
Abstract
The Wnt/β-catenin signaling pathway typically shows aberrant activation in various cancer cells, especially colorectal cancer cells. This signaling pathway regulates the expression of a variety of tumor-related proteins, including c-myc and cyclin D1, and plays essential roles in tumorigenesis and in the development of many cancers. Small molecules that block the interactions between β-catenin and Tcf4, a downstream stage of activation of the Wnt/β-catenin signaling pathway, could efficiently cut off this signal transduction and thereby act as a novel class of anticancer drugs. This paper reviews the currently reported inhibitors that target β-catenin/Tcf4 interactions, focusing on the discovery approaches taken in the design of these inhibitors and their bioactivities. A brief perspective is then shared on the future discovery and development of this class of inhibitors.
Impact statement
This mini-review summarized the current knowledge of inhibitors of interactions of beta-catenin/Tcf4 published to date according to their discovery approaches, and discussed their in vitro and in vivo activities, selectivities, and pharmacokinetic properties. Several reviews presently available now in this field describe modulators of the Wnt/beta-catenin pathway, but are generally focused on the bioactivities of these inhibitors. By contrast, this review focused on the drug discovery approaches taken in identifying these types of inhibitors and provided our perspective on further strategies for future drug discoveries. This review also integrated many recently published and important works on highly selective inhibitors as well as rational drug design. We believe that the findings and strategies summarized in this review have broad implications and will be of interest throughout the biochemical and pharmaceutical research community.
Keywords
Essential role of β-catenin in the Wnt/β-catenin signaling pathway
β-Catenin, first discovered by McCrea, is a component of the cell adhesion complex that binds to the cell adhesion molecule E-cadherin. 1 In mammals, β-catenin is both structurally and functionally homologous to the product of the armadillo gene in Drosophila. β-Catenin plays a well-known role in cell–cell adhesion; 2 however, this protein is now receiving increasing attention from researchers in a variety of fields because of its essential role in the Wnt/β-catenin signaling pathway. This signaling is involved in various developmental/maintenance processes in animals and shows a close relationship with tumorigenesis and the development of a wide range of cancers.3–7
The Wnt/β-catenin signaling pathway, also called the canonical Wnt signaling pathway, is quite ancient and evolutionarily conserved. Wnt signaling via β-catenin regulates many cell behaviors, such as proliferation, by shutting down β-catenin degradation, which causes β-catenin accumulation and subsequent activation of Tcf/Lef transcription factors. In the absence of the extracellular messenger Wnt, β-catenin undergoes degradation by forming a destruction complex consisting of adenomatous polyposis coli protein (APC), glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α), and Axin.
8
CK1α and GSK3β phosphorylate β-catenin, making it recognizable by an E3 ubiquitin ligase, β-TrCP, which then promotes the ubiquitination and proteasomal degradation of β-catenin (Figure 1(a)).
9
Consequently, β-catenin is maintained at quite a low level in the cytoplasm and does not readily enter the nucleus to interact with transcription factors, like Tcf and Lef. Under these circumstances, Tcf/Lef are bound by repressors, such as CREB-binding protein (CBP) or p300. Therefore, in normal conditions, the transcription of β-catenin target genes is repressed.
10
By contrast, when extracellular Wnt is present and binds to its transmembrane receptors, frizzled (FZD) and LDL receptor-related proteins (LRP5/6), an intracellular protein, Dishevelled protein (DVL), is activated.10,11 Activated DVL disrupts the destruction complex, thereby preventing β-catenin degradation and causing its accumulation in the cytoplasm. This β-catenin then enters the nucleus and activates Tcf or Lef (Figure 1(b)). These, in turn, activate the transcription of Wnt/β-catenin target genes such as c-myc,
