
Abstract
The ataxia telangiectasia mutated (ATM) kinase is a key transducer of the cellular response to DNA double strand breaks and its deficiency causes ataxia-telangiectasia (A-T), a pleiotropic genetic disorder primarily characterized by cerebellar neuropathy, immunodeficiency and cancer predisposition. While enormous progress has been achieved in elucidating the biochemical and functional regulation of ATM in DNA damage response, and more recently in redox signalling and antioxidant defence, the factors that make neurons in A-T extremely vulnerable remain unclear. Given also that ATM knockout mice do not recapitulate the central nervous system phenotype, a number of human neural stem cell (hNSC) model systems have been developed to provide insights into the mechanisms of neurodegeneration associated with ATM dysfunction. Here we review the hNSC systems developed by us an others to model A-T.
Introduction
Ataxia-telangiectasia (A-T) is a hereditary neurodegenerative and cancer-predisposing syndrome arising from inactivating mutations in the ataxia telangiectasia mutated (ATM) gene, which encodes a nuclear protein kinase member of the phosphoinositide 3-kinase (PI3K)-related protein kinase (PIKK) family.1,2 In response to DNA double-strand breaks (DSBs), ATM becomes rapidly activated and phos-phorylates a plethora of substrates (.1000) implicated in cell cycle checkpoint arrest, DNA repair, chromatin re-modelling and apoptosis. 3
Due to the central role of ATM in signalling and responding to DNA DSBs, cells from A-T patients are hypersensitive to ionizing radiation and defective in cell cycle checkpoint arrest.1,2 The most prominent neuropathological finding in A-T is cerebellar ataxia, due to progressive loss of Purkinje and granule cells, and of neurons in striatum and substantia nigra. 2 Although the neurodegenerative phenotype has been attributed to a defective DDR in pre- and postmitotic neurons, oxidative stress and reduced antioxidant defence may also play a role. For instance, A-T patients show persistent oxidative stress at cellular level, and also brains and DNA from ATM knock-out mice exhibit increased signatures of reactive oxygen species.4,5 Furthermore, dietary supplementation of NAC (N-Acetyl-Cysteine) ameliorates oxidative DNA damage 6 and protects Purkinje cells from redox-induced death. 7 How ATM deficiency augments oxi-dative stress was unclear until recently, when it was demonstrated that oxidants induce disulfide bond formation in ATM causing its activation 8 through a mechanism clearly distinct from that elicited by DNA breaks. Hence, ATM works in redox sensing and signalling, and the loss of redox balance may be central to the neuropathogenesis of A-T. Another event which could be particularly noxious to Purkinje and granule cells in A-T is the unscheduled re-entry of postmitotic neurons into the cell cycle, as this initiates apoptosis and can occur in response to stimuli like chronic inflammation, hypoxia and oxidative stress. 9 Moreover, in neurons, a fraction of ATM is localized in the cytoplasm 10 where it interacts with the synaptic proteins VAMP2 and Synapsin-1 11 and this evidence raises the possibility that the neurodegenerative phenotype in A-T may also reflect an impaired cytoplasmic activity of ATM separate from its nuclear activity. Most recently, neurodegeneration in A-T has been linked to the loss of the histone deacetylase HDAC4 in the cytoplasm and accumulation in the nucleus with consequent suppression of neuronal gene expression. 12 Notably, ATM deficiency also severely impairs glial cell functionality and vascular integrity, suggesting that cerebellar degeneration in A-T is the result of a dysfunctional neuro-astro-vascular unit. 13 In summary, several factors are implicated in the neurodegenerative phenotype of A-T, but which of them plays the most crucial role is still debated, primarily because of the unavailability of human model systems able to re-capitulate the neurological disease in vitro.
Here, we review the human neural stem cell (hNSC) systems that have been established as an attempt to study the outcome of ATM inactivation in relation to the neuro-glial differentiation and response to genotoxic agents and oxidative-stress, with the overall goal to get an insight into the molecular mechanisms of neurodegeneration in A-T.
