
Abstract
Physiologically, the human and murine hearing systems are very similar, justifying the extensive use of mice in experimental models for hearing impairment (HI). About 340 murine HI genes have been reported; however, whether variants in all human-mouse ortholog genes contribute to HI has been rarely investigated. In humans, nearly 120 HI genes have been identified to date, with GJB2 and GJB6 variants accounting for half of congenital HI cases, of genetic origin, in populations of European and Asian ancestries, but not in most African populations. The contribution of variants in other known genes of HI among the populations of African ancestry is poorly studied and displays the lowest pick-up rate. We used whole exome sequencing (WES) to investigate pathogenic and likely pathogenic (PLP) variants in 34 novel human-mouse orthologs HI genes, in 40 individuals from Cameroon and South Africa diagnosed with non-syndromic hearing impairment (NSHI), and compared the data to WES data of 129 ethnically matched controls. In addition, protein modeling for selected PLP gene variants, gene enrichment, and network analyses were performed. A total of 4/38 murine genes, d6wsu163e, zfp719, grp152 and minar2, had no human orthologs. WES identified three rare PLP variants in 3/34 human-mouse orthologs genes in three unrelated Cameroonian patients, namely: OCM2, c.227G>C p.(Arg76Thr) and LRGI1, c.1657G>A p.(Gly533Arg) in a heterozygous state, and a PLP variant MCPH1, c.2311C>G p.(Pro771Ala) in a homozygous state. In silico functional analyses suggest that these human-mouse ortholog genes functionally co-expressed interactions with well-established HI genes: GJB2 and GJB6. The study found one homozygous variant in MCPH1, likely to explain HI in one patient, and suggests that human-mouse ortholog variants could contribute to the understanding of the physiology of hearing in humans.
Impact statement
Despite, human and murine hearing system being very similar, the contribution of variants in relevant mouse-ortholog genes to hearing impairment (HI) has not been fully investigated. The contribution of variants in the known non-syndromic hearing impairment (NSHI) genes among Africans is poorly studied, suggesting that the novel gene(s) and mutations are yet to be discovered in NSHI in the African populations. Using whole exome sequencing (WES), this study identified rare candidate pathogenic and likely pathogenic (PLP) variants in 3/34 novel human-mouse ortholog genes in 3/40 individuals, with one homozygous variant, MCPH1, c.2311C>G p.(Pro771Ala), likely to explain HI in one patient. In silico functional analyses suggest that these human-mouse ortholog genes could contribute to the understanding of the physiology of hearing in humans and thus the variants identified in those genes deserve additional investigations.
Introduction
Hearing impairment (HI) is one of the leading causes of disability globally. 1 A disabling HI refers to hearing loss ≥40 decibels (dB) in adult (15 years or older) and ≥30 dB in children (0 to14 years). Congenital HI affects 1–2 in 1000 new-borns globally. 1 A higher prevalence is recorded in sub-Saharan Africa (about 6 out of 1000 live births) compared to the high income countries (about 1 out of 1000 live births). 2 In most African settings, the actual population prevalence of HI is not entirely known. Observational studies reported relatively high prevalence of 2.0% to 21.3% among children from the rural and urban settings in Nigeria, Ghana, Cameroon, Sudan, Kenya, Tanzanian, Zimbabwe, Uganda, Angola, and South Africa. 3
In most populations, genetic factors contribute to about half of cases of congenital HI, of which about 70% are non-syndromic hearing impairment (NSHI). 4 In Cameroon, in a total of 582 patients, with limited molecular investigations, the genetic causes of HI were estimated at 14.8%, environmental factors at 52.6% and unknown causes estimated at 32.6%. 5 The inheritance pattern of NSHI of genetic origin is predominantly autosomal recessive (80%). 6 Among the populations of European and Asian ancestries, common mutations associated with NSHI are being identified in the GJB2 and GJB6 connexin genes. 6 But mutations in these connexins genes have been rarely found in HI in the populations of African ancestries.7–9
An exception is the founder mutation p.R143W in the GJB2 gene reported in Ghana in 1998, 10 that accounts for nearly 25% of familial cases and 8% of isolated cases of NSHI in the Ghanaian populations. 11 HI is genetically highly heterogeneous with nearly 120 genes identified, to date.12–14 Nevertheless, targeted panel sequencing has demonstrated a consistently lower pick-up rate in individuals of African ancestry.15,16 Moreover, the prevalence of autosomal recessive non-syndromic hearing impairment (ARNSHI) due to pathogenic and likely pathogenic (PLP) variants, selected from the ClinVar and Deafness Variation Databases, and gnomAD database, was estimated at 5.2 per 100,000 individuals for Africans/African Americans, compared to the highest prevalence of 96.9 per 100,000 individuals for Ashkenazi Jews. 17 Therefore, there is an urgent need to investigate HI in populations of African ancestry in order to address this knowledge deficit that is likely to hinder our current understanding of the HI pathophysiology.
