
Abstract
Galectin-3 (gal-3) is a β-galactoside-binding lectin, which regulates cell–cell and extracellular interactions during self/non-self-antigen recognition and cellular activation, proliferation, differentiation, migration and apoptosis. It plays a significant role in cellular and tissue pathophysiology by organizing niches that drive inflammation and immune responses. Gal-3 has some therapeutic potential in several diseases, including chronic inflammatory disorders, cancer and autoimmune diseases. Gal-3 exerts a broad spectrum of functions which differs according to its intra- or extracellular localization. Recombinant gal-3 strategy has been used to identify potential mode of action of gal-3; however, exogenous gal-3 may not reproduce the functions of the endogenous gal-3. Notably, gal-3 induces monocyte–macrophage differentiation, interferes with dendritic cell fate decision, regulates apoptosis on T lymphocytes and inhibits B-lymphocyte differentiation into immunoglobulin secreting plasma cells. Considering the influence of these cell populations in the pathogenesis of several autoimmune diseases, gal-3 seems to play a role in development of autoimmunity. Gal-3 has been suggested as a potential therapeutic agent in patients affected with some autoimmune disorders. However, the precise role of gal-3 in driving the inflammatory process in autoimmune or immune-mediated disorders remains elusive. Here, we reviewed the involvement of gal-3 in cellular and tissue events during autoimmune and immune-mediated inflammatory diseases.
Introduction
Galectin-3 (gal-3) is a β-galactoside-binding protein which regulates cell–cell and cell–extracellular matrix interactions affecting cell proliferation, migration, adhesion, differentiation and apoptosis. 1 Gal-3 is produced by macrophages, monocytes, dendritic cells (DCs), eosinophils, mast cells, NK cells and activated T and B cells.2,3 During the past decade, gal-3 has attracted the attention of researchers due to its regulatory role in immune response, inflammation and fibrosis.4,5
The biological activities are structurally linked to C-terminal carbohydrate recognition domain (CRD)) and N-terminal domain enriched by proline and glycine (protein recognition domain) responsible for specific interactions. 1 Moreover, gal-3 regulates the expression of several regulatory genes 6 and forms lattices with cell-surface glycoprotein receptors and senses self-derived or pathogen glycoconjugates when located on cell surfaces. 7 Indeed, gal-3 controls immune responses through damage-associated molecular pattern (DAMP) ) and pathogen-associated molecular pattern (PAMP) pathways. 8 It is able to recognize endogenous (“self”) and exogenous (“non-self”) carbohydrate compounds eliciting complex immune and autoimmune responses. 9
Galectin-3 and the immune response: An overview
