
Abstract
In an EBM Minireview published in 2010, we proposed that the heat shock protein (Hsp)90/Hsp70-based chaperone machinery played a major role in determining the selection of proteins that have undergone oxidative or other toxic damage for ubiquitination and proteasomal degradation. The proposal was based on a model in which the Hsp90 chaperone machinery regulates signaling by modulating ligand-binding clefts. The model provides a framework for thinking about the development of neuroprotective therapies for protein-folding diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), and the polyglutamine expansion disorders, such as Huntington’s disease (HD) and spinal and bulbar muscular atrophy (SBMA). Major aberrant proteins that misfold and accumulate in these diseases are “client” proteins of the abundant and ubiquitous stress chaperone Hsp90. These Hsp90 client proteins include tau (AD), α-synuclein (PD), huntingtin (HD), and the expanded glutamine androgen receptor (polyQ AR) (SBMA). In this Minireview, we update our model in which Hsp90 acts on protein-folding clefts and show how it forms a rational basis for developing drugs that promote the targeted elimination of these aberrant proteins.
Keywords
Introduction
The function, trafficking, and turnover of a wide variety of proteins (many of them signaling proteins) are regulated by heat shock protein 90 (Hsp90). 1 The regulation is adenosine triphosphate (ATP)-dependent and dynamic in that the proteins are continuously cycling into and out of heterocomplex with Hsp90. Some proteins form heterocomplexes with Hsp90 that are stable enough to be isolated and analyzed biochemically. For want of a better term, we call this “stable” cycling, and the classic Hsp90 clients, like steroid receptors, Src, and a variety of other protein kinases, cycle with Hsp90 in this manner. 2 In contrast, other proteins (e.g., nitric oxide synthases [NOS], epidermal growth factor receptor) cycle much more dynamically with Hsp90 in heterocomplexes that rapidly disassemble such that no (or only trace amounts of) Hsp90 heterocomplexes can be observed in cell lysates. 2 Although the greatest interest has focused on clients that undergo stable cycling with Hsp90, it is important to recognize that there is a range extending from stable to very dynamic cycling. In general, it can be said that the turnover of client proteins that undergo stable cycling is more stringently regulated by Hsp90. When Hsp90 cycling is inhibited by specific inhibitors like geldanamycin or radicicol, these classic clients rapidly unfold and are rapidly ubiquitinated and degraded by the proteasome. In contrast, the more dynamically cycling proteins are more intrinsically stable, and there is much less difference in their half-lives in the presence and absence of Hsp90 inhibition. There are a number of examples of mutational switching of client protein between stable and dynamic cycling with Hsp90.2,3
The mechanism of Hsp90 heterocomplex assembly has been worked out almost exclusively with steroid receptors, and it is not yet known if stable and dynamic cycling with Hsp90 utilizes the same assembly mechanism. A system of five purified proteins—Hsp90, Hsp70, Hsp organizer protein (Hop), Hsp40, and p23—has been shown to be sufficient for efficient assembly of Hsp90 heterocomplexes with glucocorticoid and progesterone receptors. 1 Hsp90 and Hsp70 are both required for assembly, and they are brought together in a machinery by Hop. The Hsp70 co-chaperone Hsp40 and the Hsp90 co-chaperone p23 interact dynamically during (Hsp40) or at the end (p23) of the assembly process. For details of the assembly mechanism and original citations, see Pratt and Toft. 1
Like a number of protein chaperones, Hsp90 has been shown in vitro to assist refolding of partially unfolded proteins to a properly folded, active conformation. However, it is quite clear that Hsp90 is not required for de novo protein folding, 4 and it does not associate with the glucocorticoid receptor (GR) until termination of translation. 5 In contrast to the in vitro experiments on unfolded substrates, the Hsp90/Hsp70-based chaperone machinery acts on proteins that are in their native (or near native) conformation. There is no amino acid sequence specificity governing Hsp90 interaction with substrate. Rather, surface features, such as the αC-ß4 loop region of the catalytic domain of many protein kinases, define the capacity of the chaperone machinery to recognize clients. 6 From our experience with the interaction of the chaperone machinery with the GR, both hydrophobic and electrostatic surface interactions are involved during the heterocomplex assembly process. In our work with the GR, we have noted that there is a very focal site of attack that lies on the surface of the ligand-binding domain at the opening of the hydrophobic steroid-binding cleft.7,8 Such regions where hydrophobic clefts open onto the charged protein surface are a general topological feature of virtually all proteins in native conformation. In previous reviews, we have suggested that the openings of ligand-binding clefts onto the protein surface form the general surface feature recognized by the Hsp70/Hsp90-based chaperone machinery.1,2
The cleft model
Our focus on ligand-binding clefts started when Bresnick et al. 9 demonstrated a direct relationship between the amount of GR-associated Hsp90 and high-affinity glucocorticoid-binding capacity of immunopurified receptors. Conditions (e.g., salt) that cause Hsp90 to dissociate eliminate the steroid-binding capacity of the GR, even if it is kept at 0℃. 10 The Hsp90-free GR can be converted back to the Hsp90-bound form by incubating it with reticulocyte lysate, and this is accompanied by restoration of steroid-binding activity. 11 There is a linear relationship between the number of GR-Hsp90 heterocomplexes that are assembled in this cell-free system and the amount of steroid-binding activity. 12 Subsequently, it was shown that only the Hsp90-Hop-Hsp70 core machinery is required to open the ligand-binding cleft to allow steroid binding, 13 but that the Hsp90 co-chaperone p23 is required for stable GR-Hsp90 heterocomplex assembly. 14 Thus, in the absence of p23, there is dynamic assembly with accompanying cleft opening, and in the presence of p23, which binds to Hsp90 only after it achieves in its ATP-bound conformation, there is stable cleft opening.