Physiologically, the human and murine hearing systems are very similar, 18 supporting the widely used approach of studying the genetics of HI through murine models. About 340 murine HI genes have been reported 18 ; but whether all human-mouse ortholog genes have some roles in human HI remains to be fully investigated. A recent study identified 38 novel murine HI-associated genes in a cohort of newly generated mouse mutants and found among these genes, one human-mouse ortholog (SPNS2) that harbors PLP in childhood HI in humans. 19
In the present study, taking into account the huge diversity of African populations,20,21 and the low pick-up rate of known HI genes in African cohorts with HI,15,16 we hypothesized that investigating novel human-mouse ortholog genes in African populations may reveal important findings that could contribute to the knowledge of NSHI genetic causes, and ultimately to genetic medicine practice.
We used whole exome sequencing (WES) to investigate PLP variants in selected novel human-mouse ortholog HI genes, associated with HI in an autosomal recessive manner, among individuals affected with NSHI from Cameroon and South Africa.
Materials and methods
Ethical approval
The study was performed following the Declaration of Helsinki. The study was approved in Cameroon by the Institutional Research Ethics Committee for Human Health of the Gynaeco-Obstetric and Paediatric Hospital of Yaoundé, Cameroon (Ethics approval number No723/CIERSH/DM/2018) and in South Africa by the Human Research Ethics Committee (Ethics approval number HREC Ref: 104/2018) of the University of Cape Town, South Africa. Written informed consent was obtained from all participants if they were 18 years or older, or from the parents/guardians with verbal assent from the children.
Participants
HI diagnoses and phenotypic classifications were done by audiologists and medical geneticists. Only individuals with NSHI and individuals with prelingual onset (0–3 years) were included. A total of 40 unrelated patients selected for WES were included: 18 from Cameroon and 22 from South Africa with Xhosa ethno-linguistic ancestry. A structured questionnaire to eliminate syndromic and environmental causes of HI was described previously. 7 A total of 28 patients were sporadic cases of HI, while 12 patients' clinical and pedigree profiles were compatible with ARNSHI. All the patients did not have PLP variants in GJB2 and GJB6 genes, as described previously. 7
In addition to the WES study of the 40 NSHI individuals, 15 unrelated Cameroonians and 10 South Africans diagnosed with NSHI, and 26 ethnolinguistically matched control individuals with no personal or family history of HI were selected and investigated for the targeted mutations previously found in childhood HI in the human-mouse ortholog: SPNS2. 19
Whole exome sequencing
Exome library preparation and WES were carried out by Omega Bioservices (Norcross, GA, USA). DNA concentration was measured using the QuantiFluor dsDNA System on a Quantus Fluorometer (Promega, Madison, WI, USA). An Illumina Nextera Rapid Capture Exome kit (Illumina, San Diego, CA, USA) was used for exome library preparation. Briefly, 50 ng of genomic DNA was fragmented using the Nextera transposomes. The resulting libraries were hybridized with a 37 Mb probe pool to enrich exome sequences. The libraries were then hybridized with a 37 M probe pool to enrich the sequences and were then sent for WES using the Illumina HiSeq 2500 (Illumina), 100 bp run format, with an average read depth of 30×.
Genotyping of targeted variants in SPNS2
For the selected 51 additional individuals (25 patients with NSHI, irrespective of the mode of inheritance, i.e. sporadic or autosomal recessive; and 26 ethnolinguistically matched controls), we used the Sanger sequencing technique to screen for the two deletion mutations (c.1066_1067delCCinsT and c.955_957delTCC) associated with childhood HI in the SPNS2 human-mouse ortholog genes previously reported. 19 Specific primers targeting exons 5 and 6 were designed using Primer3 input and NCBI BLAST (Supplementary Table 1). The polymerase chain reaction (PCR) for the amplification of the specific exons was done using the PCR thermocycler machine. The optimal annealing temperature for the PCR was 60°C. The DNA sequencing was performed using the Sanger sequencing reaction on a BigDye Terminator v 3.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA). Sequenced chromatograms obtained from the reaction were analyzed using the BioEdit software to identify the mutations.
Variants’ annotation
The FASTQ files generated from the sequencer were processed to discard reads too short and the low-quality sequences. We checked for the high sequenced duplication rates, GC content, and Nextera adapter contaminations. Trimmomatic v0.39 was used for removing the Nextera adapters, 22 by trimming off three low-quality bases from the 5ʹ end and 30 low-quality bases at the 3ʹend of each read. Post sequencing reads were aligned to the build 37 of the human genome using the Burrows-Wheeler Aligner (BWA v0.7.15). 23 The sequencing quality check and data reports were done with the FastQC v0.11.5 and MultiQC v1.7 tools. 24 The GATKv4 MarkDuplicates command was used for marking and removing duplicate reads. 25 The GATKv3.8 IndelRealigner was used for the realignment of INDELs using the known sites on the 1000 Genomes Phase 1. The GATKv4 BaseRecalibrator and ApplyBQSR commands were used for the computational recalibration. The base quality score recalibration was done on the dbsnp 138. Variants were called from aligned sequence data using the GATK HaplotyperCaller to generate individual gVCF files. Supplementary Figure 1 illustrates the pipelines implemented for the variant calling. The variants were annotated with ANNOVAR, 26 SnpEff v4.3, 27 and ENSEMBL’s variant effect predictor v98.2 28 utility tools. The variants’ rs IDs were then updated on the dbSNP151 database using the Bcftools v1.9 (SAMtools package). 29 The variant pathogenicity predictions were based on the SIFT, PolyPhen2, mutation assessor, mutation taster, MetaLR, CADD, and LR algorithmic scores.
