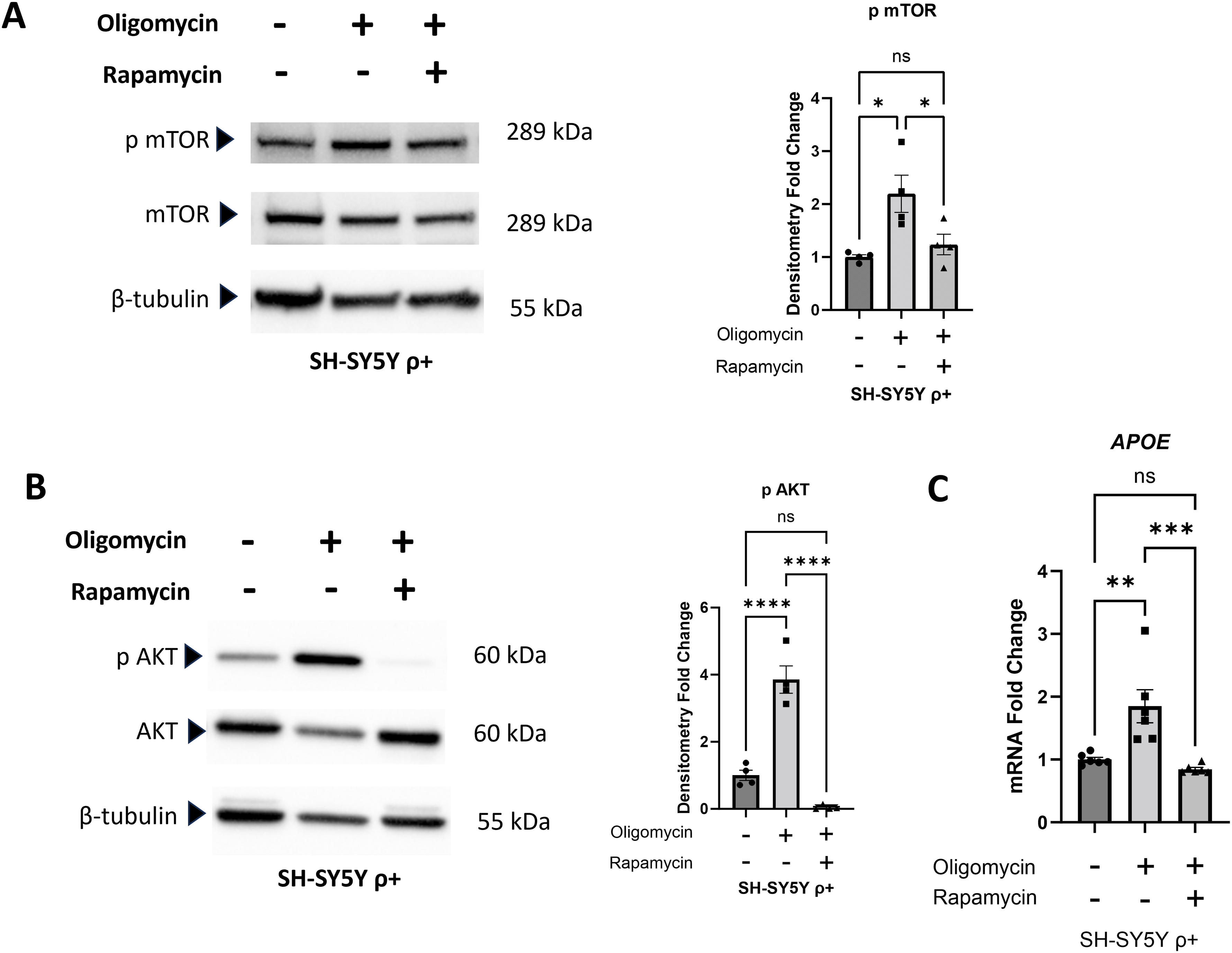
Effect of oligomycin and oligomycin plus rapamycin on SH-SY5Y ρ+ cells. (A) A 24-h oligomycin exposure increased mTOR serine 2448 phosphorylation, and rapamycin, added 24 h prior to the oligomycin and continued during the oligomycin exposure, prevented that increase. (B) The oligomycin increased Akt serine 473 phosphorylation, and rapamycin prevented that increase. (C) Oligomycin increased the APOE mRNA level, and rapamycin prevented that increase. Statistics reflect one-way ANOVA followed by a Tukey test. Error bars represent SEM. ns = not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Because oligomycin increases mitochondrial free radical production to increase oxidative stress, 44 oxidative stress activates the AKT pathway by promoting Akt serine 473 phosphorylation, 45 and Akt affects mTOR function, 46 we assessed the state of AKT pathway activation by measuring its level of Akt serine 473 phosphorylation. Oligomycin robustly increased SH-SY5Y ρ+ cell Akt serine 473 phosphorylation, while rapamycin completely blocked this effect (Figure 3B). This finding suggests rapamycin, at least in this experiment in which SH-SY5Y ρ+ cells were maintained in rapamycin over a 2-day period, also reduced mTORC1 signaling in part by acting upstream of mTORC1. Oligomycin by itself increased SH-SY5Y ρ+ cell APOE mRNA levels, while rapamycin was able to block that effect (Figure 3C).