There is considerable difference in the way different steroid-binding clefts behave with respect to Hsp90. Like the unliganded GR, the mineralocorticoid and dioxin receptors immediately lose all steroid-binding activity at 0℃ when Hsp90 dissociates. 10 In contrast, the unliganded progesterone receptors may be dissociated from Hsp90 without loss of steroid-binding activity as long as they are maintained at 0℃. 15 However, if it is heated the progesterone receptor loses steroid-binding activity, and this loss is mitigated by the chaperone machinery of reticulocyte lysate. 15 In greater contrast, the estrogen receptor requires the chaperone machinery to achieve its steroid-binding conformation, 16 but the receptor can be purified extensively in the Hsp90-free state and maintain steroid-binding activity. Once it is opened by the chaperone machinery, the steroid-binding cleft of the estrogen receptor is likely stabilized by water, leaving a quite stable open cleft where the binding of ligands is diffusion limited. In contrast, the steroid-binding cleft of the GR is very unstable in the absence of chaperone, and the binding of ligands by the GR-Hsp90 complex is 2–3 orders of magnitude slower than the ER. This suggests that for the GR, the cleft opening is much narrower or that the cleft spends much less time in the open state, even when the receptor is in heterocomplex with Hsp90.
Hsp90 binding to a client protein is targeted and limited. Hsp90 is biologically active only as a homodimer, and two molecules of Hsp90 are bound to one molecule of receptor.
10
Hsp90 and Hsp70 bind selectively to domains of client proteins that contain ligand-binding clefts. For example, both chaperones bind selectively to the hormone-binding domains of steroid receptors
10
and, as illustrated in Figure 1(a), to the oxygenase domain of neuronal nitric oxide synthase (nNOS), which is the domain containing the heme/substrate-binding cleft.
17
Both pulldown and peptide competition data suggest that a region in the oxygenase domain of endothelial NOS is important for Hsp90 binding.18,19 Hsp90 also interacts with the catalytic domains containing the ATP-binding clefts of protein kinase clients, such as Raf
20
and ErbB2.
21
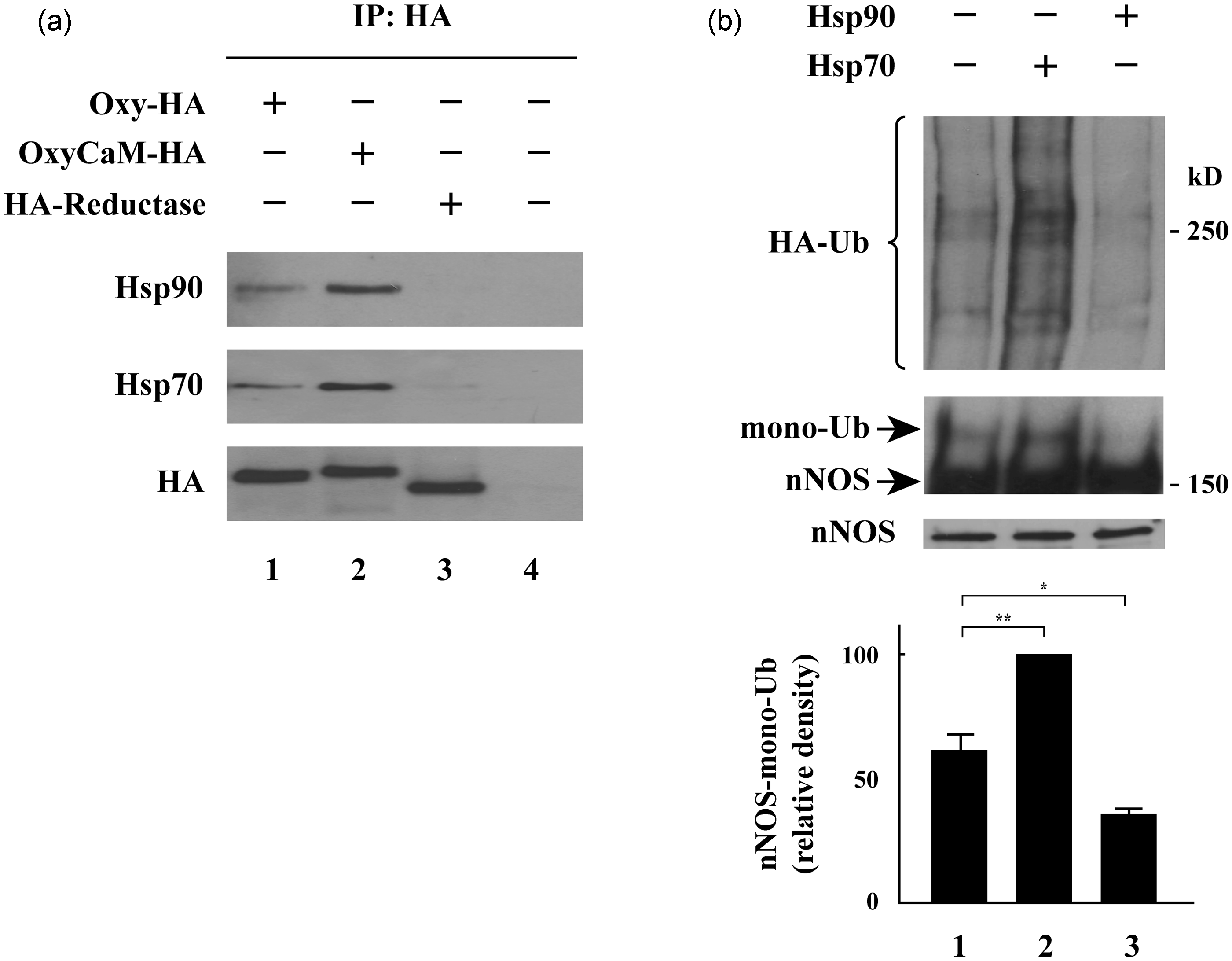
Hsp90 and Hsp70 bind to the oxygenase domain of nNOS, with Hsp90 inhibiting and Hsp70 stimulating nNOS ubiquitination by CHIP. (a) HEK293T cells were transfected with plasmids encoding HA-tagged oxygenase domain (Oxy-HA), the combined oxygenase-calmodulin segment (OxyCaM-HA), or the reductase domain (HA-Reductase) of nNOS as indicated. Approximately 48 h later, cells were cross-linked with dithiobis[succinimidylpropionate] (DSP), and lysates were immunoprecipitated with anti-HA. The presence of Hsp70 and Hsp90 was determined under each condition. (b) Cells were transfected with cDNAs for His-HA-ubiquitin, CHIP, nNOS, and Hsp90 or Hsp70 or vector plasmid. After 48 h, lysates were immunoprecipitated with anti-nNOS antibody and western blotted with anti-nNOS and anti-HA. The upper panel shows the region above mono-ubiquitinated nNOS blotted with anti-HA antibody. The lower panel shows a short exposure for unmodified nNOS protein, and the middle panel shows a long exposure for the mono-ubiquitinated nNOS. The bar graphs show the relative densities of mono-ubiquitinated nNOS bands expressed as means ± SE for three separate experiments. **P < 0.01, *P < 0.05. HA-Ub, HA-ubiquitin. This research was originally published in The Journal of Biological Chemistry. Peng HM, Morishima Y, Pratt WB, Osawa Y. Modulation of heme/substrate binding cleft of neuronal nitric oxide synthase (nNOS) regulates binding of Hsp90 and Hsp70 proteins and nNOS ubiquitination. J Biol Chem 2012; 287:1556–1565. ©the American Society for Biochemistry and Molecular Biology.