12
cyclin D1,
13
and Bcl-w,
14
many of which have been known to be important in tumorigenesis and cancer progression.
Wnt/β-catenin signaling pathway. (a) Inactive state (extracellular messenger Wnt is not involved); (b) Active state (Wnt is actively involved). (A color version of this figure is available in the online journal.)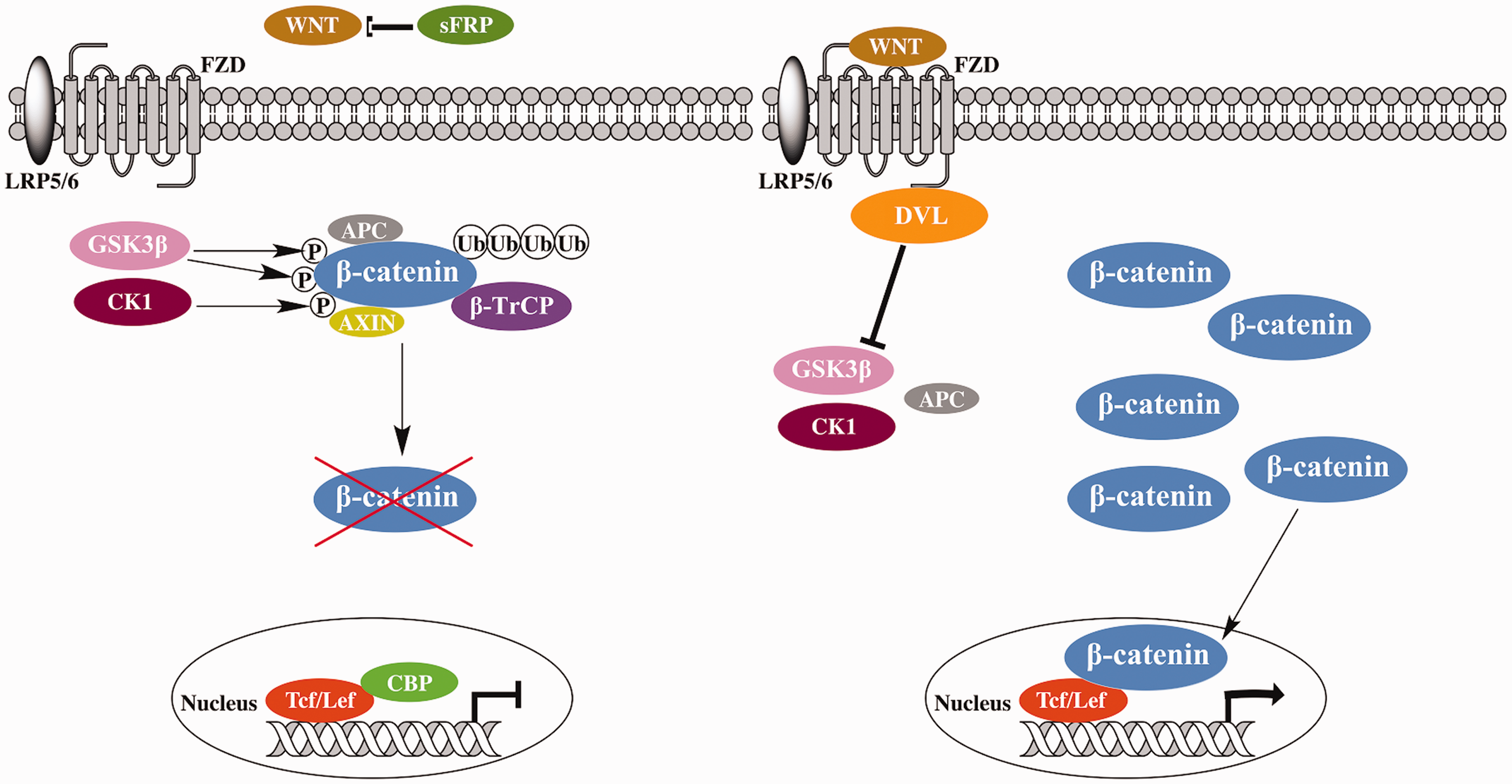
Structural characterization of β-catenin
β-Catenin contains 781 amino acids with a core of 12 highly conserved armadillo repeats in the center; each armadillo repeat consists of 40 amino acids in three α-helices (Figure 2).15,16 The farthest N- and C-termini are highly unstructured and not as conserved as the armadillo repeats. Both terminal domains are subject to protease digestion due to the presence of a TEV protease site, making them unlikely to undergo stable folding on their own. However, they mediate protein–protein interactions.
17
In the N-terminal region, the first armadillo repeat is structurally unique and features an extended helix with a kink, formed by the fusion of α-helices 1 and 2.17,18 This fusion creates a curvature of the armadillo domain, which aids in the binding of the β-catenin partners, such as α-catenin.
18
The N-terminal region also has a conserved motif for E3 ubiquitin ligase binding for β-catenin degradation.
19
The C-terminal region (residues 665–781), which consists of the α helix of β-catenin C-terminal domain (residues 667–683), is designated as helix C. The hydrophobic residues of the second and third helices of armadillo repeat 12 are capped tightly by the hydrophobic side chains from helix C. Together with the first armadillo repeat and armadillo repeat 11 to the C-terminus, helix C contributes to transcriptional regulation by recruiting both effectors and inhibitors.10,20 The central armadillo repeats, which are very conserved and well structured, are responsible for binding and interacting with most partners/coactivators, including Tcf, Lef, APC, and E-cadherin, as well as small molecule ligands.
A simplified diagram of β-catenin structure. The central armadillo repeats and several important motifs in N- and C-termini are shown. (A color version of this figure is available in the online journal.)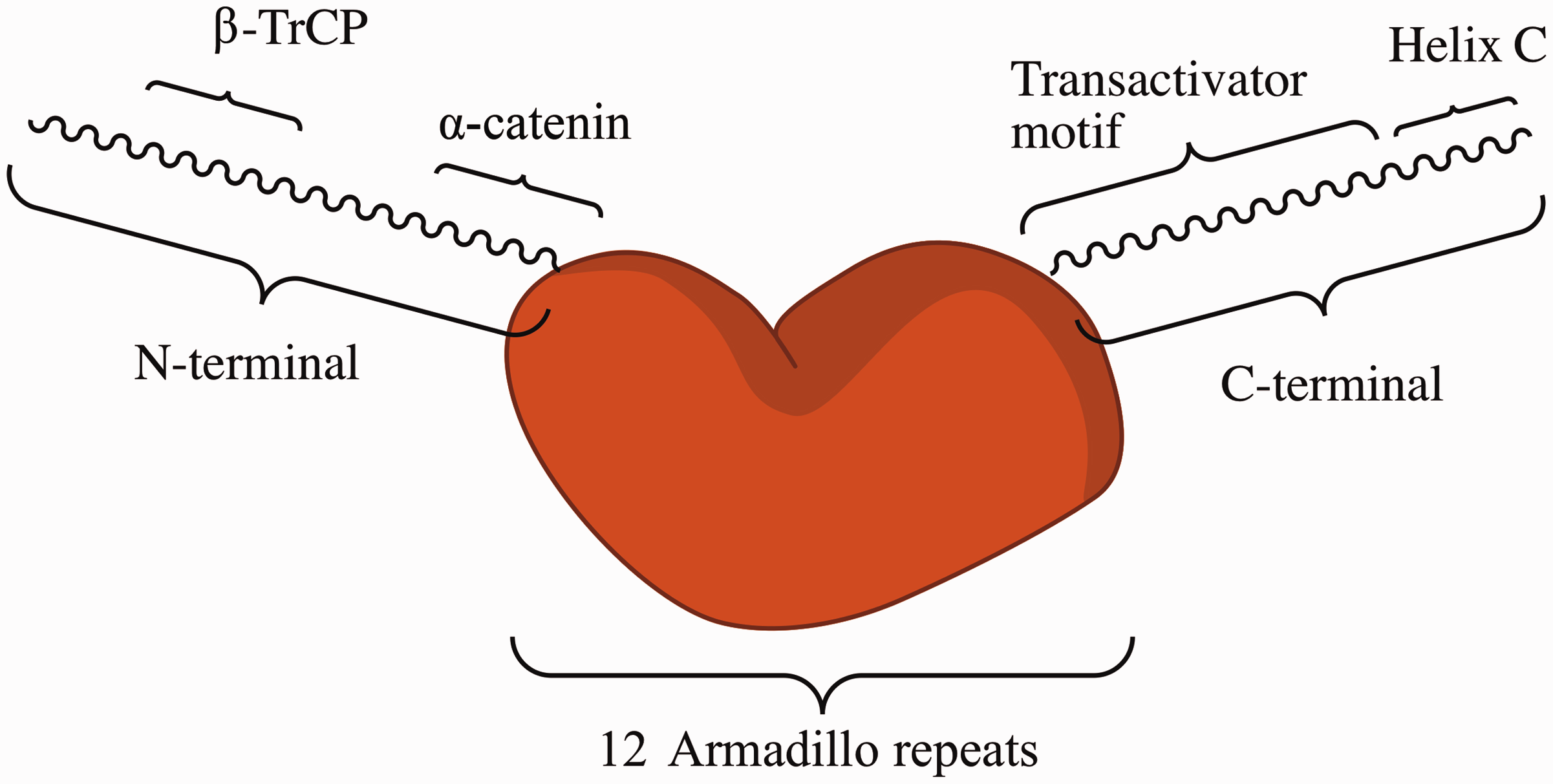
Inhibitors of the Wnt/β-catenin signaling pathway for cancer therapy
The Wnt/β-catenin signaling pathway plays a pivotal role in the regulation of transcription of many oncogenes; thus, various types of cancer show aberrant activations of this signaling pathway.21,22 Inhibitors of the Wnt/β-catenin signaling pathway could therefore efficiently disrupt signal transduction by this pathway and suppress the transcription of its target oncogenes, making these inhibitors potentially powerful anticancer drugs.21,23,24 Over the past decades, many inhibitors of the Wnt/β-catenin signaling pathway have been discovered, including biologics and small molecules that have shown promising activities in cancer therapy. The biologics include antibodies and RNA interference molecules that target Wnt protein25,26 and recombinant proteins that target extracellular modulators of the Wnt/β-catenin pathway, like Wnt inhibitory factor 1 (WIF-1) and secreted frizzled-related proteins (SFRPs). 27 The small molecule inhibitors can be divided into four categories according to their mechanism of action: (1) inhibitors of β-catenin/Tcf interactions, (2) antagonists of transcriptional co-activators (CBP, p300, BCL9, etc.), (3) molecules that bind to the PDZ domain of DVL, and (4) inhibitors based on other mechanisms (e.g. stabilization of the Axin protein). 28 Notably, a few existing clinical drugs are also inhibitors/modulators of the Wnt/β-catenin signaling pathway, including some nonsteroidal anti-inflammatory drugs (such as aspirin and sulindac), retinoids, and Vitamin D derivatives; their clinical values in cancer treatments are still being investigated. 24
Discovery of inhibitors that directly target β-catenin/Tcf interactions
Small molecule inhibitors targeting β-catenin/Tcf4 interactions

Names starting with “TMP” were assigned temporally by authors of this review.