In vitro NSC models of A-T
Neural stem cells (NSCs) are self-renewing, multipotent precursors localized in specialized regions of the embryonic and adult central nervous system (CNS) and participate in its homeostasis. Genetically stable NSCs have been established from multiple locations within the mammalian brain and shown to retain in vitro self renewal capacity and differentiation potential into neurons, astrocytes and oligodendrocytes.14,15 hNSCs derived from anatomically distinct fetal brain regions such as midbrain and cortex can be maintained in vitro as neurospheres in the presence of epidermal growth factor and fibroblast growth factor-2, and can be induced to differentiate into the three major CNS cell types, neurons, astrocytes and oligodendrocytes, in proportions that mirror their physiological distribution. In particular, they can generate neurons with a GABAergic, glutamatergic or dopaminergic phenotype and electrophysiological action potentials, and upon transplantation these cells can improve sensorimotor functions and generate synaptic junctions.16–18 To get a long-term supply of cells, primary hNSCs have also been immortalized with v-myc (ihNSCs) and shown to retain self-renewal and multipotential capacity.16–18 Examples of ihNSCs derived from different fetal brain regions and denoted ReNcellVM, 16 ReNcellCX 17 and ihNSC, 18 are shown in Figure 1a. It can be seen that after 7–17 days of in vitro differentiation these hNSCs give rise to 5–20% neurons positive for β-Tubulin III and microtubule associated protein (MAP2), 1–5% galactocerebroside (GalC) and 2'3'-clyclic nucleotide 3'-phosphodiesterase (CNP) positive oli-godendrocytes and a large proportion (60–75%) of glial fibrillary acidic protein (GFAP)-positive astrocytes. The expression of these proteins during differentiation of ihNSC and ReNcell VM is shown in the Western blots in Figure 1b. The impact of ATM on hNSCs has been generally studied in two ways, either by chemical inhibition of ATM with KU-55933 19 or by stable transduction of lentiviral vectors carrying shRNAs designed to knock down ATM, as shown in Figure 1c and in a previous study. 15 Another model system useful for assessing the effects of loss or inactivation of ATM consists in human embryonic stem cells (hESCs), which possess self-renewal capacity and ability to differentiate into all tissues of the organism, and in particular can be directed to differentiate into neural progenitors and mature neurons and astrocytes. The role of ATM in hESCs and their neuronal progenitors and mature descendents has been studied in vitro through both stable knock down of ATM with lentiviral shRNAs20,21 and chemical inhibition with KU-55933. 22
In vitro NSC models of A-T. (a) Human NSCs denoted
immortalization of human neural stem cells (ihNSC), ReNcellVM and
ReNcellCX were induced to differentiate for 14–17 d without EGF and
FGF-2 into the three major central nervous system cell types, identified
by immunofluorescence with antibodies specific for neurons (β-Tubulin
III and MAP2), astrocytes (GFAP), and oligodendrocytes (GalC). In (b),
the expression of Nestin (marker of multipotent neural stem cells) and
of differentiation markers was analysed by Western blots at different
days of differentiation of ihNSC and ReNcellVM. In (c) ihNSC and
ReNcellVM stably transduced with lentiviral vectors carrying shRNAs
designed to knock down ATM or a control sequence, were assessed for ATM
protein expression. Protein loading per lane was verified with
anti-β-Actin antibody. A-T, ataxia-telangiectasia; NSC, neural stem
cells; ihNSC, immortalized human neural stem cells; EGF, epidermal
growth factor; FGF-2, fibroblast growth factor (A color version of this
figure is available in the online Journal)
Interestingly, neuronal models of A-T based on the use of patients’ tissues are currently being developed. In one case, olfactory mucosa-derived neurospheres have recently been established from several A-T patients and these cells have been shown to differentiate in vitro into neurons, astrocytes and oligodendrocytes. 23
A major breakthrough in stem cell research came from the discovery that human somatic cells can be re-programed into induced pluripotent stem cells (iPSCs). Owing to their capacity to differentiate into pure populations of specific cell types, iPSCs provide a unique opportunity for disease modeling. Taking advantage of this technology, we and others have re-programed fibroblasts from A-T patients and normal donors24,25 obtaining iPSCs from which we have established proliferating neural precursor cells (NPCs) that can differentiate into neurons expressing GABAergic and glutamatergic markers and which possess electrophysiological function upon maturation. 24 The phe-notypic characterization of the re-programed cells, which express the pluripotency markers Oct3/4, SSEA4 and Tra-1-81 as well a cartoon depicting the protocol to generate the NPCs is reported in Figure 2a. Quite significantly, upon differentiation the iPSC-derived NPCs yield a large proportion of β-Tubulin III and MAP2-positive neuronal cells (80–95%), the remaining being oligodendrocytes and astrocytes (Figure 2a, bottom). This evidence is also confirmed by Western blot analysis showing that NPCs express, as expected, Nestin but not Tra-1-81 (Figure 2b) and upon differentiation dramatically upregulate the levels of β-Tubulin III, while GFAP concentrations remain minimal. The characterization of an A-T derived hiPSC clone, negative for ATM protein expression, is shown in Figure 2c.