We extended our analyses to include human iPSC-derived, APOE3-homozygous forebrain neurons. ISRIB, which can increase protein synthesis, 47 when administered for 5 days boosted neuron APOE mRNA expression (Figure 4A).
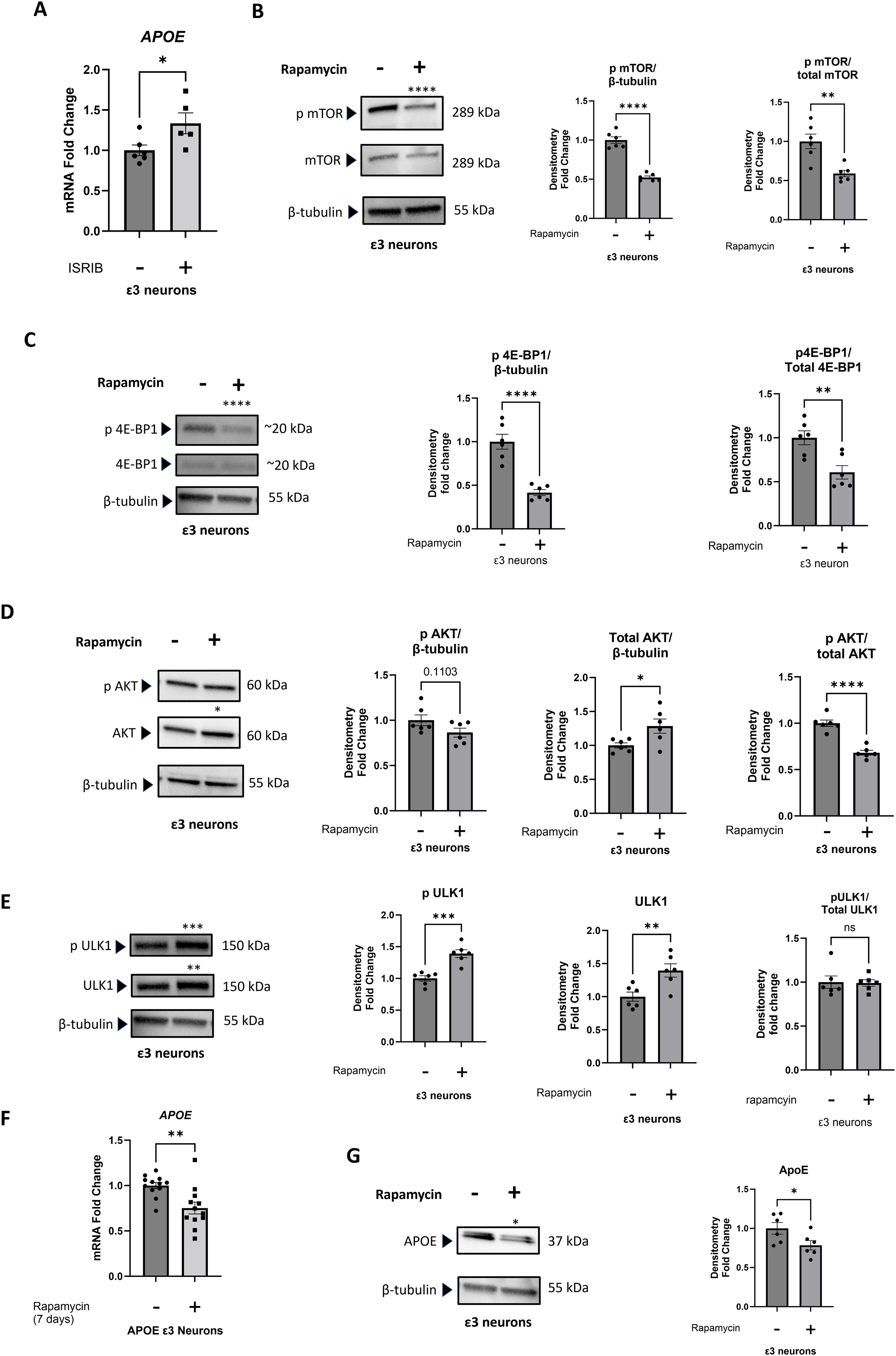
Effect of ISRIB and of rapamycin on iPSC-derived neurons. (A) A 5-day ISRIB exposure increased APOE mRNA. (B) A 7-day rapamycin exposure decreased mTOR serine 2448 phosphorylation and the phosphorylated mTOR/total mTOR ratio. (C) The 7-day rapamycin exposure decreased 4E-BP1 threonine 70 phosphorylation and the phosphorylated 4E-BP1/total 4E-BP1 ratio. (D) The 7-day rapamycin exposure increased the total Akt protein, and the phosphorylated Akt serine 473/total Akt ratio decreased. (E) The 7-day rapamycin exposure increased levels of both serine 757 phosphorylated ULK1 and total ULK1, with no change in the ratio between the two. (F) The 7-day rapamycin exposure decreased the amount of APOE mRNA. (G) The 7-day rapamycin treatment decreased the amount of APOE protein. Statistics reflect unpaired, two-tailed Student's t-tests. Error bars represent SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
To evaluate rapamycin's impact on the iPSC-derived neurons, we confirmed target engagement by the 20 nM concentration. As was the case with the SH-SY5Y ρ0 cells, 20 nM of rapamycin, administered over 7 days, decreased mTOR serine 2448 phosphorylation (when normalized to tubulin or referenced to total mTOR protein) and 4E-BP1 phosphorylation (when normalized to tubulin or referenced to total 4E-BP1 protein) (Figure 4B, C). As was the case with the SH-SY5Y ρ0 cells, rapamycin also decreased the phosphorylated serine 473 Akt to total Akt ratio; in the iPSC-derived neurons this was partly driven by an increase in the total Akt protein level (Figure 4D).