T-cell factor 4 (Tcf4), also called Transcription Factor 7-like 2 (TCF7L2), is the most important transcription factor in the Tcf/Lef family (which includes Tcf1, Tcf3, Tcf4, and Lef1 in humans). Tcf4 binds to DNA and regulates the transcription of a variety of target genes. Interactions between Tcf4 and β-catenin are responsible for activation of transcription of many tumor-related proteins, including c-myc, 12 cyclin D1, 13 IL-8, 46 MDR1, 47 Bcl-w, 14 and ZEB1, 48 and play essential roles in tumorigenesis and development of many cancers. Therefore, the discovery of β-catenin/Tcf4 inhibitors has made these compounds a focal point for studies aimed at interrupting the Wnt/β-catenin signaling pathway, as well as for the development of anticancer drugs.
The human form of Tcf4 consists of 619 amino acids. It binds to DNA with its HMG-box domain (residues 350–418), whereas it binds to β-catenin with its 56 N-terminal residues. The 3D X-ray crystallography structures of β-catenin complexed with the Tcf4 N-terminal segment [PDB entry 1JPW; 49 2GL7 16 ] revealed that Tcf4 binds with its extended unstructured region (residues 13–25) to a positively charged groove on β-catenin, made up of consecutive armadillo repeats, while it binds with a short α-helix (residues 40–50) to the armadillo repeats 3–5 of β-catenin. Although interface of such protein-protein interactions is rather large (∼4800 Å2), experimental and computational approaches have demonstrated the presence of “hot spots” on this protein–protein interface.37,50 A hot spot is a small region on a protein whose contribution is significant or critical to the whole protein-protein binding affinity. The existence of hot spots makes it possible to use small molecules as protein–protein interaction inhibitors. If a small molecule binds tightly to a hot spot, it may efficiently disrupt the binding of β-catenin with other protein(s).
The binding site on β-catenin for Tcf4 also significantly overlaps with the binding sites for APC and E-cadherin. (Please refer to co-crystal structures of β-catenin with E-cadherin and APC.51,52) Binding between β-catenin and APC is necessary for β-catenin degradation, while binding between β-catenin and E-cadherin is essential for cell adhesion and normal stem cell functions. Therefore, a good inhibitor should have a high selectivity for the interactions of β-catenin/Tcf4 over those of β-catenin/APC and β-catenin/E-cadherin to avoid potential side effects. However, the β-catenin binding sites and binding modes of Tcf4, APC, and E-cadherin are quite similar, so this selectivity has been difficult to achieve. Nevertheless, recent studies using rational drug design strategies have made breakthroughs in selectivity and seem to indicate directions for discovering inhibitors with high potency and selectivity.
To date, many of small molecule inhibitors have been documented (Table 1), and their discovery approaches have included high throughput screening (HTS), conventional assay screening, in silico virtual screening, structural optimization of lead compound, and rational drug design. The following is a brief discussion of these approaches.
Discovering inhibitors through HTS
Modern drug discovery and medicinal chemistry now utilize HTS as one of the key approaches for identifying lead compounds. Lepourcelet et al. 29 reported an HTS of ∼7000 natural compounds using an ELISA method, which led to the discovery of six hits that showed IC50 values lower than 10 µM, including PKF115-584 (IC50 = 3.2 µM), PKF222-815 (4.1 µM), CGP049090 (8.7 µM), PKF118-744 (2.4 µM), PKF118-310 (0.8 µM), and ZTM000990 (0.64 µM) (Table 1). All six of these compounds came from microbial sources. The same HTS was also performed on a collection of 45,000 synthetic compounds, but no active hits were detected. The inhibitory activities of the hits were then confirmed by further biochemical assays. The interactions of β-catenin/E-cadherin were basically retained, but the interactions of β-catenin/APC were almost eliminated by the lead compounds PKF222-815, PKF115-584 and CGP049090. PKF115-584 and PKF222-815 also disrupted the binding of Tcf proteins to DNA. To our knowledge, this study is the first to report small molecule inhibitors of β-catenin/Tcf4 interactions with Tcf/E-cadherin/APC selectivities. Unfortunately, these selectivities were not presented in many studies reported thereafter.