The effects of gal-3 clearly depend on cellular or tissue localization. In order to identify therapeutic targets, recombinant gal-3 has been used in distinct experimental models, including heart failure and liver diseases where it can mimic the extracellular functions of this molecule.5,10 Functions of endogenous gal-3 have been widely studied in tumors, where gal-3 seems to play a role in cell transformation, proliferation, metastasis and apoptosis. 11 Recombinant gal-3 may not affect the functions of the endogenous protein, which is also influenced by gal-3 antagonists, including GCS-100, which dampens tumor growth and metastasis in distinct experimental models. 12 The identification of the cell compartment where gal-3 is detected is crucial in order to understand the effects of this molecule on cell function. 11
In mice, gal-3 is poorly expressed by resting T and B cells, being up-regulated after activation.3,13,14 On the other hand, in humans, gal-3 is constitutively expressed by regulatory (Treg) and CD4+ memory T cells. 15 Extracellular gal-3 induces T-cell apoptosis, 16 migration of CD4+/CD8+ thymocytes on laminin, 17 and T-cell activation resulting in intracellular calcium influx. 18 Intracellular gal-3 inhibits apoptosis 19 and interleukin (IL)-5 expression, 20 promotes cell growth and negatively regulates TCR-mediated CD4 T-cell activation at immunological synapses. 21
As far as B lymphocyte subpopulations are concerned, gal-3 regulates B220+ and CD138+ cell niches in lymphoid tissues 22 and inhibits plasma cell differentiation in vitro 3 and in vivo in the bone marrow, mesenteric lymph nodes and spleen.22–24 Moreover, gal-3 inhibits B-cell differentiation into plasma cells in distinct lymphoid tissues,23–25 plays anti-apoptotic functions on proliferative B-cell neoplasms 26 and maintains B-cell anergy. 27
Galectin-3 expression, association with different autoimmune diseases and potential mechanisms of action
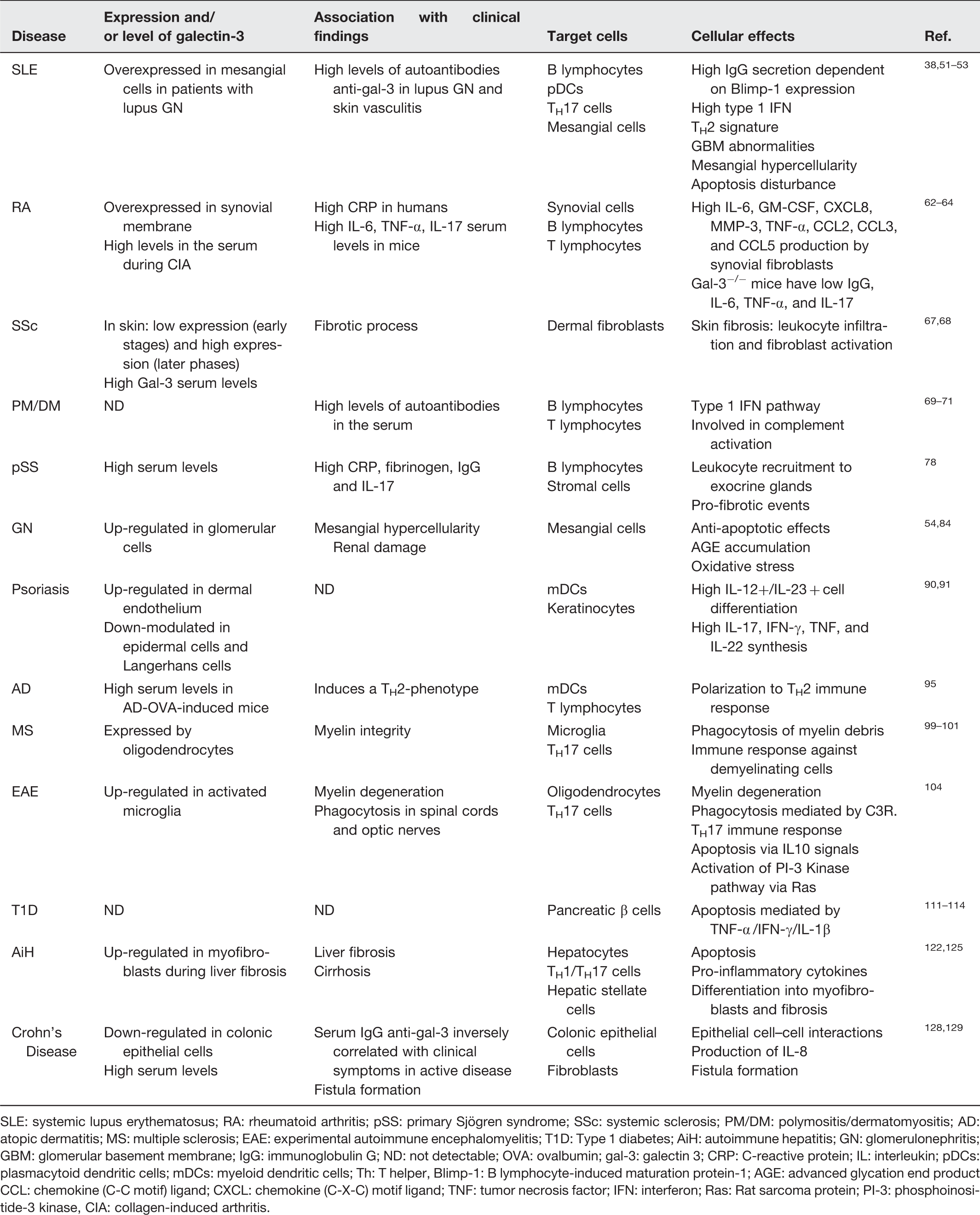
SLE: systemic lupus erythematosus; RA: rheumatoid arthritis; pSS: primary Sjögren syndrome; SSc: systemic sclerosis; PM/DM: polymositis/dermatomyositis; AD: atopic dermatitis; MS: multiple sclerosis; EAE: experimental autoimmune encephalomyelitis; T1D: Type 1 diabetes; AiH: autoimmune hepatitis; GN: glomerulonephritis; GBM: glomerular basement membrane; IgG: immunoglobulin G; ND: not detectable; OVA: ovalbumin; gal-3: galectin 3; CRP: C-reactive protein; IL: interleukin; pDCs: plasmacytoid dendritic cells; mDCs: myeloid dendritic cells; Th: T helper, Blimp-1: B lymphocyte-induced maturation protein-1; AGE: advanced glycation end product CCL: chemokine (C-C motif) ligand; CXCL: chemokine (C-X-C) motif ligand; TNF: tumor necrosis factor; IFN: interferon; Ras: Rat sarcoma protein; PI-3: phosphoinositide-3 kinase, CIA: collagen-induced arthritis.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a complex autoimmune multi-organ disorder in which genetic predisposition, environmental triggers and imbalanced immune response lead to autoantibody secretion and tissue injury.33,34 Beside autoreactive B lymphocytes and autoantibody-secreting plasma cells, Treg cells, NKT cells, mesenchymal stem cells, DCs and autoreactive T cells are also involved in the immunopathogenesis of SLE.34–36