Characterization and neural differentiation of (hiPSCs). (a) Human
primary fibroblasts re-programed using a polycistronic lentivirus
expressing the OKSM transcription factors give rise to iPSC colonies
which express the pluripotency markers Oct3/4, SSEA4 and Tra-1-81. The
cartoon shows how, through the formation of embryoid bodies and of
neural rosettes, iPSCs generate proliferating NPCs which, upon 14 d of
differentiation, give rise to a large fraction of β-Tubulin III and
microtubule associated protein (MAP2)-positive neurons and a small
fraction of GalC-positive oligodendrocytes and glial fibrillary acidic
protein (GFAP)-positive astrocytes. (b) The Western blot shows the
downregulation of the pluripotency markers Oct3/4 and Tra-1-81 in
iPSC-derived NPCs and the upregulation of the neural markers Nestin
(D0), which is downregulated during further differentiation, β-Tubulin
III and GFAP (D14). In (c) iPSCs derived from fibroblasts of an A-T
patient do not express the ATM protein, but they express the same levels
of Oct3/4 as normal control (Ctrl) iPSCs. NPCs, neural precursor cells;
ATM, ataxia telangiectasia mutated (A color version of this figure is
available in the online Journal)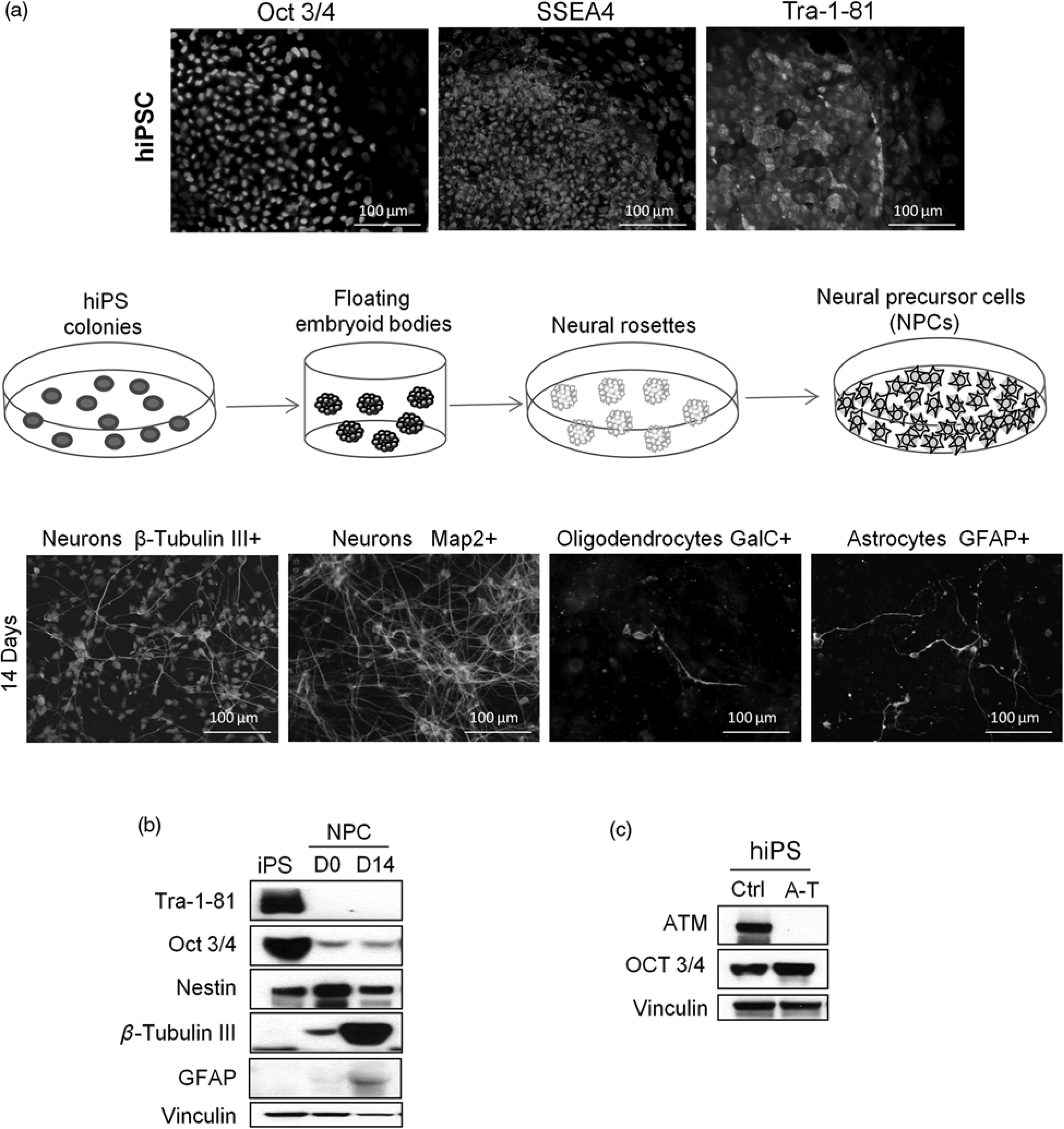
The DDR in pre- and postmitotic neural cells
NSCs are rapidly dividing, but when terminally differentiated they exit the cell cycle and become postmitotic. As the repair of DNA DSBs occurs through either the error-free homologous recombination (HR) or the error-prone non-homologous end joining, the former operating in dividing cells while the latter also in non-dividing cells, 26 the genotoxic response may differ according to the cell cycle phase. Moreover, in human neuron-like cells the levels of ATM and of other DDR components, e.g. ATR and NBS1, are downregulated during neuronal differentiation, with consequent attenuation of the damage response in non-dividing versus dividing cells. 27 Clearly, the availability of hNSCs offers the opportunity to study these phenomena in relation to ATM deficiency.