Gonsalves et al. 30 presented HTS results using RNAi-based modifier screening method and reported the discovery of three small molecule inhibitors, iCRT3, iCRT5, and iCRT14. The primary screening used a Wnt responsive luciferase reporter, dTF12, to screen 14,977 small molecules, which included three collections of know bioactive compounds, fungal extracts, and a subset of a synthetic library with high structural diversity. This screening identified 34 inhibitory hits and 8 stimulatory hits for the Wnt pathway. After a secondary screening of 23 of inhibitory hits using an epistasis analysis in Drosophila cells, 21 compounds were found to inhibit the dTF12 reporter downstream of Slimb/βTrCP (a negative regulator downstream of the Axin/APC/GSK-3β complex). The dTF12 activity in Cl8 cells transfected with degradation-resistant S37A-β-catenin was strongly inhibited by iCRT3, iCRT5, and iCRT14. These mechanistic studies demonstrated that the inhibitory effect of these compounds was independent of β-catenin degradation/accumulation. Protein–protein binding assays and coimmunoprecipitation assays further confirmed that these three compounds, at micromolar or slightly higher concentrations, directly inhibited association of β-catenin with Tcf4. Coimmunoprecipitation studies also showed that these three compounds had very weak effects on the association between β-catenin and either E-cadherin or α-catenin. However, their effects on β-catenin/APC interactions were not reported in that study.
Wang et al. 31 identified a derivative of a natural diterpenoid, NC043, as a β-catenin/Tcf4 inhibitor by HTS of a library of 4000 small molecules. This HTS consisted of three steps: (1) A TOP-flash reporter assay activated by Wnt1; (2) A LiCl-induced TOP-flash reporter assay; and (3) A TOP-flash reporter assay using SW480 and Caco-2 cancer cells (in which the transcription of β-catenin target genes is constitutively activated due to APC truncation). NC043 can be easily synthesized through oxidation of spiramilactone by active MnO2; a preliminary SAR study showed that the 15-carbonyl group (which was newly introduced by oxidation) might be necessary for its inhibitory activity. NC043 did not affect β-catenin expression and distribution, but it significantly inhibited the interactions of β-catenin with Tcf4 in HEK293T and SW480 cells when supplied at 7.5 µM and 3.75 µM, respectively, as determined by immunoprecipitation and Western blots. Immunoprecipitation also showed that NC043 at concentrations up to 15 µM did not affect the interactions of β-catenin with E-cadherin in SW480 cells. Tumor inhibition studies also revealed exciting in vitro and in vivo activities of NC043 in SW480 colon cancer cells. The addition of 3.75 µM and 7.5 µM NC043 caused G2/M phase arrest of SW480 cells and led to apoptosis. Administration of NC043 (90 µg/kg for 17 days) to mice bearing SW480 xenograft tumors significantly decreased both the tumor weight and the tumor volume, but showed no effect on mouse body weight.
CWP232228, a newly discovered inhibitor of Wnt/β-catenin signaling, was found by HTS using a cell-based reporter assay.32,33 This compound antagonized the interactions of β-catenin with Tcf in the nucleus, and down-regulated a subset of the target genes of the Wnt/β-catenin pathway. CWP232228 has displayed promising activities in breast cancer stem cells and liver cancer stem cells in both in vitro and in vivo studies. Its prominent inhibitory effects on breast cancer stem cells may be attributed to disruption of IGF-I signaling. 32 CWP232228 has an ionizable structure and seems to have difficulty penetrating biomembranes for entry into the cells; therefore, this compound might work as a water-soluble prodrug that could be converted to a membrane permeant phosphate or phenol form after administration.
Fang et al. 34 recently reported the discovery of LF3, a small molecule with thiourea and sulfonamide groups. An Alpha Screen-based HTS was performed on 16,000 synthetic compounds using GST-tagged β-catenin (armadillo repeat domain, residues 134–668) and His-tagged Tcf4 (N-terminus, residues 1–79); 132 compounds were identified from primary screening, which then underwent examination of Alpha Screen and ELISA to give five hits (LF1 ∼ LF5). Of these five hits, LF3 exhibited good inhibitory activity, with IC50 < 2 µM in both assays. LF3 disrupted the interactions of β-catenin with both Tcf4 and Lef1 in a dose-dependent manner, as determined by binding assays using endogenous proteins from HCT116 cells. Its inhibitory effects on Wnt/β-catenin signaling were confirmed using stable and conventional TOP-flash assays. LF3, even at 60 µM, did not affect binding of β-catenin to E-cadherin, nor did it interrupt cell–cell adhesion mediated by β-catenin/E-cadherin interactions. It suppressed the expression of Wnt/β-catenin target genes, like Axin2, c-myc and cyclin D1, in various colon cancer cell lines, and it disrupted the self-renewal of cancer stem cells with high Wnt signaling and induced the differentiation. Most importantly, LF3 administered at 50 mg/kg inhibited the growth of SW480 xenotransplanted tumors in mice and promoted tumor differentiation without systemic toxicity.