Mice susceptible to autoimmune diseases, including lupus, have abnormal germinal center reactions, which seem essential in triggering autoimmune signals. 37 Spontaneously generated splenic germinal centers were associated with autoantibody production and atypical distribution of B220+ PNA+ (peanut agglutinin) GL-7+ (Ly77 or B-/T-cell activation marker) B cells and M1+ (Murine 1) follicular DCs. 38 Splenic follicular gal-3+ cells have DC morphology, 39 but it is not clear whether follicular macrophages also express gal-3 within lymphoid follicles. 22 In gal-3−/− mice chronically infected by helminthes, such as Schistosoma mansoni, niches of CD138+ plasma cells and B220+ B lymphocytes were severely disturbed in the spleen and mesenteric lymph nodes, affecting the activation and survival of these cells. Moreover, the high number of follicular apoptotic bodies indicates that gal-3 is an important mediator of apoptosis and/or clearance in these lymphoid organs. 22 Notably, both mechanisms are involved in the pathogenesis of SLE and other autoimmune diseases. 40
In the thymus, exogenous gal-3 plays a de-adhesive role favoring intrathymic migration of CD4/CD8 T cells during differentiation events. 41 By contrast, intracellular gal-3 suppresses TH17 phenotype by inhibiting IL-23/IL-17-axis in DCs. 42
In gal-3−/− mice, the serum levels of IL-23, transforming growth factor (TGF)-β1 and IL-1β were significantly increased leading to a decrease in IL-12, interferon (IFN)-gamma and TH1 cells. 43 Since SLE progression is critically associated with pro-inflammatory TH17 cells secreting IL-17, IL-1, IL-6 and IL-23, 44 it is likely that gal-3 plays a regulatory role in the pathogenesis of SLE by suppressing TH17 cell differentiation.
SLE patients with predominant type I IFN signature have a high rate of macrophage differentiation and autoantibody production leading to a more severe disease. 45 Plasmacytoid DCs (pDCs) responsible for type I IFN signature are activated by self-nucleic acid complexes via CD32 and toll-like receptor (TLR)-9. 46 Self-nucleic acid complexes are abundantly released by apoptotic cells and by neutrophils during neutrophil extracellular traps formation. 47
On peripheral blood mononuclear cells, gal-3/CD32 (low-affinity IgG receptor—Fcgamma RII) interaction inhibits TH2 cytokine responses and favors a type I IFN phenotype. 18 It is well known that the inhibition of type I IFN signals leads to a delayed symptoms in experimental SLE 48 ; thus, gal-3 could promote SLE by increasing type I IFN expression.