Concordant with previous findings 15 and as illustrated in Figures 3a and b, the radiation-induced DDR of ATM-deficient ihNSCs is markedly attenuated relative to the ATM-proficient cell counterparts, as demonstrated by the reduced phosphorylation, both in time- and IR-dose dependent assays, of several direct target substrates of ATM, such as Smc1-S966, Chk2-T68 and p53-S15. We have also shown that the resolution of γ-H2AX, an indicator of ongoing DSB repair, is markedly delayed in shATM cells 15 indicating that ATM deficiency, like in other dividing cell types, delays the DNA repair also in ihNSCs. Conversely, terminally differentiated non-proliferating neural cells devoid of ATM exhibit IR-induced γ-H2AX fluorescent foci of greater size and intensity than cells expressing ATM 24 although the time-dependent resolution of the foci in the former is only modestly delayed, suggesting a minor dependence on ATM for DSB repair. It should be noted that compared with ATM-proficient neurons, non-dividing neurons from ATM-deficient neural progenitors obtained from hESCs are virtually negative for γ-H2AX expression. 20
Response of immortalized human neural stem cells (ihNSC) and ReNcell VM
to ionizing radiation (IR)-induced DNA damage. The response to
IR-induced DNA damage was analyzed by Western blot on proliferating
cells. In (a) proliferating ihNSC with ablated expression of ATM (shATM)
or a control (shCon) were irradiated with escalating doses of IR and
harvested 45 min later. The autophosphorylation of ATM-S1981 and
phosphorylation of the ATM targets Smc1-S966, Chk2-T68 and p53-S15 were
analyzed. In (b) proliferating RenCellVM with ablated expression of ATM
(shATM) or a control (shCon) were treated with 5Gy IR and analyzed at
different time points by Western blot for the autophosphorylation of
ATM-S1981 and phosphorylation of the ATM targets Smc1-S966, and p53-S15.