From the initial predicted variants, we extracted those candidate variants predicted to be pathogenic in the list of 34 genes (Table 1) with a directed python command. Then, we excluded all the positions with the minor allele frequency (MAF) >5% in all the populations cataloged on the gnomAD database, and as well as the MAF with the AF ≤ 0.1% on the ExAC database. BEAGLEv5.0 was used for the data phasing for the site-specific heterozygosity, nucleotide diversity, and genotype frequencies computed using a python package, pyVCF. Selected candidate variants were queried against available WES data from Cameroonian control individuals (n = 129) without personal and family history of HI.
Novel HI murine genes investigated.

N/A: not available; AR: autosomal recessive; AD: autosomal dominant.
Mode of inheritance was retrieved from OMIM database https://omim.org/ and GeneCards https://www.genecards.org/ based on clinical and functional information available on the condition/disorders associated with these genes.
Principal component analyses
The population structures of the cohorts were determined on the principal component analysis (PCA) chart using the smartpca tool.30,31 The WES data (participants and controls) were merged and computed with data extracted from the 1000 genomes Phase3 32 : 186 Yoruba (YRI) in Nigeria, 173 Esan (ESN) in Nigeria, 280 from Western Division in Gambia (GWD), 116 Luhya (LWK) in Webuye, Kenya, 112 from African ancestry in Southwest US (ASW), 128 Mende (MSL) in Sierra Leone and 123 from African Caribbean in Barbados (ACB), and an available control dataset from the individuals from Democratic Republic of Congo. PCA was plotted and annotated using Genesis2 tool.
Protein modeling
Protein modeling of MCPH1 p.Pro771Ala
The protein sequence of MCPH1 isoform 1 was retrieved from NCBI and queried on the Protein Data Bank database. The ‘A’ chains of both templates were retrieved, using the PyMol’s indicate chain command. The MCHP1 protein sequence was truncated to obtain the region covering the ‘A’ chains of the templates (including the mutated region). The rs369802722 (c.2311C>G, p.P771A) mutation was introduced into the appropriate position. A multiple template-based modeling strategy was used for the modeling of the 3D structures for the wild type (wt) and mutant (mt) using Modeller v9.23. 33 Top five models were selected based on their Discrete Optimized Protein Energy score evaluated on the Galaxy, 34 Rampage, and ProsA web services. The best model for both wt and mt structures was then retained based on its Galaxy energy and the proportion of residues in a Ramachandran plot, and ProsA Z-score.
RNA thermodynamic structure prediction of SPNS2, c867C>A p.(Pro289Gln)
The DNA sequence of exon 6 of SPNS2 was retrieved from Ensembl (www.ensembl.org). Positional substitution for SPNS2, c867C>A was done in the FASTA file. Using the Vienna RNA Package, the RNAfold server (http://rna.tbi.univie.ac.at/) was used to predict the minimum free energy structures and base pairing probabilities of the secondary structures of the single stranded RNA. The base pairing probability matrix in addition to the minimum free energy structure of the RNA for both the substituted and non-substituted SPNS2, c867C>A DNA sequences were generated and interactively studied.
Gene enrichment and network analyses
The gene enrichment analyses of the novel human-mouse ortholog genes were done using the g.Profiler tool (https://biit.cs.ut.ee). g.Profiler mapped the selected genes to their functional data sources and detected statistically significant enriched biological processes, pathways, regulatory motifs, and protein complexes. 35 Only the adjusted enrichment at threshold >16,000 for the biological terms, P values of 0.05, and Bonferroni correction threshold were considered. The gene network analyses were constructed, using STRING (v10), with the threshold at 0.4. The connected genes within the network were derived. All non-zero weighted edges were considered, and the fully connected components were visualized. Further analyses to understand the co-expression, physical interactions, shared protein domains, co-localization, and pathways shared by the selected genes MCPH1, OCM (i.e. OCM1 and OCM2) and LRIG1 and the well-established HI genes GJB2 and GJB6; as well as gene-to-gene network interaction was analyzed using GENEMANIA. 36
Results
Patients description
All the study participants had profound prelingual HI. The hearing acuities ranged from 71 to 120 dB. The percentage of male to female was 70% (28) to 30% (12). The median age at onset for HI was one year (range: 1–4 years). Twenty-eight individuals had isolated non-familial HI of likely genetic origin, and 12 had a familial history of HI inherited in an autosomal recessive non-syndromic pattern (Figure 1).

Study participants’ demographic and phenotypic information. (A color version of this figure is available in the online journal.)
Identifications of sequenced variants
Up to 95% of the WES data were paired reads, and 97% of the targeted regions had ≥10× coverage. Up to 95% WES were paired reads, and 97% of targeted regions had ≥10× coverage. Four murine genes d6wsu163e, zfp719, grp152, and minar2 have no human ortholog, therefore, 34/38 novel human-mouse ortholog genes were studied.
The average PHRED score for all the samples was 35 (Supplementary Figure 2). The percentage GC content of the coding regions was 45–50% (Supplementary Figure 3). The WES statistics showed that the quality was good (Supplementary Table 2).