Another mechanism potentially involved in gal-3 inducing SLE development consists in the interaction with transcription factors that induce B-/T-cell differentiation and hypergammaglobulinemia, such as Blimp-1 (B lymphocyte-induced maturation protein-1) which follows DC activation. 49 In MRL.Faslpr mice, the absence of DCs leads to milder SLE symptoms, reduced T-/B-cell expansion and lower production of autoantibodies. 50 Although poorly explored in SLE, polymorphisms in Blimp-1 have been proposed as a “risk factor” for SLE. 49 Inhibition/deletion of gal-3 in vivo leads to up-regulation of Blimp-1 affecting plasma cell behavior, survival and synthesis of IgG and IgE.3,24
IgG anti-galectins have been reported during the early stage and progression of SLE, especially anti-gal-2, −3, −7, −8 and −9. 51 In humans, anti-gal-3 autoantibody serum levels were significantly higher in SLE patients than in healthy donors or in patients with rheumatoid arthritis (RA), primary Sjögren's syndrome (pSS) and systemic sclerosis (SSc). 52
As far as clinical manifestations are concerned, Shi et al. found an association between anti-gal-3 antibodies and SLE cutaneous vasculitis. Indeed, SLE patients bearing skin lesions have high serum levels of anti-gal-3 autoantibodies, which are localized on the vessel walls. Notably, the injection of anti-human gal-3 antibodies in BALB/c mice evoked a dermal vasculitis 36 h post injection. 53
Gal-3 and anti-gal-3 antibodies might also be involved in development of lupus nephritis, which affects approximately 50–60% of SLE patients. 54 During the early phases of lupus nephritis, autoimmune T cells respond to tissue injuries reacting against chromatin or nucleosomes derived from cell death, including apoptosis, necrosis, necroptosis, autophagy and possible disturbances in the clearance of dying cells, leading to differentiation of autoimmune lymphocytes.55,56
Interestingly, SLE patients affected with lupus nephritis were seen to display higher serum levels of anti-gal-3 antibodies compared with controls; additionally, gal-3 stained in glomeruli in the majority of patients, while it was absent in controls. 38 Notably, gal-3 was expressed in distal tubules of normal kidneys. 38 Gal-3 expression scores correlated with anti-double stranded (ds) DNA antibody titers, renal histological activity indexes and complement consumption in SLE patients. 38
In rats, gal-3 modulates mesangial cell proliferation and matrix synthesis during experimental glomerulonephritis. Moreover, it was demonstrated that gal-3 is significantly secreted by infiltrating macrophages and distal tubular epithelium. 57 Recently, Nielsen et al. 58 revealed an increased population of gal-3-binding protein (G3BP)-positive microparticles either in sera or in electron-dense deposits from kidney biopsies of SLE patients affected with lupus nephritis, suggesting that gal-3 co-localizes in immune complex deposits. These observations support the potential role of gal-3 as a putative autoantigen in development of specific lupus manifestations.
Taking into account these data, whether gal-3 is involved in fueling or dampening SLE pathways is currently unclear (Table 1).