Protein loading per lane was verified with anti-β-Actin antibody. ATM,
ataxia telangiectasia mutated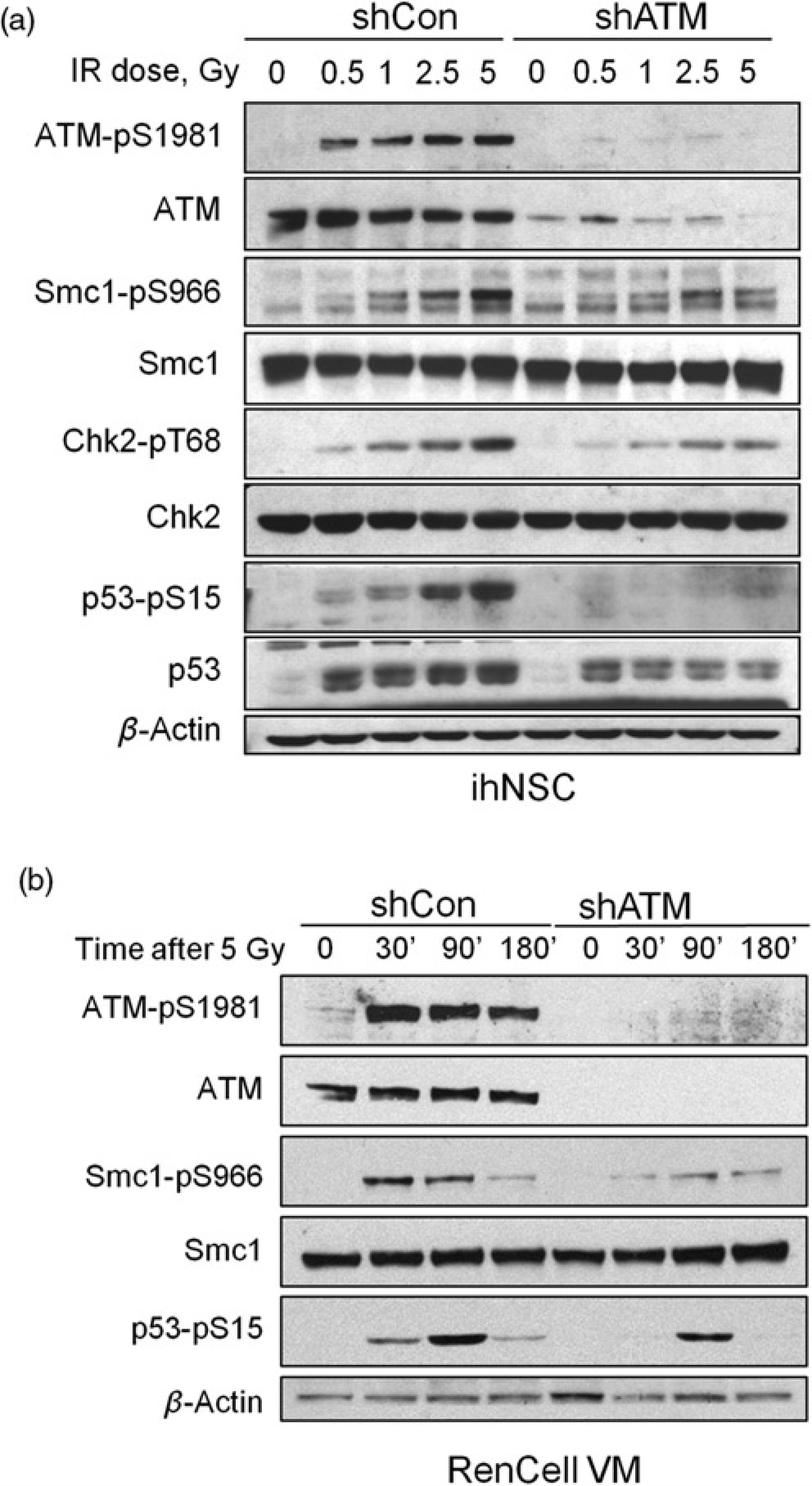
The DDR has also been analyzed in iPSCs derived from A-T patients with truncating mutations in the ATM gene and undetectable ATM protein. A recent study 25 showed that in control iPSCs exposure to radiation induces a vigorous autophosphorylation of ATM on Ser1981 and phosphorylation of several downstream substrates, including Smc1-S957, Kap1-S824, Chk2-T68 and p53-S15, while in A-T iPSCs these phosphorylations are undetected. Likewise, the DNA damage S-phase and mitotic checkpoints in A-T iPSCs are dysfunctional since the cells fail to transiently arrest in these phases of the cell cycle following radiation. 25 We have compared the response of terminally differentiated postmitotic neurons derived from control and A-T iPSCs and found that the phosphorylation of the ATM substrates Kap1-S824, Chk2-T68 and p53-S15 is repressed in A-T neurons (Figure 4). This in turn results, in the case of Chk2, in a lack of activation, as seen by the poor phosphorylation of Kap1-S473, an in vivo substrate of Chk2. 28
DNA damage response by postmitotic neurons derived from
induced-pluripotent stem cells (iPSCs). neural precursor cells from iPSC
of a control and an ataxia-telangiectasia case were differentiated for
14 days, at which time-point are non-dividing and express to >90% the
neuron-specific markers β-Tubulin III and microtubule associated
protein2. These postmitotic neurons were then analyzed by Western blot
at different time-points after 4Gy IR. The phosphorylation of the ataxia
telangiectasia mutated targets Kap1-S824, Chk2-T68 and p53-S15, the the
Chk2 target Kap1-S423 and the apoptotic marker Cleaved poly (ADP ribose)
polymer-ase-1 (PARP-1) were analyzed. Protein loading per lane was
verified with anti-β-Actin antibody
ATM deficiency and neural differentiation
When induced to differentiate, NSCs generate both neur-onal and glial progeny. We have shown that upon differentiation, ATM-proficient and ATM-deficient ihNSCs yield a similar number of neurons expressing the MAP2 and β-Tubulin III markers. By contrast, the yield of neurons exhibiting a GABAergic phenotype (γ-aminobutiric acid positive) is consistently attenuated by ATM deficiency. Of note, GABA signaling plays an important role in adult neurogenesis, through a mechanism in which GABA induces the phosphorylation of H2AX by ATM/ATR, and this epigenetic change within the subventricular zone niche restricts the proliferation of NSCs and neuronal output.29,30 Whether the reduced yield of GABAergic neurons in ATM-deficient ihNSCs arises from a dysfunctional GABA signaling owing to ATM deficiency and somehow limiting a specific neuronal output remains unknown. Nevertheless, this in vitro result agrees with pathological and clinical findings showing a GABA deficiency in the cerebellum of an A-T patient, 31 and amelioration of the ataxia manifestation by treatment with a GABA-analog. 32