17
Ligand-binding clefts and protein stability
Ligand-binding clefts are hydrophobic clefts that must open to allow access of ligands, such as steroids, ATP, and heme, to their binding sites within the protein interior. In the absence of cycling with the chaperone machinery, ligand-binding clefts are often dynamic, shifting to varying extents between closed and open states. When clefts are open, hydrophobic residues of the protein’s interior are exposed to solvent, and continued opening may progress to protein unfolding. Therefore, the extent to which the ligand-binding cleft is open determines ligand access and thus protein function, but clefts are inherent sites of conformational instability. With the steroid receptors, the chaperone machinery assists cleft opening, and Hsp90 binding stabilizes the open state of the cleft preventing further unfolding and Hsp70-dependent ubiquitination. Although we have focused on ligand-binding clefts as the target of the chaperone machinery, major protein-folding clefts that are similarly metastable but do not bind ligands would provide the same requirements for interaction with and stabilization with Hsp90. Indeed, three of the major Hsp90 clients that are critical targets for intervention in neurodegenerative diseases—huntingtin, α-synuclein, tau—do not bind any known ligands in the protein interior. 22 It is not known where Hsp90 interacts with these clients, but their status as Hsp90 client proteins is clearly established. 22
One important criterion for assigning client protein status is that the protein of interest undergoes degradation, most often via the ubiquitin/proteasome pathway, when cycling with Hsp90 is blocked by treating cells with an Hsp90 inhibitor. In the case of the Hsp90 clients, the key determinant of ubiquitination is carboxyl-terminus of Hsc70-interacting protein (CHIP), a ubiquitin E3 ligase that binds via a tetratricopeptide repeat (TPR) domain to Hsc/Hsp70.23,24 When stabilization of the client protein by Hsp90 is blocked, Hsp70 interacts with the unfolding client, and CHIP functions as an Hsp70 co-chaperone to direct the ubiquitin-charged E2 enzyme to transfer ubiquitin to Hsp70-bound client. As shown with both a purified CHIP-dependent ubiquitinating system 25 and in cells expressing CHIP, 17 Hsp90 and Hsp70 have opposing effects on CHIP-dependent ubiquitination—with Hsp90 inhibiting and Hsp70 promoting the process (see Figure 1(b)). Correspondingly, overexpression of Hsp90 increases the level of client protein, and overexpression of Hsp70 decreases the level of client protein. 17
The opposing roles of the two essential components of the chaperone machinery are essential to its role in the triage of damaged and aberrant client proteins. When clefts open (unfold) to the point where Hsp90 can no longer interact with the client to stabilize it, Hsp70/CHIP-dependent ubiquitination can proceed unopposed, as summarized in the scheme of Figure 2. Although we have focused on CHIP in this description of the protein quality control function of the chaperone machinery, parkin, another Hsp70-dependent E3 ligase, can also act redundantly on some Hsp90 clients.