We additionally assessed the status of ULK1, a protein kinase that regulates autophagy. 48 ULK1 is phosphorylated at its serine 757 position by mTORC1. 49 ULK1 serine 757 phosphorylation prevents its interaction with AMPK, which independently phosphorylates ULK1 at a different site to activate autophagy. 50 mTORC1-mediated ULK1 serine 757 phosphorylation, therefore, inhibits autophagy. Rapamycin's effect on ULK1 was complex. With rapamycin the absolute amount of ULK1 serine 757 phosphorylation unexpectedly increased (when normalized to tubulin), although this occurred within the context of increased total ULK1 protein and the ratio of serine 757 phosphorylated to unphosphorylated ULK1 did not change (Figure 4E).
The 1-week rapamycin treatment decreased the iPSC-derived neuron APOE mRNA level (Figure 4F). The neuron APOE protein level also decreased (Figure 4G).
mTOR serine 2448 phosphorylation trended higher in neurons treated with chloroquine for 24 h, but this was not a significant increase (p = 0.1). A 24-h pretreatment with rapamycin, added before the addition of the chloroquine, resulted in an mTOR serine 2448 phosphorylation level that was less than it was in both the chloroquine-treated and untreated neurons (Figure 5A). The chloroquine treatment increased neuron APOE mRNA and protein levels, while rapamycin prevented those increases (Figure 5B, C).