Interestingly, oxygen levels also affect the differentiation of neural progenitor cells. 33 Concordant with this, we have observed that the amount of neurons expressing β-Tubulin III is markedly reduced in ihNSCs differentiating under hypoxia, although to the same extent in ATM-deficient and -proficient cells. Hypoxia also affects the yield of GABAergic neurons, but to a greater extent in ATM-deficient than in control ihNSCs, and the expression of potassium channel-interacting protein-1 (KCHIP-1), predominantly present in GABAergic neurons, 34 also appears to be markedly dependent on oxygen tension, since the fraction of positive cells falls substantially under hypoxia, and to a higher degree in ATM-deficient ihNSCs.
The lack of ATM also attenuates the yield of GalC- and CNP-expressing oligodendrocytes 15 and furthermore, when ihNSCs are induced to differentiate under oxidative stress conditions achieved by the glutathione-depleting agent buthionine sulfoximine or by hypoxia, a remarkable vulnerability of oligodendrocytes to ATM deficiency is observed. Quite significantly, these results indicate that ATM deficiency does not appear to impair overall neuro-genesis, though it partially affects oligodendrogenesis.
Concluding remarks
As ATM knockout mouse models of A-T do not fully recapitulate the progressive neurodegeneration seen in the human syndrome35,36 attempts have been made to establish NSC lines from human brain tissues to study the underlying mechanisms of neurodegeneration. hNSCs are a renewable source of undifferentiated cells, can be induced to differentiate into functional neurons and glia, and can be genetically manipulated. Studies with such cells are providing important clues on the role of ATM in DDR and neuronal differentiation progression and maturation, as well as on the vulnerability to genotoxic and oxidative agents. However, since the characteristics of hNSCs either derived from ESCs or from different regions of the brain can differ, possibly reflecting normal developmental patterns, it must be recognized that despite many advantages, the use of these stem cells for disease modeling has limitations. 37 Human iPSCs re-programed from A-T fibroblasts and capable of generating a variety of functional neurons offer an additional tool to study neurodegeneration especially in relation to specific disease-associated ATM mutations. We have shown that the human fetal brain-derived NSCs give rise to a large proportion of astrocytes, whereas the iPS-derived NPC yield a large proportion of neurons, and whether this difference affects the phenotype associated with ATM dysfunction remains to be elucidated.
An evident limitation of these in vitro models is represented by the current lack of protocols to generate Purkinje neurons, the cells whose demise in the cerebellum is responsible for ataxia in A-T.
Footnotes
Acknowledgements
This work was financially supported by the Italian Telethon Foundation (grant GGP10066) and Italian Association for Cancer Research (AIRC grant IG10248).