Rheumatoid arthritis
RA is an autoimmune disease characterized by an aberrant immune response against citrullinated self-peptides and progressive joint destruction. 59 Failure in CD4+ Treg-mediated self-tolerance and increase in TH17 cells seem to be critical in the immunopathogenesis of RA. 60 Indeed, in RA synovitis, the inflammatory process is orchestrated by activated monocytes (secreting IL-1β, IL-6, IL-7 and tumor necrosis factor-alpha (TNF)-α), fibroblast-like synoviocytes (synthesizing metalloproteases, chemokines and cytokines, mainly IL-15), and dysregulated osteoclasts (due to high levels of TNF-α and IL-17). 61
Gal-3 seems to play a pro-inflammatory role in RA 62 and high serum levels of gal-3 have been found in mice during collagen-induced arthritis. 63 In humans, gal-3 mRNA and protein are overexpressed in the synovial membrane close to joint destruction and are highly correlated with C-reactive protein (CRP) serum levels and IL-6, granulocyte–macrophage colony stimulating factor (GM–CSF), chemokine (C-X-C) motif ligand 8 (CXCL8), metalloproteinase-3 (MMP-3), TNF-α, chemokine (C-C motif) ligand 2 (CCL2), CCL3 and CCL5 secretion by synovial fibroblasts. 64 The lack of gal-3 prevented antigen-induced arthritis in mice which showed lower levels of antigen-specific IgG, IL-6, TNF-α and IL-17. 62
Genetic polymorphisms in gal-3 gene seem to be involved in RA susceptibility. Indeed, it has recently been shown that gal-3 gene (LGALS3) + 292C allele is associated with RA in Taiwanese population. 65 Notably, recombinant gal-3 in gal-3−/− mice can restore RA symptoms and cytokine production. 62 Finally, gal-3 short hairpin RNA (shRNA) injected in inflamed joints reduced collagen-induced arthritis in rats. 63 Thus, gal-3 is likely to play a critical role in RA development by regulating synovial fibroblast functions (Table 1).
Systemic sclerosis
SSc is a heterogeneous autoimmune disease characterized by progressive fibrosis of skin and internal organs due to an abnormal and chronic activation of fibroblasts. 66 Gal-3 has been suggested to play a key role in inducing fibrosis in different tissues. 5 Gal-3 serum levels are substantially increased in SSc patients where they seem to be correlated with the fibrotic process; in fact, gal-3 seems to regulate fibroblast activation, one of the major pathways responsible for fibrosis in SSc. 67 Indeed, gal-3 serum levels were not increased in the early stages of diffuse SSc but they progressively increased over disease course. 68 Thus, gal-3 represents an interesting molecule, potentially involved in the pathogenesis of SSc, which can help us to better understand the mechanisms of fibrogenesis in experimental models and humans (Table 1). It might also represent a therapeutic target in SSc.
Polymyositis/dermatomyositis
Polymyositis and dermatomyositis are considered the two major subsets of idiopathic inflammatory myopathies. 69 Polymyositis is characterized by a CD8+ T-cell response against muscle antigens presented by MHC Class I molecules. On the other hand, dermatomyositis is marked by a complement-mediated microangiopathy, typically mediated by type I IFN phenotype and autoantibodies with distinct specificities70,71; however, the exact mechanism involved in complement activation remains unclear. 72 Current biological treatments in polymyositis/dermatomyositis include blockade of cells and cytokines, including B-cells by rituximab, 73 T cells by abatacept and cytokines by anti-IL-1, anti-IL-6 or anti-IFNα monoclonal antibodies. 74
Autoantibodies to gal-3 were found more frequently in the sera of patients with polymyositis/dermatomyositis than in those with other autoimmune diseases, including RA, pSS, SSc and SLE. 52 However, the analysis of sensitivity and specificity of these autoantibodies in large cohorts of patients is critical in order to determine whether anti-gal-3 could be used as diagnostic marker in patients with polymyositis/dermatomyositis.
Primary Sjogren syndrome
pSS is an autoimmune disease well defined by mononuclear cell infiltrate in the exocrine glands, primarily lacrimal and salivary glands. 75 The inflammatory infiltrate leads to acinar atrophy, which is correlated with ectopic follicles (tertiary lymphoid tissues) composed by lymphocytes, plasma cells and stromal fibrosis. 76 In the follicles, autoantigens are presented to T and B cells and there are germinal centers where autoantibodies can be generated. 77 Gal-3 serum levels are significantly increased in pSS patients where they are correlated with serum levels of plasma CRP, fibrinogen, IgG and IL-17. 78 However, the cellular and molecular mechanisms of gal-3 in pSS have been poorly investigated so far (Table 1).