In the Cameroonian cohort, a total of 4,490,688 single nucleotide variants (SNVs) were identified. Therefore, positions with read depth (DP) < 8, and genotype quality (GQ) < 20 were excluded resulting to a total of 152,289 SNVs (3.4% of the initial number) with a total genotyping rate of 99.8%. Of these, 143,489 (94.2%) were single nucleotide polymorphisms (SNPs), 3255 (2.1%) were insertions, and 5545 (3.6%) were deletions. The transition/transversion (Ti/Tv) ratio (2.46) of the variants fell within the expected range for good quality data. This ratio was even better for known variants only (3.50) which made up 1.5% (2279) of the total filtered variants based on dbsnp138. Most of the variants observed were silent (98,220—51.3%), while missense variants made up 48.2% (92,151) and nonsense mutations constituted 0.52% (998).
In South African cohort, a total of 4,297,880 variants were called, 620 (1 multi-allelic) of which passed the following filters: minimum depth (DP) = 5, minimum GQ = 10. Of the 620 SNVs, 478 (77.2%) were SNPs. Then, 61 (9.85%) were insertions, while 81 (13.08%) were deletions. Missense mutations made up 44.83% of the variants while the remaining 55.17% were silent mutations. As expected, most of the variants (48.56%) were intronic, with the next most frequent category being intergenic variants (19.42%) which usually confer no significant effect. Also, as expected, exonic (1.68%), 5ʹ UTR (1.08%), and intragenic (0.78%) variants made up the least frequent categories.
Most of the annotated variants had the MAFs that ranged from 0.0 to 0.09 (Figure 2). The average number of annotated SNPs identified per patient was 7971 and 6522 in the Cameroonian and South African cohorts, respectively (Figure 2).
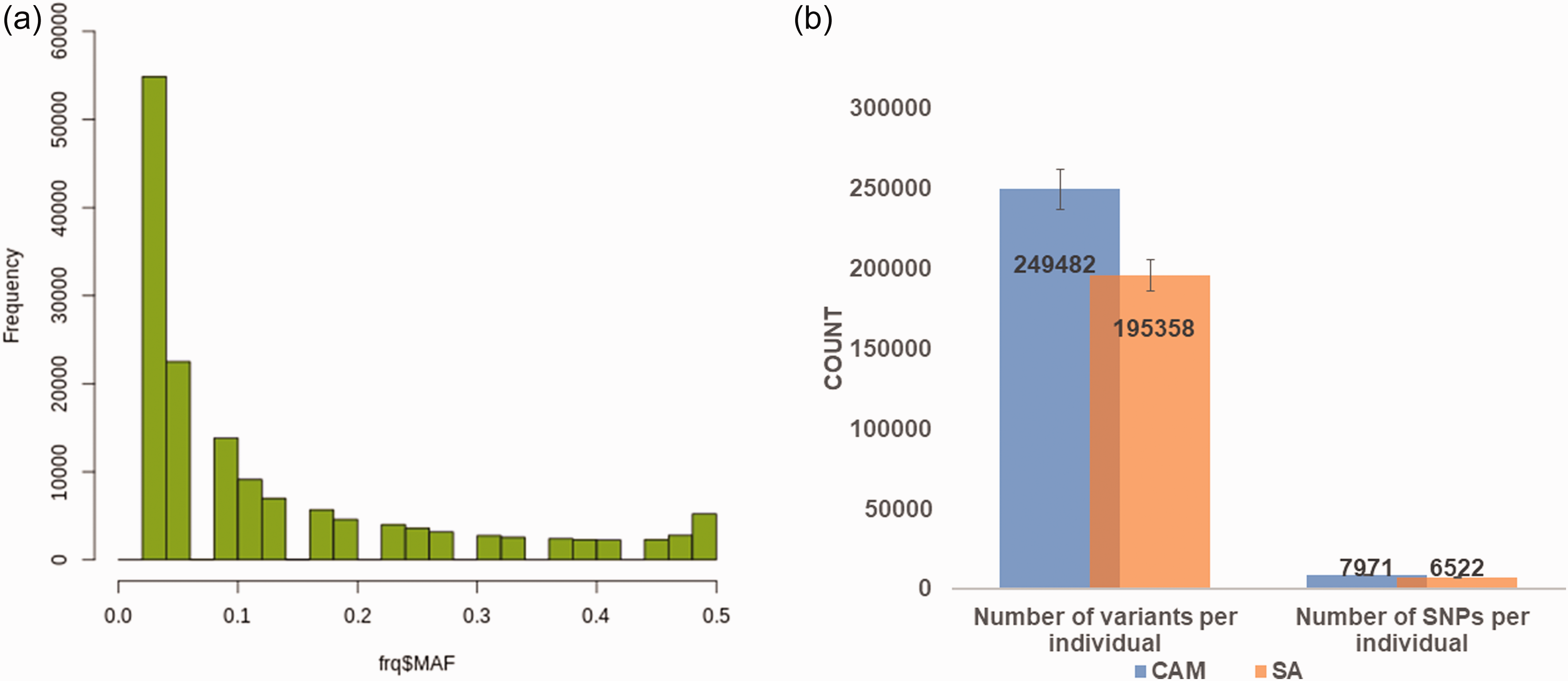
(a) Average number of variants and commonly reported SNPs identified per HI individuals (estimate of 40 samples) in the WES study. (b) The MAF distributions of all variants annotated on the gnomad database. (A color version of this figure is available in the online journal.)