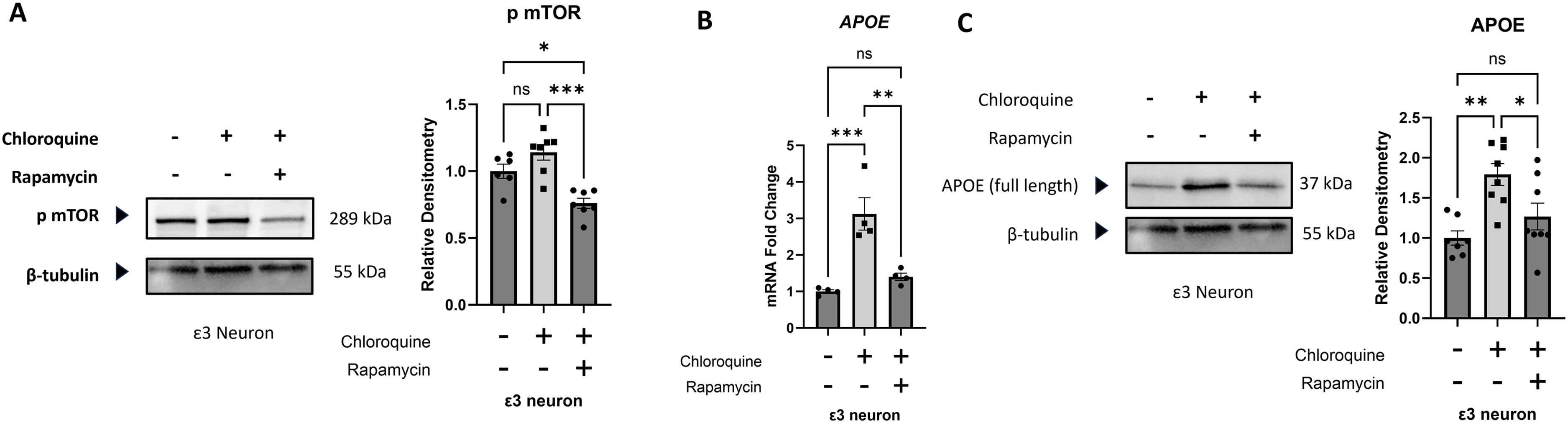
Effect of chloroquine and chloroquine plus rapamycin on iPSC-derived neurons. (A) Rapamycin, given 24 h prior to adding chloroquine and then continued for the next 24 h in the presence of chloroquine, reduced mTOR serine 2448 phosphorylation to a level that was below the level seen in both chloroquine treated and untreated iPSC-derived neurons. (B) The 24-h chloroquine exposure increased APOE mRNA, and the 24-h rapamycin pretreatment with continued treatment during the chloroquine exposure prevented that increase. (C) The 24-h chloroquine exposure increased the neuron APOE protein level, and the 24-h rapamycin pretreatment with continued treatment during the chloroquine exposure prevented that increase. Statistics reflect one-way ANOVA followed by a Tukey test. Error bars represent SEM. ns = not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Discussion
Neuron and neuronal cell APOE expression increases with stress, but the reasons for this are unclear. 21 APOE is implicated in AD, 19 a disease that is notable for protein stress, so we assessed whether protein stress might impact APOE expression. We considered different ways to manipulate protein stress, including an induction of protein misfolding through heat shock, increasing protein production by enhancing translation, and decreasing autophagy-dependent protein clearance. We further looked at the converse impact associated with decreasing protein stress through rapamycin. These are relatively dirty approaches that could have multiple effects, but which share an impact on protein stress. The aggregate of our findings suggests protein stress increases neuron and neuronal cell APOE mRNA expression.
We were interested in linking this theme to mitochondrial dysfunction, as mitochondrial dysfunction occurs in AD,11–14 and we previously demonstrated in SH-SY5Y cells that mitochondrial dysfunction, including mitochondrial dysfunction induced by oligomycin, initiates APOE expression.23,25 Mitochondrial dysfunction also causes protein stress,8,9 and our current finding that rapamycin attenuates oligomycin-induced APOE expression in SH-SY5Y cells suggests protein stress may at least partly mediate oligomycin-induced APOE expression.
What mitochondria-induced increases in APOE expression are intended to accomplish is unclear and further complicated by the fact that neuron-generated APOE is handled differently than it is in cells that normally express it, such as astrocytes. For example, APOE secretion by neurons is less efficient than it is in astrocytes, 51 which may facilitate its retention and a subsequent targeting of the protein or its cleavage products to mitochondria. 52 At mitochondria, APOE or its cleavage products reduce respiratory chain function. 53 APOE-induced respiratory chain inhibition of normal mitochondria would seem to reflect an inherently pathological event, but a mitochondrial dysfunction-initiated APOE attack on failing mitochondria raises the possibility that neuron APOE expression evolved as an adaptation-intended response. Although APOE is characterized as a stress-induced protein in neurons, 21 our observed activation of APOE expression by the ISR inhibitor ISRIB nevertheless suggests APOE is potentially not an ISR-induced stress protein.