26
It is likely that some other E3 ligases are functionally redundant with CHIP, as classic Hsp90 clients, like the GR, the estrogen receptor, and the polyQ AR, are degraded at the same rate in CHIP–/– cells as in CHIP+/+ cells.26,27 Nevertheless, CHIP is regarded as the most important E3 ligase involved in chaperone-dependent ubiquitination and degradation of damaged and aberrant proteins.
Triage of nNOS by the Hsp90/Hsp70-based chaperone machinery. Normally, the client protein is stabilized by cycling into a heterocomplex with Hsp90. The mechanism of Hsp90 assembly with nNOS is not worked out, and the steps of assembly shown at the top are abbreviation of those defined for the GR.
2
Cycling with Hsp90 facilitates nNOS binding of heme in the heme/substrate-binding cleft. Fe, heme iron. Mechanism-based inactivation of nNOS by certain substrates leads to unfolding of the heme/substrate-binding cleft to a degree that Hsp90 cannot cycle with the enzyme to inhibit ubiquitination by Hsp70-dependent E3 ligases, such as CHIP. The solid crescents represent the Hop and CHIP TPR domains. These are dynamic processes in which the nNOS is cycling into and out of heterocomplex with Hsp90 and ubiquitination is countered by deubiquitinating activity. Thus, the scheme is very much a simplification of both the stabilization and degradation events in the triage process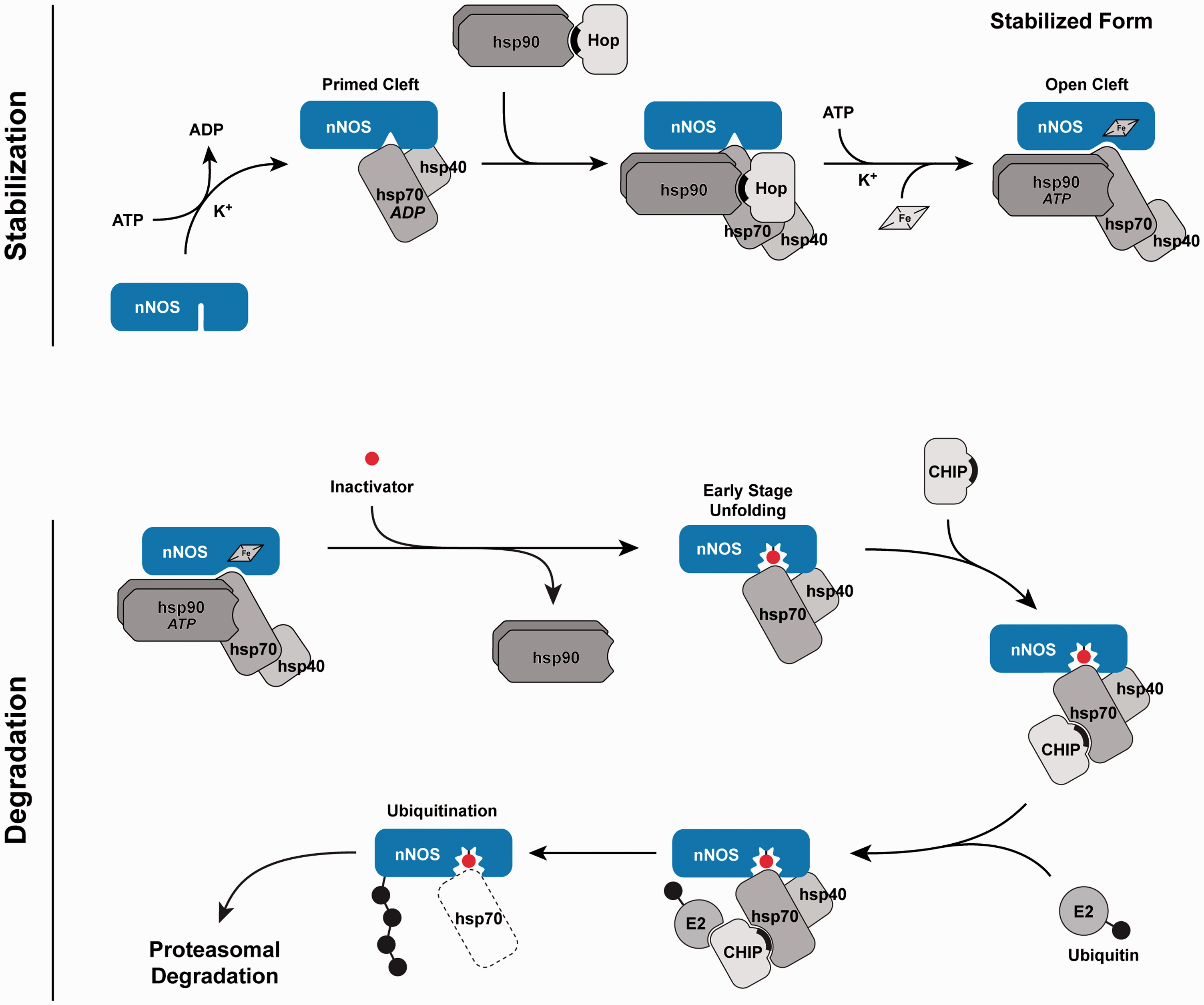
Targeting ligand-binding clefts to promote client protein ubiquitination and degradation
Site-specific inactivation is an example of toxic damage that is targeted to the ligand-binding cleft and triggers client protein ubiquitination. As examples among many, site-specific inactivators trigger ubiquitination of nNOS, 25 ErbB2/HER2, 28 and the estrogen receptor, 29 all of which are Hsp90 clients that are ubiquitinated by CHIP.30–32 Because site-specific inactivation serves as a good way to link events occurring within a protein-folding cleft to the protein quality surveillance function of the chaperone machinery, we will use the example of nNOS to illustrate the model.
The NOS enzymes function as cytochrome P450-type hemoproteins to catalyze the formation of nitric oxide and citrulline from arginine, O2, and nicotinamide adenine dinucleotide phosphate (NADPH).
33
The prosthetic heme is the site of oxygen activation, which is required for the metabolism of
Because metabolism-based inactivators such as NG-amino-