IgA nephropaty
Glomerulonephritis refers to an immune inflammatory process within renal glomeruli, which is sometimes associated with autoantibodies, eventually leading to end-stage renal disease. 55 The first step in the development of glomerulonephritis is the activation of TLRs+ and nod-like receptors (NLRs)+ cells by PAMPs and DAMPs. 79 A combined TH1- and TH2-cytokine response induces CD4+ T-cell differentiation into TGF-β+IL-10+CTLA4+ Treg cells via TGF-β induction, INF-γ+TNF-α+ TH1 cells via IL-12 induction, IL-4+IL-5+IL13+ TH2 cells via IL-2, IL-4 and IL-13 induction, or IL-17+IL-21+ TH17 cells via TGF-β, IL-6 and IL-17, all of them involved in glomerular injury. 80
The most prevalent glomerulonephritis in humans is the immunoglobulin A (IgA) nephropathy, which is characterized by huge glomerular deposition of IgA immune complexes leading to renal injury. 81 IgA nephropathy is hallmarked by high levels of IgA in the serum and poorly galactosylated polymeric IgA1 is widely deposited in the mesangium, following secretion by plasma cells in mucosa-associated lymphoid tissues (MALT). 82 Since gal-3 was shown to control mesangial cell proliferation and extracellular matrix compounds in a rat model of glomerulonephritis induced by passive transfer of anti-Thy1 antibodies, 57 it is possible that gal-3 is involved in the pathogenesis of the IgA nephropathy.
Gal-3 protects renal tubules from chronic injury by limiting apoptosis. 83 Consistently, gal-3−/− mice develop pronounced changes in renal function and structure in age-related renal disease 84 ; however, it is not clear whether and how gal-3 regulates renal function. Furthermore, no relationship between gal-3 and renal disease associated with IgA deposition has been demonstrated yet. Recently, Oliveira et al. 85 demonstrated that IgA expression and secretion are up-regulated in serum and peritoneal fluid in the absence of gal-3, suggesting that IgA-differentiating plasma cells and IgA secretion are at least in part regulated by gal-3. Although gal-3 and IgA nephropathy are not correlated, these observations deserve further studies.
Psoriasis
Psoriasis is characterized by red and scaly patches in the skin, ridged fingernails, hair loss, painful joints and systemic findings. 86
Myeloid DCs (mDCs) drives the immunopathogenesis of this immune-mediated inflammatory disease by releasing IL-23 and IL-12, 87 which are responsible for the activation of TH17, TH1 and TH22 cells which produce the “psoriatic cytokines” IL-17, IFN-γ, TNF and IL-22 affecting keratinocyte functions. 88 In contrast, Treg cells control immune system by preventing autoimmune responses against self-antigens. 89
The role of gal-3 in the pathogenesis of psoriasis is still poorly understood; however, it has been shown that it is up-regulated in dermal endothelial cells and down-modulated in the epidermis 90 and Langerhans cells. 91 The mechanisms involved in these modifications of gal-3 expression have not been studied yet.
Atopic dermatitis
Atopic dermatitis (AD) is an autoimmune disorder characterized by defective skin barrier, which becomes permissive to penetrating allergens with systemic sensitization 92 and IgE and IgG anti-nuclear autoantibody production. 93 It is first driven by a TH2-cytokine phenotype which is then switched to a mix TH1/TH2 phenotype. 94 In mouse models, gal-3 polarizes immune responses towards a TH2 phenotype. 43 AD-ovalbumin (OVA)-induced mice had high levels of serum gal-3 whereas OVA-sensitized gal-3−/− mice developed a TH1-polarized immune response associated with a decrease in inflammatory markers, 95 suggesting that gal-3 may act as an immunomodulatory molecule during early stage of AD.
Multiple sclerosis
Multiple sclerosis (MS) is the most prevalent autoimmune disease of the central nervous system (CNS) and can be mimicked by experimental autoimmune encephalomyelitis in animal models. The pathogenesis of MS is marked by a potent TH17 immune response against demyelinating degenerative cells. 96 Apoptotic oligodendrocytes are engulfed by microglia in damaged tissues, which are infiltrated by macrophages, T/B lymphocytes, NKT cells and DCs.97,98
The involvement of gal-3 in the pathogenesis of MS may be coupled with oligodendrocyte differentiation, since gal-3 contributes to myelin integrity and function. 99 In gal-3−/− mice, myelinated axons are numerically reduced and the myelin is severely disorganized around axons. 99 However, during wallerian degeneration, sciatic nerve was rapidly regenerated by Schwann cell activities and the clearance of the myelin debris was significantly efficient in the absence of gal-3.100,101 Although the reason is still unclear, gal-3 may play a regulatory role in the development of MS, at least in part by affecting phagocytosis of myelin debris.
Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) partially reproduces human MS in experimental animals and is classified as a neuroinflammatory disease orchestrated by self-antigens recognized by autoreactive T cells. TH17 and TH9 cells have currently been considered pivotal in the pathogenesis of EAE, 102 besides the removal of degenerated myelin via phagocytosis mediated by microglia. 103 In EAE mice, gal-3 is up-regulated by activated microglia during myelin degeneration and phagocytosis in spinal cords and optic nerves. 104 In these activated cells, gal-3 activates PI-3 kinase pathway via rat sarcoma protein (Ras), phagocytosis mediated by complement receptor-3 and promotes a high-affinity interaction between degenerated myelin (myelin debris) and phagocytosis-receptors during tissue repair.105,106
In EAE, gal-3 prevents apoptosis and IL-10 production on the one hand and induces TH17 phenotype and IFN-γ synthesis on the other hand. In gal-3−/− mice, EAE was marked by a reduction in monocyte/macrophage infiltration and an increase in apoptotic cells. 107 Gal-3 regulates T-cell function forming complexes in TCR with Mgat5 (beta1,6 N-acetylglucosaminyltransferase V), an enzyme which exposes N-acetylgalactosamine domain, the binding site of gal-3. 108 Gal-3 inhibition phenocopies Mgat5−/− TCR clustering and increases the susceptibility to EAE. Thus, gal-3 can be involved in EAE development.
Type I diabetes (T1D)
The autoimmune type 1 diabetes (T1D) is caused by genetic predisposition and immune dysregulation leading to abnormal autoreactive T lymphocyte differentiation, pancreatic β cell apoptosis and deficient insulin production. 109 According to genome-wide association studies, pro-apoptotic genes contribute to insulitis and β-cell destruction. 110 Gal-3 protects pancreatic β cells against IL-1-mediated apoptosis. 111 On the other hand, inhibition or deletion of galectin-3 renders pancreatic β-cells more resistant to TNF-α/IFN-γ/IL-1β apoptosis. 112 Probably, extracellular and intracellular gal-3 have a different effect in T1D pathogenesis. Approximately 50% of pancreatic cells express intracellular gal-3 in healthy tissues. 113
Gal-3−/− mice are resistant to multiple low-dose streptozotocin-induced diabetes (MLD-STZ) showing modest mononuclear cell recruitment to pancreatic islets, low expression of IFN-γ and inducible nitric oxide synthesis (iNOS) and absence of expression of TNF-α and IL-17 in comparison with wild type mice. 114 The ablation of gal-3 was associated with up-regulation of “anti-apoptotic” genes, suggesting that gal-3 induces apoptosis of islet β-cells upon pro-inflammatory condition and exposition of autoantigens driving autoimmune responses during early T1D. The mechanism involved in the control of apoptosis on pancreatic β-cells by gal-3 has not been fully elucidated yet. However, gal-3 seems to play a role in the development of T1D and it could become a promising therapeutic target in T1D patients.
Autoimmune hepatitis (AiH)
Autoimmune hepatitis (AiH) is a chronic disease in which apoptotic hepatocytes and portal inflammatory reaction induce a collapse of hepatic lobules. 115 The pathogenesis of AiH is initially driven by a TH1/TH17 phenotype marked by infiltrating CD8+ T cell,116,117 and is subsequently switched to TH2 signature which promotes clonal expansion of autoantibody-secreting plasma cells and inhibits TH1/TH17 functions. 118 During the dampening of tissue inflammation, Treg cells attenuate the immune response 119 and NKT cells drive a fibrotic response. 120 Gal-3 is an interesting molecule due to its ability to induce hepatocyte apoptosis 121 and liver fibrosis, 122 both orchestrating the activation and differentiation of hepatic stellate cells (HSCs) into myofibroblasts. 123
Concanavalin-A (Con A)-induced hepatitis is an experimental model that mimics pathological changes observed in AiH patients. 124 The induction of hepatitis by Con A in gal-3−/− mice resulted in a weak liver injury, marked by low levels of pro-inflammatory cytokines secreted by hypoactive lymphocytes and DCs and a high number of annexin V+ propidium-idodide+ later apoptotic cells. 32 Moreover, gal-3 induces myofibroblast activation 122 and favors phagocytosis by HSCs during liver fibrosis, 125 indicating that gal-3 plays a critical role in the pathogenesis of Con A-induced hepatitis and probably in AiH.