eIF2 phosphorylation serves as a convergence point for and mediator of the ISR. 6 eIF2α phosphorylation reduces general protein translation while strategically altering, and in some cases activating, the translation of specific proteins. Among the proteins that increase in quantity or otherwise activate during protein stress conditions are the C/EBP proteins and the C/EBP homologous protein (CHOP).6,54,55 We previously showed that CEBPA, which encodes the C/EBPα transcription factor, contributes to increased APOE expression in SH-SY5Y ρ0 cells. 23 CEBPA gene expression is influenced by the ISR, and by mTOR and rapamycin.6,55,56 While the reported impact of mTOR signaling on CEBPA is complex, 55 our current data are consistent with the view that part of the mitochondria dysfunction-triggered pathway that turns on APOE expression in SH-SY5Y ρ0 cells is activated through mTOR signaling, and that this regulation could involve CEBPA.
The question arises as to why a state of mitochondrial dysfunction, especially one that results in energy deficiency, would activate mTOR signaling. mTOR activation associates with anabolic states that promote cell growth and even cell division.57–59 Cells behave in an anabolic fashion when they are energy-rich, but oligomycin-treated cells should be energy-poor. We wonder whether respiratory chain failure, which causes energy deficiency that should trigger a catabolic response, has a perhaps unique ability to confuse the cell into thinking it is energy-rich even when it is not. For example, respiratory chain failure elevates NADH/NAD + ratios 60 which could be misinterpreted by the cell as an anabolism-supporting energy environment. If so, respiratory chain dysfunction in neurons might give rise to a “pseudo-anabolic state,” in which an energy-deficient cell activates pathways that would normally associate with being energy-rich.
It is interesting that rapamycin prevented Akt 473 phosphorylation. mTORC2 causes this phosphorylation, 61 and rapamycin was classically believed to not inhibit mTORC2. 62 Indeed, this understanding contributed to the naming of the mTORC2 associated protein Rictor, which is an acronym for “rapamycin-insensitive companion of mTOR.” 63 Subsequent data, though, showed chronic rapamycin exposure also blocks mTORC2. 64 Our data are consistent with the latter view.
We did not interrogate the relevant mechanism via which oligomycin increased Akt 473 or mTORC1 2448 phosphorylation, but current literature can guide speculation. By inhibiting the mitochondrial ATP synthase, oligomycin should decrease ATP production and increase oxidative stress. 44 Oxidative stress has been shown to induce Akt 473 phosphorylation. 45 In addition to boosting mTORC1 activity through TSC1/2 and Rheb, 17 Akt activated through serine 473 phosphorylation could then phosphorylate PRAS40, which in its unphosphorylated state binds the mTORC1 Raptor component and through this association inhibits mTORC1. 65 PRAS40 phosphorylation, therefore, would prevent PRAS40 binding to Raptor, reduce PRAS40-mediated mTORC1 inhibition, boost mTORC1 signaling, and increase mTOR serine 2448 phosphorylation.
Akt phosphorylation can occur in different contexts and could represent a sign of cell stress or of cell growth.17,46 It is hard to know from our experiments whether the oligomycin-induced increase in Akt 473 phosphorylation reflects a stress signal, growth signal, or both, but in each case that phosphorylation could conceptually reflect the presence of a pseudo-anabolic state.
When it comes to AD therapeutic development, the APOE gene and APOE are considered rational targets. 66 How to manipulate APOE or APOE, though, is unclear, with some advocating an increase in its actions to overcome a loss of function, and others a decrease in its actions to attenuate a toxic gain of function. 67 Precision approaches that consider isoform differences are increasingly proposed, and include introducing APOE2, with the hope that it will function as a trophic factor, and suppressing APOE4, which experimental data suggest has toxic properties.68,69 In further deference to the complicated nature of this question, it is necessary to consider that if APOE4 in fact does not increase AD risk by acting as a toxin but rather increases AD risk through physiologic hypofunction, 68 a case could be made for increasing its level. Regardless, rapamycin or rapamycin analogs (rapalogs) could offer a practical way to reduce APOE expression.