Population structure
The PCA analyses of common SNPs showed closer clusters between the HI samples and controls (PCA1), suggesting cases and controls population homogeneity. These clusters however differ with a significant distance from the other African populations (Figure 3). This result revealed unique genetic diversities in the African populations.
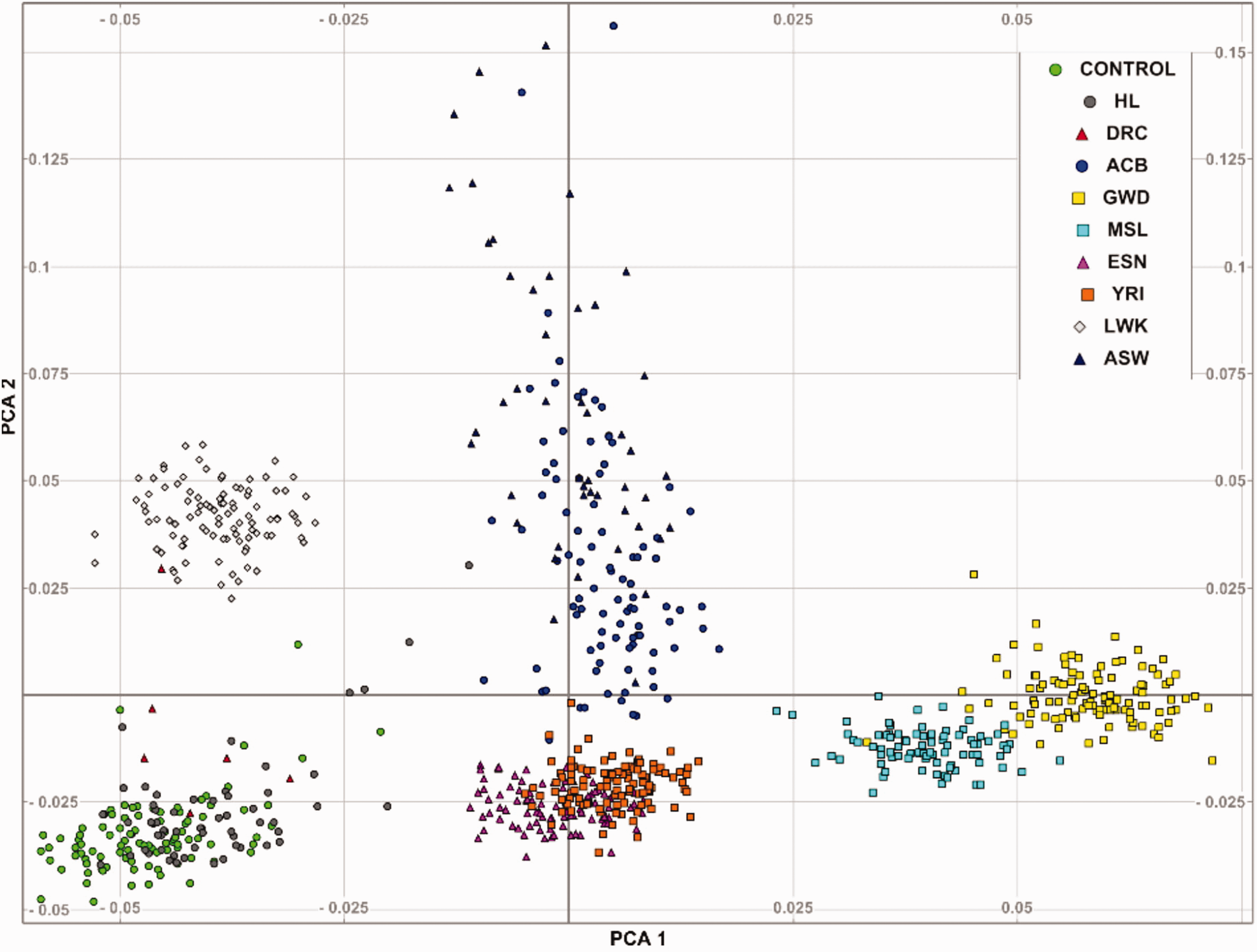
The PCA plot illustrating clusters distances of 0.075 to 0.15 that could be interpreted as disparity in allelic frequencies in the study cohorts (cases and controls) versus selected African populations in the 1000Genomes Phase 3 project.
Pathogenic and likely pathogenic variants in WES
Three rare PLP variants were found in the WES studied in three human-mouse ortholog genes, among three unrelated Cameroonian patients (Table 2). Two PLP variants are in heterozygous state, i.e. OCM2, c.227G>C(p.(Arg76Thr), RNA not analyzed), LRGI1, c.1657G>A (p.(Gly533Arg), RNA not analyzed). In one patient, the variant MCPH1, c.2311C>G p.(Pro771Ala); RNA not analyzed) is in homozygous state. No PLP variant was found among the South African patients.
Candidate risk missense variants selected in this study.