Crohn's disease
Crohn's disease is an inflammatory bowel disease of autoimmune origin in which genetic predisposition and environmental factors results in abnormal T-cell immune response. 126 Gal-3 secreted by resident macrophages and colonic epithelial cells activates atypical colonic fibroblasts in lamina propria inducing the production of IL-8 via NF-κB and driving fistula formation. 127 During the evolution of the disease, the expression of gal-3 is decreased in the colon interfering with fibroblast migration into fistulae. 128
Epithelium breakdown was significantly associated with reduced expression of gal-3 and high levels of TNF-α, 129 suggesting that gal-3 regulates epithelial cell–cell interactions in the colon. In patients affected with Crohn's disease, high serum levels of IgG anti-gal-3 autoantibodies were inversely correlated with clinical symptoms. 130 By contrast, high levels of gal-3 in the sera and a high number of CD14+ cells in the blood of patients with Crohn's disease were found during the active stages of the disease, 131 Altogether these data suggest that galactin-3 can exert a pro-inflammatory effect in Crohn's disease and may have a role in the disease development.
Conclusions and perspectives
A number of targeted therapies have been tested in patients with autoimmune diseases and some of them were successfully introduced in clinical practice since they were shown to ameliorate disease course, improving quality of life and survival of patients. However, many unmet needs still remain in patients with autoimmune diseases.
Among new molecules capable of regulating immune synapses, histological organization and tissue repair, gal-3 emerged as a multifunctional protein with immunomodulatory effects on cell-fate decisions such as apoptosis, cell activation, differentiation and migration, which are all critical in immune inflammatory responses.
So far, the precise mechanisms by which gal-3 could influence the onset and progression of an autoimmune disorder have not been fully elucidated; however, preliminary data suggest that gal-3 may play a “double-faced” role in regulation of autoimmune responses. Actually, gal-3 may either dampen or foster the development of autoimmune disturbances, depending on which mechanisms are prominent in each disease. As an example, gal-3 is able to hinder apoptosis, thereby reducing the autoantigen burden in SLE, yet it can enhance type I IFN responses, thereby worsening autoimmune reactions, which are favored by an IFN-enriched environment. In addition, the intracellular or extracellular localization of gal-3 may privilege its anti-inflammatory versus its pro-immune attitude, similarly to what was described concerning the intracellular or extracellular isoforms of other molecules involved in modulation of the immune responses. 132 Interestingly, molecules involved in cellular homeostasis may not rarely become hazardous under aberrant conditions, e.g. an altered cytokine microenvironment or exaggerated antigen exposition, especially when they can affect cellular viability at several levels, as is the case of gal-3.
Involvement of gal-3 in regulation of cell survival and apoptosis is witnessed by gal-3 expression being frequently altered in several malignancies, including lymphomas.22,133 Interestingly, several systemic autoimmune disorders display an association with neoplasms, either carcinomas or lymphoproliferative disorders, supporting the concept that intriguing molecules may link autoimmunity and tumorigenesis in the context of autoimmune diseases. Keeping into account all the aspects reviewed here, gal-3 is likely to be involved in the pathogenesis of different autoimmune diseases. Additional studies are required, which can explore the many functions gal-3 is endowed with, in order it can be tested as a therapeutic target in experimental models.
Footnotes
Authors' contribution
FLO, NB, AG and RL collected experimental data and reviewed all articles cited in the paper; FLO and MG wrote the main text; LP and AD revised the paper. All authors read and approved the final manuscript.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.