An ideal inhibitor would selectively interrupt the β-catenin/Tcf interactions without interrupting the β-catenin/E-cadherin or β-catenin/APC interactions, as these latter protein–protein interactions are necessary for the continuation of the normal cell functions, such as cell–cell adhesion and β-catenin degradation. This selectivity had been evaluated in some of the studies cited above; however, quantitative assessment of the selectivity had been difficult. Zhang et al. 35 addressed this problem by developing two high throughput selectivity assays, one based on Alpha Screen and the other on fluorescence polarization (FP), which provided a quantitative evaluation of the selectivity and more efficient identification of the selective inhibitors. Each selectivity assay was used to measure the ability of small molecules to disrupt all three protein–protein interactions, followed by calculation of two quantified selectivity indexes, Tcf/E-cadherin and Tcf/APC. For three known inhibitors, PKF115-584, CGP049090, and PKF118-310, the selectivity for Tcf/APC agreed well with previous coimmunoprecipitation results; however, none of the three compounds showed Tcf/E-cadherin selectivity. According to the authors, this difference was likely due to the different phosphorylation states of E-cadherin used in their assays and in previous cell lysate-based coimmunoprecipitation. The same two-assay approach successfully identified four hits from a small set of Sigma-Aldrich compounds (consisting of 246 compounds); the four hits had Ki ≤ 80 µM for inhibition of β-catenin/Tcf4 interactions and selectivities for Tcf/E-cadherin and Tcf/APC >1 (i.e. L338192, R360163, R999636, and T155535). The Alpha Screen assay identified all four hits, while the FP assay identified three of the four, missing L338192. Among the four hits, R360163 displayed the best selectivities in the FP assay (Tcf/E-cadherin: 16.3; Tcf/APC: 25.0), and selectivities of other three hits were between 1.1 and 7.8.
Identifying β-catenin/Tcf4 inhibitors from antitumor natural products through conventional bioassay studies
Many natural products are known to possess antitumor activities, but the molecular targets or mechanisms of action of most compounds need further clarification. For example, a well-known antitumor flavonoid, quercetin, 53 and its analogs genistein, kaempferol, isorhamnentin, and baicalein 54 are inhibitors of β-catenin/Tcf signaling; their inhibitory effects are due to the down-regulation of β-catenin and Tcf4 levels in the nucleus or interruption of binding of β-catenin/Tcf complexes to consensus DNA. Li et al. 36 reported that a diterpenoid compound, henryin, can inhibit Wnt/β-catenin signaling by interrupting β-catenin/Tcf4 binding. Henryin selectively inhibited the growth of colorectal cancer cell lines (SW480 and HCT116, whose Wnt signaling pathway is constitutively activated due to mutation of APC or β-catenin) over other cancerous or normal cell lines. The protein levels of Axin2, cyclin D1, c-myc, and survivin were also decreased after henryin treatment. Henryin at 1–4 µM significantly antagonized the binding of β-catenin to Tcf4 in in vitro binding assays. Two analogs of henryin, phyllostachysin F and oridonin, showed similar activities to henryin in a ST-Luc reporter assay. A preliminary structure-activity relationship (SAR) study showed 14β-OH and a ketone group at C-15 were necessary for bioactivity of these compounds.
Discovering inhibitors through in silico virtual screening
In recent decades, the development of computer-aided drug design (CADD) has led to the wide use of virtual screening (VS), one of the most popular and practical methods in CADD, for the discovery of lead compounds.55,56 A typical structure-based virtual screening involves docking of thousands or millions of small molecules (usually pre-screened by druglike rules, such as Lipinski's rule of five) into the ligand-binding site of a receptor protein using a molecular docking method. The compounds are then scored and ranked according to their binding affinities to the receptor, calculated usually by molecular mechanics. The top-ranked compounds, usually tens to hundreds, are purchased and subjected to biological investigations, which then can identify several active hits or lead compounds. Compared to HTS, VS may greatly increase the hit rate of the screening, and drastically reduce the expenditures of time, labor, and materials.
Trosset et al. 37 reported PNU-74654, the first inhibitor of β-catenin/Tcf4 interactions discovered through VS. As already mentioned, Tcf4 binds with its N-terminus to a long groove on β-catenin, and a few hot spots exist on the interaction interface of these two proteins. A previous alanine-scanning study of all Tcf4 residues involved in β-catenin-binding 50 revealed three hot spots on Tcf4 that were important for β-catenin-binding: D16, D11, and L41/V44/L48. D16A mutations caused the greatest (50-fold) loss of protein–protein binding affinities, indicating that the most promising strategy for discovering small molecule inhibitors might be interruption of the interaction of D16 with β-catenin. However, the N-terminus of Tcf4 is an unstructured coil with no cavities to accommodate small molecules on its surface; therefore, small molecules need to bind to the corresponding region on the β-catenin surface to block Tcf4 D16. Trosset’s study 37 evaluated the interaction interface of β-catenin/Tcf using computational programs, including PASS and FLO_QXP, and a small pocket containing K435 and R469 of β-catenin (which is exactly the interacting site of D16 of Tcf4) was finally chosen as the ligand-binding site in the next study. A library containing ∼90,000 compounds was first pre-screened using the rules of druglikeness, solubility, etc., and 17,700 compounds that passed the pre-screening were then docked into the K435/R469 hot spot selected above, using a flexible-receptor docking strategy (sidechains of C429, D390, C466, and K508 were set flexible). The top-ranked compounds underwent visual inspection and 22 candidates were finally selected for bioassay examination using NMR WaterLOGSY and isothermal calorimetry (ITC). This led to the discovery of three active hits, of which PNU-74654 was the most active. This compound exhibited a KD of 450 nM in a direct binding ITC assay, as well as inhibitory activity in a luciferase reporter assay at the cellular level.
Based on the hot-spot strategy, Tian et al. 38 performed a VS on a small library containing 1990 compounds. The three hot spots on the β-catenin surface, inferred from previous Ala-scanning studies of Tcf4, 50 were carefully inspected, and the one consisting of K435 and R469 (which was used in Trosset’s VS study) was selected as the ligand-binding site. A flexible docking was performed with AutoDock4, with sidechains of some highly polar residues (K435, R469, K508, E571, etc.) set flexible. The top-ranked compounds were visually inspected and 100 compounds were selected for biological testing with a cell-based TOPflash reporter assay; BC21 38 and BC-23 39 were finally identified as potent β-catenin/Tcf4 inhibitors. BC21 is a copper-containing coordination compound, and it can inhibit the TOP-luciferase reporter in HCT116 cells at 10 µM. BC21 can induce the death of HCT116 cells in a dose-dependent manner, and inhibited the colony-forming activity of HCT116 cells 100% at 10 µM, but showed no cytotoxicity for normal cells including HEK293 cells and HUVECs at 3-15 µM. The IC50 value of BC21 was 5 µM for the interactions of β-catenin with a Tcf4 peptide.