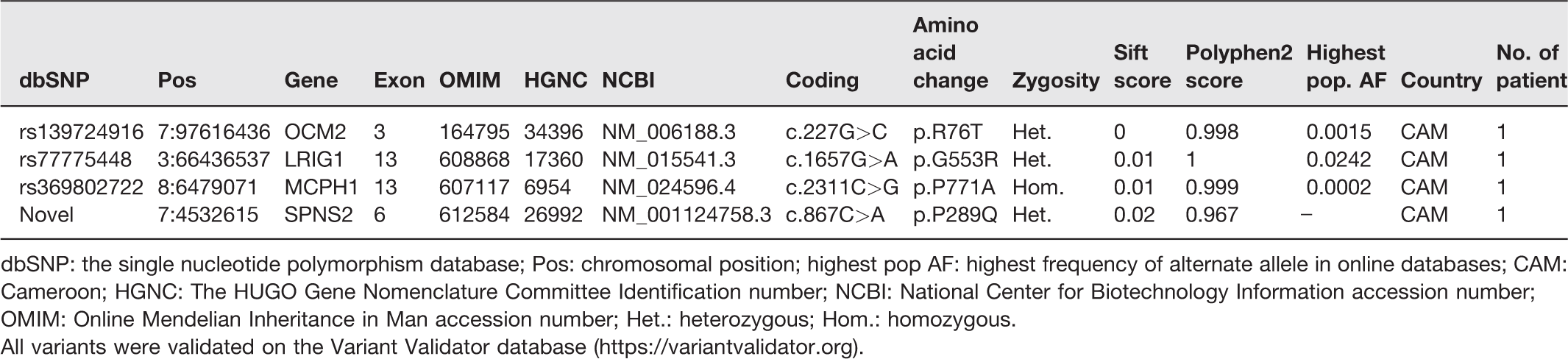
dbSNP: the single nucleotide polymorphism database; Pos: chromosomal position; highest pop AF: highest frequency of alternate allele in online databases; CAM: Cameroon; HGNC: The HUGO Gene Nomenclature Committee Identification number; NCBI: National Center for Biotechnology Information accession number; OMIM: Online Mendelian Inheritance in Man accession number; Het.: heterozygous; Hom.: homozygous.
All variants were validated on the Variant Validator database (https://variantvalidator.org).
The patient identified with biallelic PLP variant MCPH1, c.2311C>G p.(Pro771Ala), was an eight-year-old girl, presenting with congenital prelingual bilateral and symmetrical sensorineural profound hearing loss (90–100 dB), from an non-consanguineous parents, with a family history compatible with ARNSHI (Supplementary Figure 4A). She did not specifically have microcephaly, described in the mouse model with mutation in mcph1, 19 or any learning difficulty. She attended a school for the deaf, with adequate academic progress. The two other Cameroonian patients who were monoallelic with variants in LRGI1, c.1657G>A p.(Gly533Arg) and OCM2, c.227G>C p.(Arg76Thr), respectively, also presented with congenital non-syndromic sensorineural HI with family histories compatible with autosomal recessive inheritance (Supplementary Figure 4B and C). We were unable to investigate the variants in the three genes among other family members because of the non-availability of the DNA samples.
Genotyping targeted variants in SPNS2
The previously reported SPNS2 deletion mutations (c.1066_1067delCCinsT) and SPNS2 (c.955_957delTCC) were not identified in the WES data, neither were they found in the 25 NSHI patients, nor in the 26 matched controls that were Sanger sequenced. One novel variant, SPNS2, c867C>A p.(Pro289Gln), was found in heterozygous state, in one Cameroonian patient.
MCPH1 (p.Pro771Ala) protein modeling
The protein modeling of the MCPH1 (p.Pro771Ala) showed that this variant occurs on the loop in the BRCT3 domain. Considering that MCPH1 chromatin (histone A2)-binding site lies in the BRCT2 domain (residues 137–142), it is unlikely that the P771A mutation, which results in no visible change in the protein structure, would influence the overall protein function (Figure 4).