Treatment of cells with an Hsp90 inhibitor such as geldanamycin leads to nNOS degradation via the proteasome.35,45 Both inhibition of Hsp90 and site-specific inactivation by NG-amino-l-arginine promote nNOS ubiquitination by a purified CHIP-dependent ubiquitinating system. 25 Both CHIP and parkin can function as E3 ligases for nNOS ubiquitination,26,31 but CHIP accounts for all of the nNOS ubiquitinating activity 53 in the reticulocyte lysate ubiquitinating system of Hershko et al. 54 CHIP directs ubiquitination of nNOS within the CaM-binding segment. 55 Ubiquitination of nNOS by a purified CHIP-dependent ubiquitinating system25,31 and by the reticulocyte lysate system53,56 is dependent upon Hsp70. Again, Hsp90 and Hsp70 have opposing actions on CHIP-dependent ubiquitination, with Hsp70 stimulating ubiquitination and Hsp90 inhibiting ubiquitination17,25 (Figure 1(b)).
Rationale for promoting elimination of aberrant proteins
Because it is the substrate-bound Hsp70 that is mediating CHIP-dependent ubiquitination in the model of Figure 2, one can have the impression that Hsp70 makes the triage decision. But with Hsp90 client proteins, such as a polyglutamine protein, α-synuclein, or tau, it is the Hsp90 interaction with the unfolding substrate that determines whether ubiquitination will proceed at any moment or not, and the opposing effects of the two chaperones on ubiquitination determine protein quality control by the chaperone machinery.
In the adult-onset neurodegenerative diseases, it is the soluble oligomer form of unfolded protein that is neurotoxic, and the goal of neuroprotective drug therapy is to promote unfolded protein degradation before oligomer formation. 22 Currently, there are three ways in which drugs can promote aberrant protein degradation in the model of Figure 2. The approach that has been examined most extensively is to inhibit their stabilization by inhibiting client protein-Hsp90 heterocomplex assembly with specific Hsp90 inhibitors. Because many oncoproteins that are critical in the genesis and maintenance of malignancy are Hsp90 clients, a variety of Hsp90 inhibitors have been developed as potential anticancer drugs and a number are currently in clinical trial.57–59 Some of these Hsp90 inhibitors can be administered orally and some pass through the blood–brain barrier, both of which are conditions that must be met if they are to be considered for treatment of neurodegenerative diseases. A major drawback of this approach is that all Hsp90 client proteins will be affected, and these are toxic drugs. Although toxicity may be accommodated in relatively short-term cancer chemotherapy protocols, a neuroprotective drug treatment would have to continue for many years for diseases such as Parkinson’s disease (PD) and Alzheimer’s disease (AD), and it is highly unlikely that Hsp90 inhibitors could be tolerated on such a chronic basis.