Compared to BC21, BC-23 is a compound with greater druglikeness and more space for structural optimization. This compound potently inhibited the interactions between β-catenin and Tcf4 peptide in an FP-based competitive binding assay (IC50 = 1.7 µM), and it showed strong inhibitory activity in a TOP-flash reporter assay in H1299 NSCLC (non-small cell lung cancer) cells (IC50 = 2.3 µM). BC-23 also significantly down-regulated the expression of cyclin D1 and c-myc, two important target genes of Wnt/β-catenin signaling. Interestingly, BC-23 could significantly enhance the sensitivity of H1299 NSCLC cells to radiation treatment. A combined treatment of 10 Gy radiation and BC-23 (3 µM) administered to H1299 cells resulted in two orders of magnitude higher clonogenic death when compared with 10 Gy radiation treatment alone. Therefore, this compound might be a promising candidate or lead compound for radiation enhancers in cancer treatment.
Optimization of lead compound structure
Following its discovery in a drug development study, a lead compound usually needs to undergo structural modifications (“lead optimization”) to increase its activity, reduce its side effects, and improve the properties of the ADME (absorption, distribution, metabolism, and elimination) before it can enter clinical trials as a drug candidate and finally become a drug. However, when compared to that abundance of lead optimization studies in many fields of anticancer drug discovery, such as those conducted for kinase inhibitors, far fewer lead optimization studies have been reported for β-catenin/Tcf4 inhibitors.
Chen et al.
40
reported their lead optimization work on TMP-A-1 (Scheme 1), a 2,4-diamino-quinazoline compound discovered through a high throughput screening (HTS) of their compound library. This lead compound had good activities in a cell-based reporter assay using Tcf22C11 cells (IC50 = 0.60 µM); however, it exhibited unfavorable pharmacokinetic properties, including poor water solubility and poor metabolic stability. A preliminary structural modification and SAR investigation replaced the biphenyl group with a sidechain containing an N-benzylpiperidinecarboxamide. The resulting compound (TMP-A-2) had higher water solubility and greater metabolic stability than TMP-A-1, so it represented a good starting point for further optimizations. TMP-A-2 was used as a template to design and synthesize two series of derivatives: TMP-A-7 (7a–7k) was obtained by altering substituents on the 2-amino group to enhance the bioactivity, whereas TMP-A-16 (16a–16p) was designed to resolve the problem of metabolic N-debenzylation. A new derivative, TMP-A-16k (R = 4-fluorobenzyl), was found to exhibit high potency in Tcf4-luciferase reporter assays (IC50: 0.22 µM in 22C11 cells, 0.19 µM in 33.13 cells) as well as improved pharmacokinetics properties, including solubility and bioavailability; however, neither TMP-A-16 k nor TMP-A-2 showed in vivo inhibitory activities in HCT116 or in HT29 xenograft models.
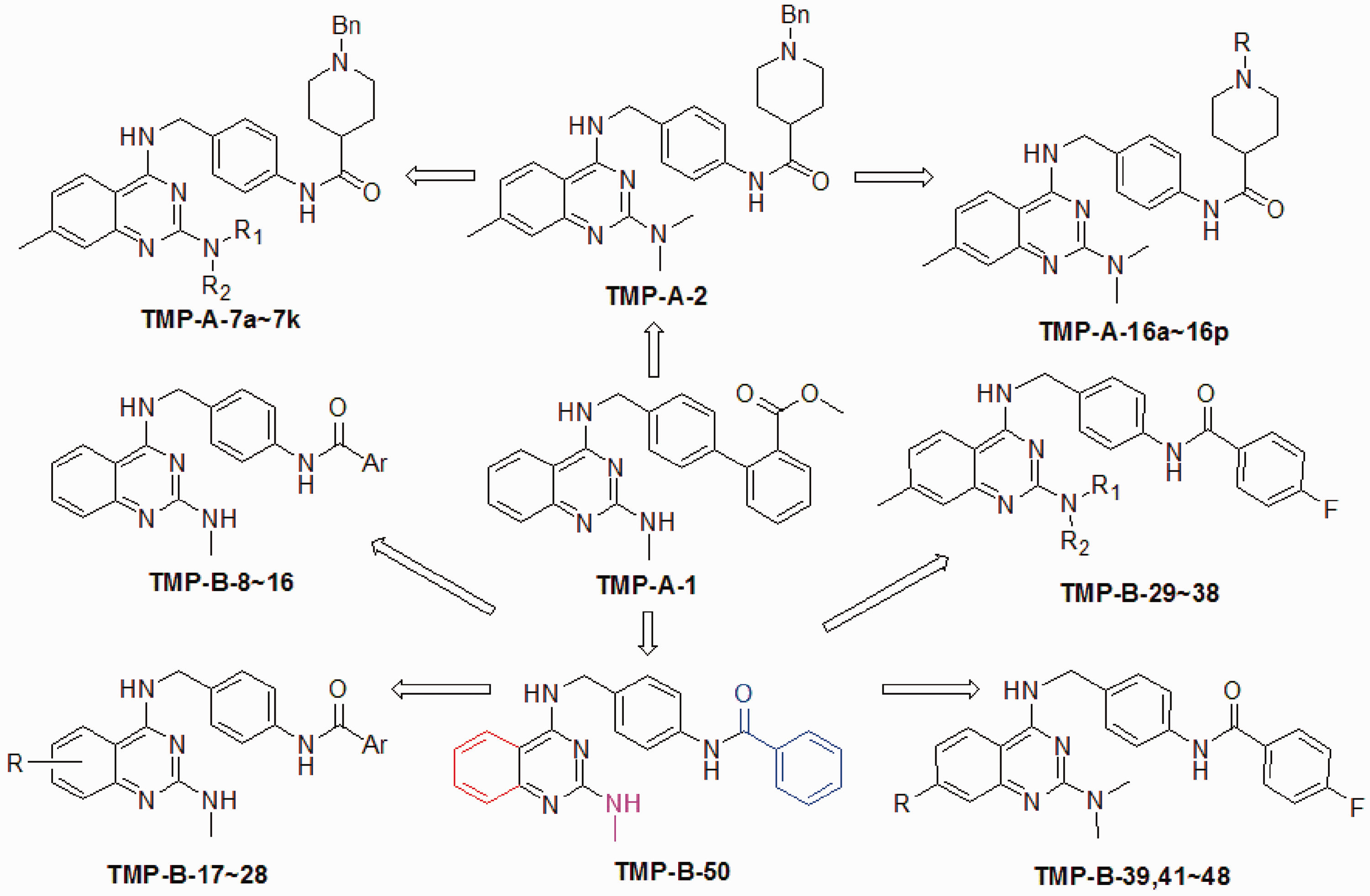
Subsequent lead optimization studies40,41 in ligand-based drug design by the same research group led to the introduction of compound TMP-B-50 (Scheme 1), which had a similar molecular shape to TMP-A-1, but supposedly better pharmacokinetic properties, including cLogP, water solubility, and metabolic stability, making it a good template for designing novel and potent inhibitors. Alteration of the substituents on the phenyl ring of quinazoline, the 2-amino group, and the terminal acyl group (marked in red, magenta, and blue in Scheme 1) led to the design and synthesis of four series of derivatives for biological evaluation using a Tcf4-luciferase reporter assay in 33.13 and 22C11 cells. Two derivatives selected for further evaluation, TMP-B-9 and TMP-B-19, exhibited high inhibitory activity on four colon cancer cell lines, HT29, DLD1, LoVo, and 33.13 (IC50 447–1739 nM). However, both compounds displayed very low drug concentrations in the plasma and tumors after oral administration, presumably due to the low membrane permeability of TMP-B-9 and the low solubility and metabolic stability of TMP-B-19. When TMP-B-9 was administered intraperitoneally to mice bearing β-catenin/RK3E xenograft tumors, it exhibited potent in vivo antitumor activities (150 mg/kg, once a week, 66% tumor growth inhibition; 18.75 mg/kg per day, 7 days, >50% inhibition).