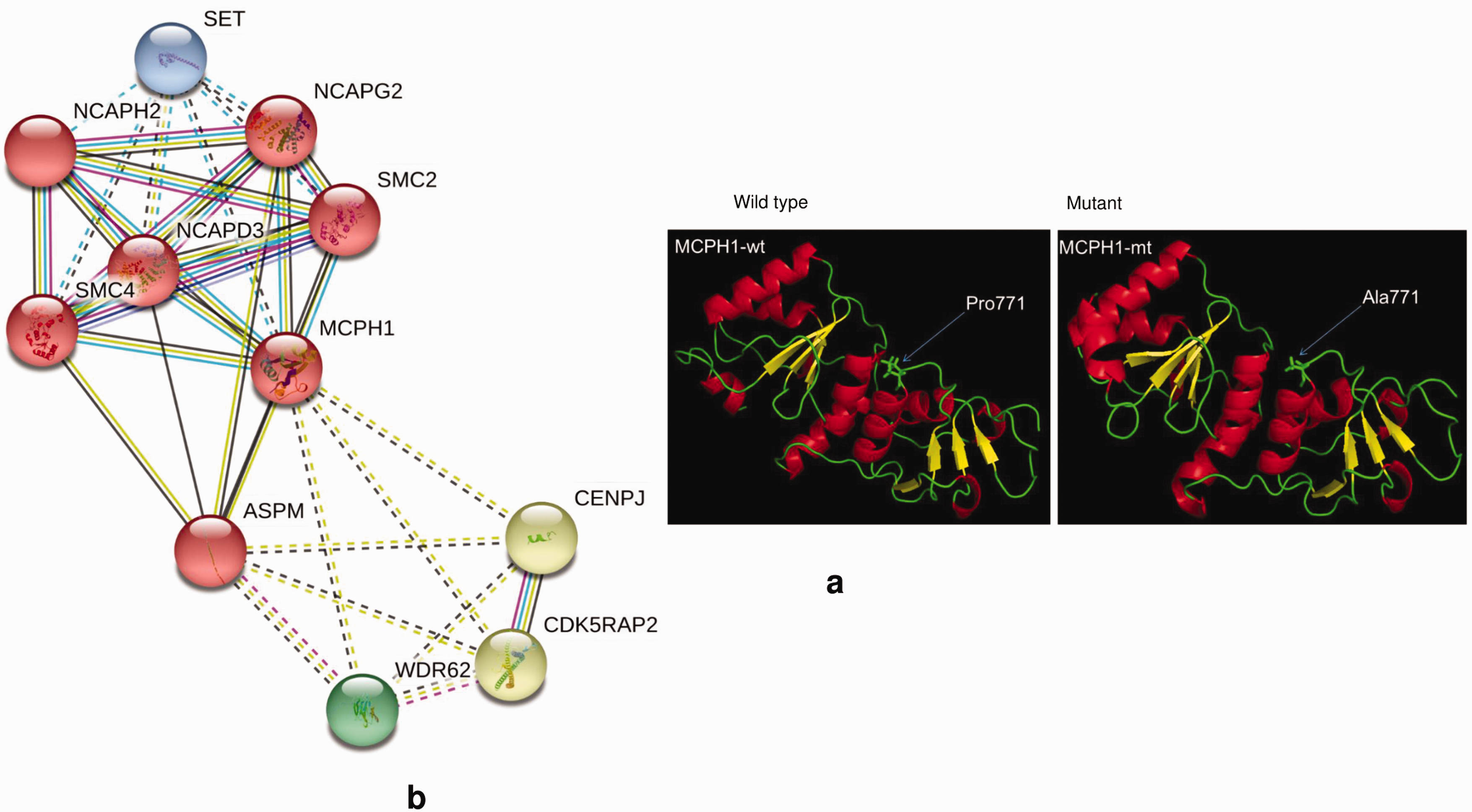
(a) Three-dimensional structures of both wild type (wt) and mutant (mt) of MCPH1 P771A variant. The variant occurs on a loop in the BRCT3 domain. Considering that MCPH1 chromatin (histone A2)-binding site lies in the BRCT2 domain (residues 137–142), it is unlikely that the P771A mutation, which results in no visible change in the protein structure, would influence the overall protein function. (b) MCPH1 STRING network.
SPNS2, c867C>A p.(Pro289Gln) RNA thermodynamic structure
The SPNS2, c867C>A p.(Pro289Gln) RNA thermodynamic structure prediction showed that the variant is located at the D loop arm of tRNA and is likely to disrupt the intramolecular base-pairing which may in turn impair the tRNA metabolism as shown in Supplementary Figure 5.
Network interactions
The STRING network analyses showed functional interactions between the MCPH1 gene and 10 non-HI genes (Figure 4). Of these, NCAPG2, SMC2, NCAPHZ, NCAPD3, and SMC4 genes were derived in the weighted interactions, while SET, ASPM, CENPJ, CDK5RAP2, and WDR62 genes were identified in the non-zero-weighted edges as shown in (Figure 4). The in silico functional interactions suggest a common biological pathway that represents systemic activities for hearing. Little is known about the functions of these associated non-HI genes in hearing. However, mutations in NCAPG2 37 and NCAPHZ 38 were previously associated with a spectrum of neurodevelopment syndromes and pregnancy-associated abnormalities.
Furthermore, the GENEMANIA network analyses of these 34 human-mouse ortholog genes showed a biochemical association that functionally co-expressed and interacted with GJB2 and GJB6 (Figure 5). The gene enrichments results showed four significant systemic activities related to molecular function, nucleic acid binding, protein binding, and catalytic activities as shown in Figure 6. These four systemic activities representing the genes’ functions pose active biological roles in the membrane-bonded organelles and intracellular/cellular compartments. Furthermore, CORUM significantly (P value 4.895 × 10−2) identified six functional complexes (CXCR2-NHERF1-PLC beta-3 complex, TS2-HERC1, RAF1-MAP2K1-YWHAE, PAX9-MSX1, HSF1-YWHAE and MLL1-WDR5) which can be implicated in the various biological responses such as cell biogenesis and transcription factors (Figure 6).