Hsp90 inhibitors prevent cycling of a not-yet-unfolded client protein with Hsp90 to promote its degradation. In the neurodegenerative disorders, the goal is to promote degradation of the already-unfolded client protein before it aggregates to form the oligomers that are thought to be responsible for the toxic gain of function that generates the pathology. 22 One way in which the degradation of unfolded proteins might be promoted is by inhibiting their deubiquitination. Protein ubiquitination is a reversible process in which deubiquitinating enzymes (DUBs) cleave the isopeptide bond at the C-terminal ubiquitin. 60 The human genome encodes at least 98 DUBs, and several DUBs are involved in cancer progression, prompting the development of DUB inhibitors for anticancer therapies.61,62 Assuming that specific DUBs are particularly active on individual aberrant proteins involved in neurodegenerative disorders, as is the case with proteins involved in cancer progression, then DUB inhibitors may prove useful in neuroprotective treatment protocols. Like the protein kinases, DUBs offer many drug targets, and inhibitors of specific DUBs are being developed for cancer treatment.61–63
A third way to promote aberrant protein degradation in the model of Figure 2 is to promote the action of Hsp70. In cellular, yeast, animal, and fly models of neurodegenerative disorders, overexpression of Hsp70, or its co-chaperone Hsp40, or CHIP has been shown to decrease the level of the aberrant target proteins, and where appropriate to ameliorate toxicity. 22 Thus, Hsp70/CHIP-dependent ubiquitination would be boosted by drugs that promote Hsp70 activity. To date, all of the drug development with respect to Hsp70 has focused on developing Hsp70 inhibitors as potential anticancer agents. 64 As predicted from the model, inhibition of Hsp70 promotes accumulation of aggregated species in a cellular model of spinal and bulbar muscular atrophy (SBMA), 56 whereas promotion of Hsp70 increases polyQ AR clearance. 65
Agents that promote Hsp70 activity
Hsc70-interacting protein (Hip) is a co-chaperone of Hsp70 that prevents the accumulation of polyglutamine inclusions in a cellular model of polyglutamine aggregation.
66
Hip overexpression also decreases formation of α-synuclein fibrils, and its knock-down in Caenorhabditis elegans increases α-synuclein aggregation.
67
Because Hip stabilizes Hsp70 in its adenosine diphosphate (ADP)-bound conformation,
68
the conformation that recognizes unfolding substrates with high affinity,
69
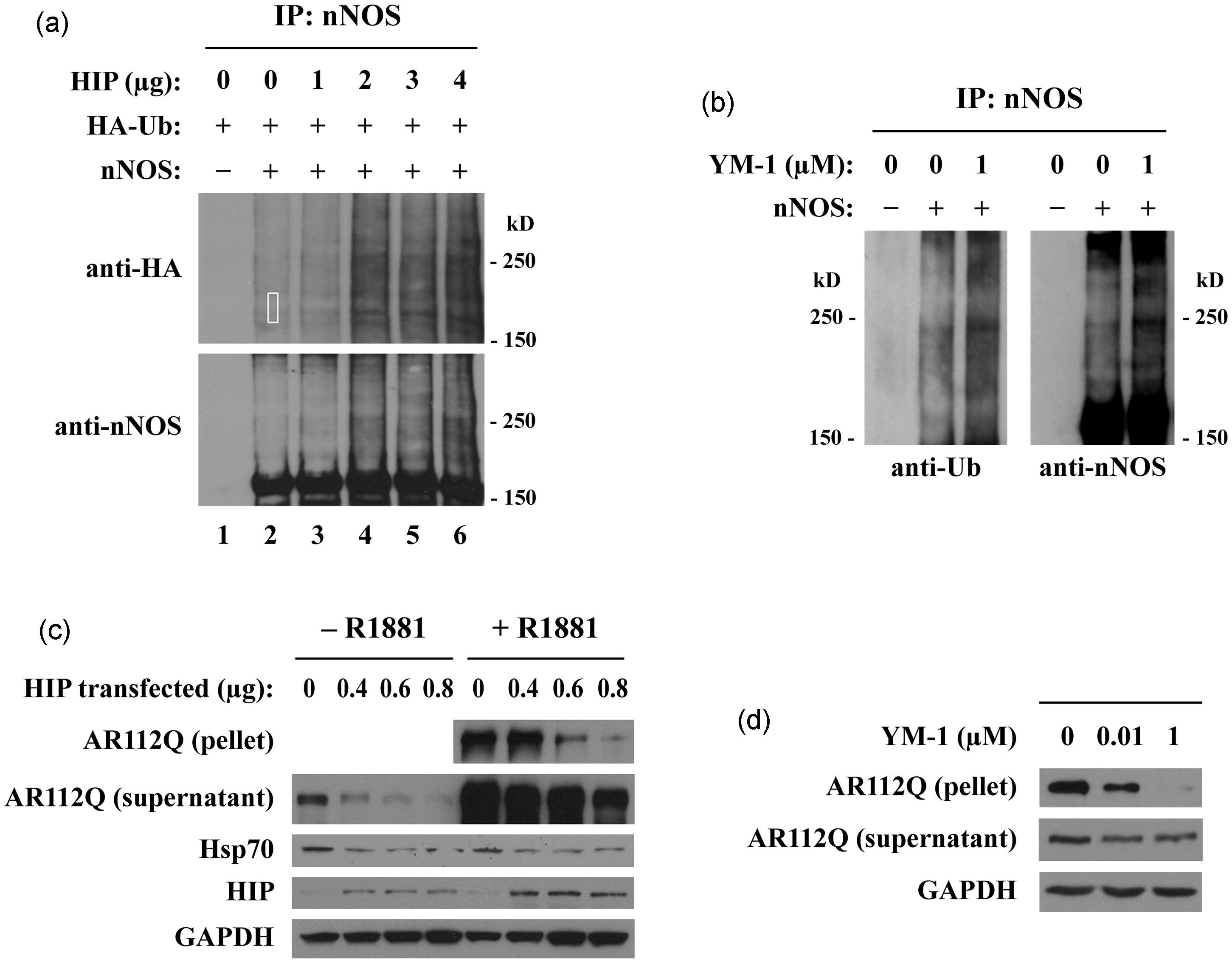
the model of Figure 2 suggested that Hip overexpression would facilitate Hsp70/CHIP-dependent ubiquitination. As shown in Figure 3(a), Hip overexpression increased ubiquitination of nNOS.
65
Hip overexpression also promoted polyQ AR clearance in a cellular model of SBMA (Figure 3(c)) and alleviated toxicity in a Drosophila model of SBMA.
65
These results support a model in which increased affinity of Hsp70 for unfolding proteins promotes their ubiquitination and degradation. Thus, small molecules that bind to Hsp70 to mimic Hip action in stabilizing its ADP-bound conformation might prove to be useful in the treatment of neurodegenerative disorders.