Mao et al.
42
examined the structure and preliminary SAR of 2,4-diamino-quinazoline compounds to initiate new explorations in lead optimization.
42
Their work included structural modifications on the 2-amino, terminal acyl, and phenyl ring of the quinazoline, as well as on other sites, like the amide linkage, the middle phenyl ring, and the quinazoline ring itself (Scheme 2). These new derivatives were synthesized and evaluated for cell growth inhibitory activities in colon HCT116 and SW480 cancer cell lines, and 20 compounds with good activities were tested for inhibition of β-catenin/Tcf4 signaling in a luciferase reporter assay in HCT116 cells. These studies revealed three potent compounds (TMP-C-74, 78 and 86), with IC50 values of 1.5–2.5 µM in reporter assays and 0.6–1.4 µM in cell growth inhibition assays. All three compounds shared the same core structure and only varied in their substituents at the terminal amide nitrogen. A subsequent 3D-QSAR (quantitative structure–activity relationship) model generated through comparative molecular field analysis (CoMFA) later illustrated the effects of the steric and electrostatic properties of the substituents on bioactivity and provided guidance in further lead optimization studies.
New derivatives designed in reference.
42
Modified parts are marked in red. (A color version of this scheme is available in the online journal.)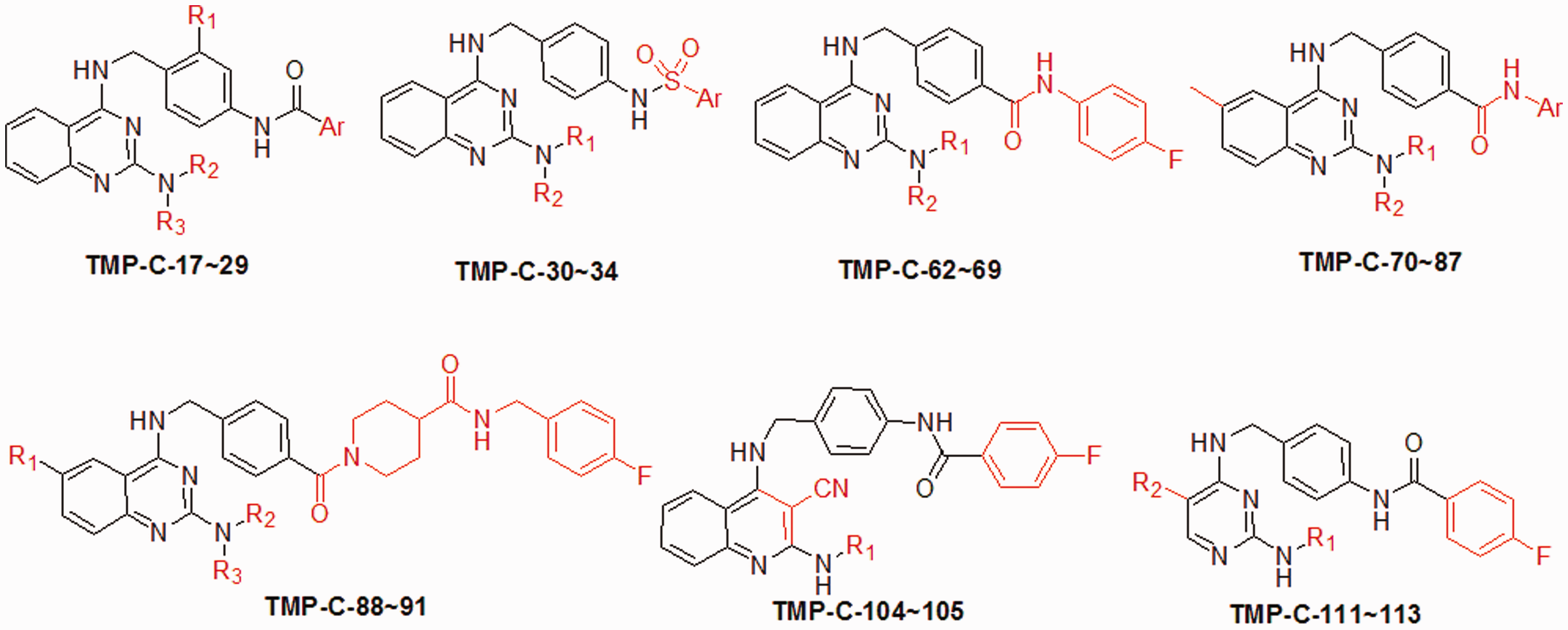
Using Alpha Screen and FP assays, Catrow et al. 43 screened several compound sets (consisting of 2093 small molecules in total) and discovered ZINC02092166. In parallel assays, ZINC02092166 exhibited comparable activity to PKF118-310, more potent than PKF115-584, CGP049090, iCRT3, iCRT5, and iCRT14. In SW480 cells, ZINC02092166 strongly inhibited expression levels of cyclin D1, c-myc, and Axin2, which are target genes of Wnt/β-catenin signaling pathway, but showed no inhibition on expression of β-catenin. Coimmunoprecipitation also confirmed its inhibitory activity on β-catenin/Tcf interactions at cell level at concentrations of 1–4 µM. However, this compound showed rather weak selectivity on interactions of β-catenin with Tcf, E-cadherin and APC (Tcf4/E-cadherin: 2.0; Tcf4/APC: 1.73). Another problem involves the acyl hydrazine group, which is known as a pan assay interference compounds (PAINS) substructure that can give promiscuous results in bioassay screenings. Prediction of the binding pose of ZINC02092166 to β-catenin by molecular docking was then used to assist subsequent structural modifications. First, the imine carbon atom was replaced with a nitrogen atom to eliminate potential chemical reactivity, and this made the whole tetracycle system aromatic. Next, the hydrazide was replaced with the amide to give TMP-D-26, from which a series of compounds (TMP-D-27–38) were designed. Three derivatives bearing a carboxylic acid group (TMP-D-29, 31, and 33) displayed high activities, probably due to salt bridge formed between the carboxylic acid and β-catenin K435 (which is crucial for interactions with D16 of Tcf4). Two derivatives with a tetrazole (a bioisostere of carboxylic acid) also had good activities. TMP-D-33 was the most potent among these compounds (Ki values were 3.4 µM in FP and 1.0 µM in the Alpha Screen). Notably, TMP-D-26, 29, 31, and 33 exhibited high β-catenin/Tcf4 selectivity (10–20 fold) against β-catenin/E-cadherin and β-catenin/APC.
Discovering inhibitors with high selectivity by rational drug design
In contrast to traditional drug discovery methods, like HTS and structural modification of lead compounds, rational drug design is based on the understanding of drug-receptor interactions and generally makes use of the three-dimensional structure of proteins and CADD. Ligands generated by rational design usually have a novel structure/scaffold, high binding affinity, and high selectivity.