Epidemiology studies that consistently show an AD-APOE isoform association frame the APOE ε3/ε3 genotype as risk-neutral but do not resolve the mechanism or mechanisms that mediate the association. 66 The observation that neurons, which don’t normally express APOE, do so under stress conditions 21 makes it necessary to consider the possibility that neuron APOE expression, when it occurs, is simply neurotoxic. Under this paradigm, any APOE isoform expressed by a neuron might harm the neuron, with APOE4 having the maximum and APOE2 the minimum adverse impact. If correct, reducing neuron APOE expression in general would theoretically make sense, with the inferred caveat that reducing APOE4 would confer a greater benefit than reducing APOE3, and reducing APOE3 would confer a greater benefit than reducing APOE2. By extension, APOE ε4/ε4 homozygotes would be expected to benefit more than APOE ε4 heterozygotes, and APOE ε3/ε3 homozygotes would be expected to benefit more than APOE ε3/ε2 heterozygotes. Given the uncertainties over how exactly APOE impacts AD risk, however, it is necessary to consider manipulating APOE biology in any form could have unintended consequences.
There is interest in using rapamycin to treat AD. 70 The rationale for this relates to the fact that AD is an aging-associated disease and rapamycin may minimize aging-related molecular changes,26–29 in conjunction with the observation that mTOR pathway activity is increased in the brains of AD patients. 18 In preclinical studies using transgenic AD modeling rapamycin mitigates pathology.71,72 Human clinical trial data are limited, but a phase 1 study could not verify orally administered rapamycin accessed the brain. 70 On the other hand, memory consolidation is believed to require new protein synthesis, 73 ISRIB was previously shown to enhance rodent memory formation, 74 and data argue ISRIB could benefit diseases such as AD that feature ISR activation.75,76 Multiple factors, such as the extent of stress present within an aging or AD brain, the underlying drivers of that stress, and effect on general versus stress protein expression may influence the impact (if any) of interventions intended to attenuate or enhance protein synthesis.
Our study has limitations. As we previously stated, the effects of our experimental interventions were not limited to increasing or decreasing protein stress. There are also modelling considerations. SH-SY5Y cells are neuronal, but grow and divide, and as is the case with tumor cells they can tolerate profound levels of mitochondrial dysfunction. 31 Recapitulating the basic SH-SY5Y findings in iPSC neurons is reassuring, but these are still cultured cells, and not brains.
Overall, our study supports the view that in neurons and neuronal cells mitochondrial dysfunction contributes to protein stress, and protein stress in turn generates its own consequences and triggers its own set of responses. One of these responses is an increase in APOE expression, which reportedly occurs during AD's initial clinical stages. 77 APOE expression by a compromised neuron may represent part of a stress response whose intent is to promote a better outcome for that neuron, but in the context of a pseudo-anabolic state arising due to respiratory chain dysfunction APOE expression could deliver a detrimental second mitochondrial hit. In essence, an APOE-mediated “braking” of mitochondria, under conditions interpreted by the cell to represent a full energy charge, could exaggerate an actual energy-deficient state. On a broader level, our findings are consistent with a mitochondrial cascade hypothesis that proposes mitochondrial dysfunction initiates a series of events that ultimately ties aging to AD, and initiates a state of protein stress that is a well-recognized feature of AD. 11 Our findings further suggest pervasive overlap exists between aging hallmark and AD biology.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261452598 - Supplemental material for An aging hallmark, Alzheimer's disease, and APOE nexus
Supplemental material, sj-docx-1-alz-10.1177_13872877261452598 for An aging hallmark, Alzheimer's disease, and APOE nexus by Alexander P. Gabrielli, Ian Weidling, Colton R. Lysaker, Lesya Novikova, Amol Ranjan, Xiaowan Wang, Heather M. Wilkins and Russell H. Swerdlow in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
This study was supported by the University of Kansas Alzheimer's Disease Research Center (P30 AG072973). RHS is supported by P30 AG072973, R01 AG061194, the Thompson Foundation, the Clune Family Foundation, the Snyder Family Foundation, the Ruble Family Foundation, and the Stop Alzheimer's Now Foundation. IW is supported by an NIH T32 fellowship (T32 AG078114).
Ethical considerations
Not applicable.
Consent to participate
Not applicable.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the University of Kansas Alzheimer's Disease Center (grant number P30 AG072973), R01 AG061194, the Alzheimer's Disease Research Center's Brain Health Training Program (grant numbers P30 AG072973, T32 AG078114), the Thompson Foundation, the Clune Family Foundation, the Snyder Family Foundation, the Ruble Family Foundation, and the Stop Alzheimer's Now Foundation.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Russell H. Swerdlow and Heather M. Wilkins are Editorial Board Members of this journal but were not involved in the peer-review process nor had access to any information regarding its peer-review. The remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data will made available upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.