Both Hip and YM-1 promote client protein ubiquitination and degradation. (a) Hip promotes nNOS ubiquitination. HEK293T cells transiently expressing nNOS, HA-ubiquitin (HA-Ub), and increasing amounts of Hip were treated with lactacystin for 24 h. nNOS was immunoprecipitated from cell lysates and probed with anti-HA (upper panel) or anti-nNOS (lower panel) antibodies. (b) YM-1 promotes nNOS ubiquitination. HEK293 cells stably expressing nNOS were treated with YM-1 and lactacystin for 24 h, and lysates were immunoprecipitated for nNOS. Western blots were probed with anti-ubiquitin (left panel) or anti-nNOS (right panel) antibodies. (c) Hip decreases AR112Q levels. HeLa cells transiently expressing AR112Q and increasing amounts of Hip were treated with the androgen R1881 (10 nM) for 24 h. Lysates were separated into supernatant and 15,000 g pellet fractions and analyzed by western blotting. (d) YM-1 decreases insoluble AR112Q. PC12 cells expressing tet-regulated AR112Q were treated with R1881 (10 nM) and YM-1 for 16 h, and lysates were immunoblotted. Data from Wang et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9:112–118, 2013. doi:10.1038/nchembio.1140 URL:http://www.nature.com/nchembio/journal/v9/n2/full/nchembio.1140.html.
65
The search for Hip mimics was facilitated by the serendipitous observation that two rhodacyanine dye compounds that inhibit Hsp70 ATPase activity substituted for Hip in promoting Hsp70-dependent nNOS maturation. 70 MKT-077 is a rhodacyanine dye that causes selective death of cancer cells, 71 reportedly by inhibiting Hsp70 (mortalin) cycling with p53, activating this tumor suppressor protein.72,73 Nuclear magnetic resonance studies established that MKT-077 binds with low micromolar affinity to the nucleotide-binding domain of ADP-bound but not ATP-bound Hsp70, stabilizing the ADP-bound state. 74 MKT-077 is concentrated in mitochondria, 71 and it was tested in phase 1 trial as a “selective mitochondrial toxin” against solid tumors, but further development was stopped because of renal toxicity. 75 A close derivative, YM-1, distributes predominantly in the cytoplasm and also selectively targets cancer cells, 76 and it was chosen for further study. 65
YM-1 increases the binding of purified Hsp70 to unfolded luciferase by stabilizing the ADP conformation of the chaperone, and the binding of YM-1 to Hsp70 is blocked by purified Hip. 65 As shown in Figure 3(b), treatment of cells with YM-1 increases CHIP-dependent ubiquitination of nNOS. YM-1 promotes polyQ AR clearance in a cellular model of SBMA (Figure 3(d)), and it alleviates toxicity in a Drosophila model of SBMA. 65 YM-1 markedly decreases polyQ AR oligomers and detergent-insoluble aggregates while having little effect on soluble polyQ AR and no effect on Hsp90 client proteins in general. 65 YM-1 reduces tau levels in cellular and primary neuronal models of tauopathy, 77 supporting further development of drugs that promote Hsp70/CHIP-dependent ubiquitination for neuroprotective treatment of AD.