Yu et al. 44 reported the rational design of UU-T01. Structure inspection disclosed several key binding features of Tcf with β-catenin, which were then used to guide a fragment-based drug design. Fragments in libraries were docked to the Tcf4-binding pocket on β-catenin to mimic these binding features, and the top-ranked fragments were then linked with a suitable chain/scaffold to form the complete molecule. This method resulted in the design and synthesis of six compounds, two of which (including UU-T01) exhibited potent activity in FP assays. UU-T01 has a novel structure and shows the lowest Ki (3.14 µM) in FP assays. These compounds also underwent structural optimization, but no derivatives showed higher activity than UU-T01 itself. Notably, UU-T01 exhibited high selectivities against β-catenin/E-cadherin and β-catenin/APC interactions (28.9 and 44.7 folds as determined by the Alpha Screen assays), 35 even though these selectivity issues had not been explicitly considered during the compound design steps.
Another rational design study was reported in 2014, by the same research group. 45 The primary goal of this later study was to design highly selective inhibitors based on the subtle differences between the binding sites and the mode of interaction between β-catenin and Tcf4, E-cadherin, and APC. Inspection of the crystal structures of β-catenin with these co-factors, as well as site-directed mutagenesis and biochemical assays, led to the proposal that the binding site of the Tcf4 G13∼E17 fragment on β-catenin was a selective binding site for Tcf4 over E-cadherin and APC, and that ligands mimicking the binding mode of the Tcf4 G13∼E17 fragment might be highly selective for β-catenin/Tcf4 interactions. Next, using a peptidomimetic strategy, seven compounds were designed and synthesized, two of which exhibited potent activity in FP assays (Ki = 5.74 and 6.63 µM, respectively). Further structural optimization led to the production of UU-T02 and UU-T03. UU-T02 exhibited high inhibitory activity on β-catenin/Tcf (Ki = 1.36 µM), and high selectivity over E-cadherin and APC (175.1 and 63.7 folds, respectively). Its diethyl ester, UU-T03, displayed high inhibitory activities in cell-based luciferase reporter assays and cell growth inhibition assays (SW480, HT29, and HCT116 colon cancer cells), while it showed no effect on β-catenin/E-cadherin or β-catenin/APC interactions when supplied at 60 µM.
Perspective
The β-catenin/Tcf4 inhibitors have been identified using most of the common approaches used in drug discovery, including conventional screening, HTS, VS, lead optimization, and rational design. When compared with the inhibitors discovered in the early stages of this type of research, the newly discovered inhibitors generally have higher activities and druglikeness, and a few inhibitors have made breakthroughs in term of selectivity for Tcf4/E-cadherin/APC. Some inhibitors have exhibited promising in vivo activities and good tolerance in xenograft tumor models of colorectal cancer cell lines (like SW480 and HCT116). However, to the best of our knowledge, none of these inhibitors have entered clinical trials.
One interesting feature is the relative rarity of lead optimization studies on β-catenin/Tcf4 inhibitors when contrasted with the number of discovered active hits or lead compounds. As indicated above, selectivity issues have been a great obstacle in the further development of many inhibitors. Several inhibitors also have not been investigated for selectivity, thereby precluding evaluation for further development or structural optimization. In our view, a selectivity investigation of these hits or leads would be invaluable, as compounds with moderate or even lower selectivities might be subjected to further structural optimization studies. Structure-based lead optimization strategies that make use of the subtle differences in interactions of β-catenin/Tcf, β-catenin/E-cadherin and β-catenin/APC would be helpful in identifying inhibitors with high specificity. Similarly, in silico virtual screening that take these subtle differences into account would also likely lead to hit with novel structures and high selectivity.
Footnotes
Authors’ contributions
All the authors participated in the literature survey and the compilation of this review. MY and GL wrote the review, and JA made revisions and supervised the entire work.
Acknowledgements
This work was supported in part by grants from the Carol M. Baldwin Breast Cancer Research Fund and the LUNGevity Foundation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.