We consider YM-1 to be an appropriate platform for developing drugs that promote ubiquitination and degradation of Hsp90 client proteins after they have unfolded and are no longer cycling with Hsp90. Modifications that improve its potency, metabolic stability, and penetration across the blood–brain barrier have already been published.78,79 Because neither Hip 80 nor YM-1 65 affects client protein-Hsp90 heterocomplex assembly, Hsp90 cycling proceeds normally to stabilize the soluble, properly folded client protein. Thus, this approach focuses on accelerating degradation of the unfolding client protein before it can form the oligomers that are deemed responsible for neurodegeneration, and it avoids the broad depletion of client proteins that would occur with Hsp90 inhibitors.
Comments on the three treatment modalities
The progression of events in formation of inclusions is first unfolding of the Hsp90 client followed by formation of soluble oligomers that in turn coalesce to form the aggregates that are visualized as inclusions. The aggregates appear to form during axonal trafficking, and the resulting inclusions that form contain Hsp70, ubiquitin, CHIP, E1 and E2 ubiquitinating enzymes, and even proteasomes. 22 There is good evidence in each disorder (AD, PD, Huntington’s disease, SBMA) that it is the soluble oligomer form that is neurotoxic, but the role of the inclusions in neurotoxicity may also be significant in the long term.
Because Hsp90 interacts with the not-yet-unfolded and not with the unfolded-and-already-aggregated client protein, Hsp90 inhibitors promote degradation of the client protein before aggregation can occur. Thus, it has been shown in a cellular model of PD that geldanamycin treatment reduces formation of α-synuclein aggregates and α-synuclein toxicity, but geldanamycin treatment of cells with pre-existing inclusions did not reduce the number of cells with inclusions. 81 If Hsp90 inhibitors can be used in the clinical setting, their primary effect might be to slow disease progression. However, if the ubiquitin-proteasome system in the inclusions is functional even at a reduced level of activity, then it is possible that over the long term, as the generation of oligomers and aggregates is blocked, some reversal of pre-existing toxicity may take place. The elimination of aggregates may be promoted by the action of the Hsp110/Hsp70/Hsp40 disaggregase machinery to extract unfolded protein from aggregates prior to Hsp70/CHIP-dependent ubiquitination. 82
Although Hsp90 and DUB inhibitors are likely to be too toxic to permit treatment of neurodegenerative disorders over a span of years, the experience gained from clinical trials in cancer treatment may suggest patient tolerance of Hsp90 inhibitors over limited time spans. Because these are slowly progressing disorders, it may be possible to treat with one of these inhibitors for a period of time, then not treat for a sustained period. The length of the intertreatment interval would be determined by the extent to which unfolded protein in pre-existing oligomers and aggregates was degraded during treatment. Such a periodic treatment might be tolerated when long-term continuous treatment would not. The use of drugs like YM-1 that promote Hsp70/CHIP-dependent ubiquitination and degradation in combination with an Hsp90 or DUB inhibitor might shorten treatment periods required to deplete oligomers and larger aggregates, reducing toxicity from the inhibitors.
We envision both monotherapy and combination drug therapy administered in intermittent protocols that allow for recovery from side effects and alternation of treatments with different side effects. It is apparent from the stepwise model of events in Figure 2 that the combination of an Hsp90 inhibitor with an Hsp70 promoter might be synergistic in yielding target protein degradation. Indeed, both classes of drug might also be synergistic with a deubiquitinase inhibitor. Preliminary results using a cellular model of a neurodegenerative disease suggest that combination treatment regimens may allow lowering the dosage of one drug, suggesting that clinical protocols could be developed that reduce drug-specific side effects.
Our goal in this Minireview has been to show how a model in which the Hsp90/Hsp70-based chaperone machinery acts on protein-folding clefts forms an important paradigm shift in the way we think about using the protein quality control system to develop neuroprotective treatments for the adult-onset neurodegenerative diseases.
Footnotes
Author contributions
All authors designed the review. Each author wrote a description of his studies to be included in the review. The sections were melded by WBP and YM. The manuscript was then reviewed and modified by all authors.
ACKNOWLEDGEMENTS
The authors’ work reported herein was funded by the National Institutes of Health (GM077430 to YO; NS055746 and NS32214 to APL; NS059690 to JEG), the Muscular Dystrophy Association (MDA238924 to APL), and the McKnight Foundation (to APL). WBP, APL, and YO are participants in The University of Michigan Medical School’s Protein Folding Diseases Initiative.