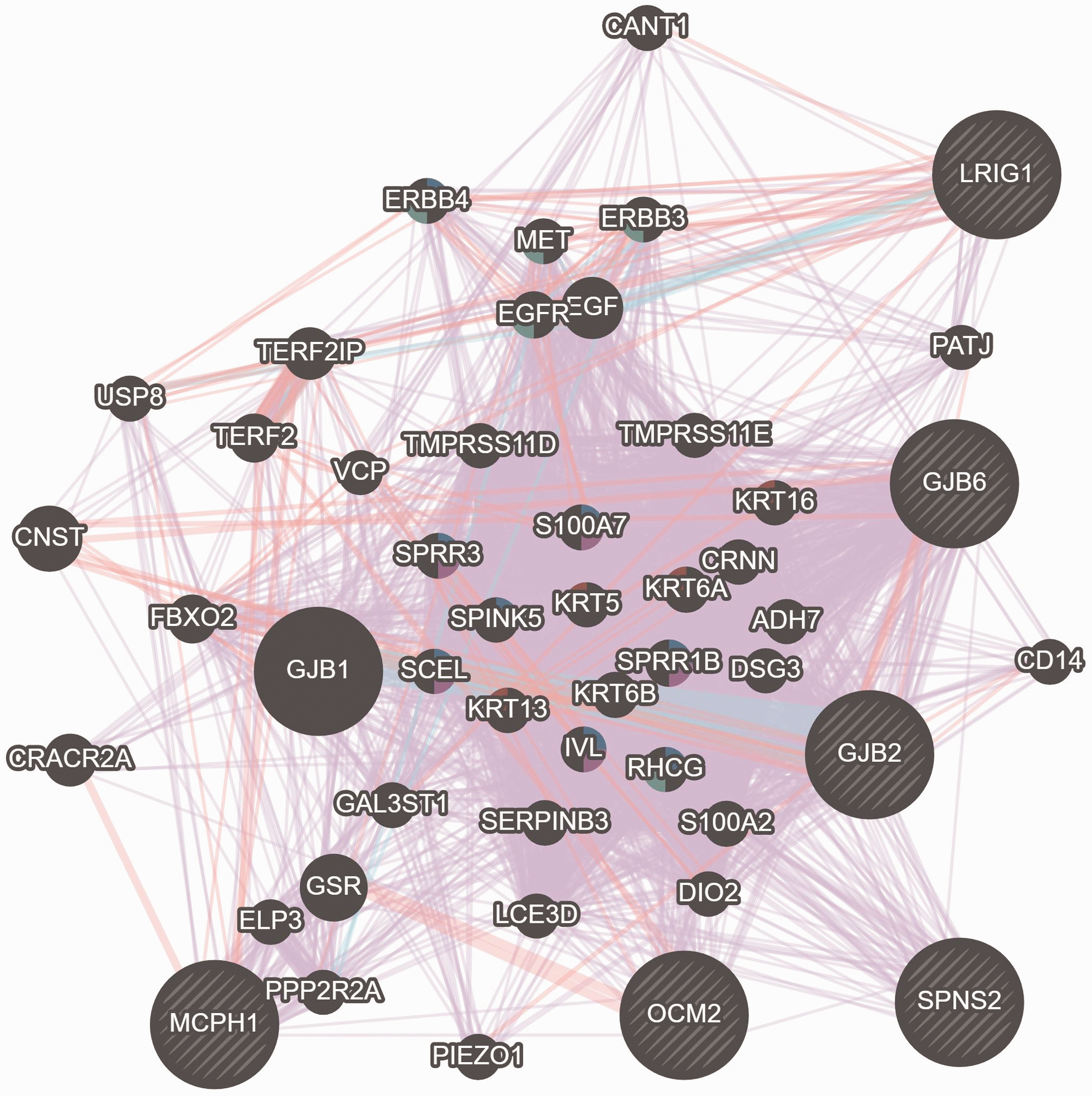
Illustration of functional co-expression interaction between the selected murine genes with the GJB2 and GJB6 genes. (A color version of this figure is available in the online journal.)

Multiquery Manhattan plot. X-axis shows the functional terms grouped and color-coded by data source. Functional enrichment terms for the 34 novel human genes. (1) Blue arrow shows four significant GO:MF terms namely (molecular function, nucleic acid binding, protein binding and catalytic activities), (2) black arrow shows strong transcriptional regulation and regulatory motif matches from TRANSFAC (TF), and (3) red arrow shows six significantly (P value 4.895 × 10−2) identified functional complexes.
Discussion
This is the first investigation of human-mouse ortholog genes in the African populations that found in a modest WES samples (n=40) a PLP variant, MCPH1, c.2311C>G p.(Pro771Ala), in a homozygous state (1/34) that could explain HI in a patient, and three additional PLP variants in 3 genes in heterozygous states. With a larger sample size, such an approach could lead to novel discovery of more novel HI PLP variants in these understudied populations, and possibly further our understanding of the pathophysiology of HI.
Indeed, the murine hearing system is very similar to that of humans 18 and had revealed important findings on the genetics of HI. Similar approaches previously identified two PLP variants in the human-mouse ortholog gene SPNS2, 19 that were not replicated in the present study. However, we found a novel heterozygous PLP variant, SPNS2 c867C>A p.(Pro289Gln), in a single individual, which is predicted to disrupt protein functions (Supplementary Figure 5). The effects of mutations in this gene were previously associated with the transporters on the P13K/Akt pathway in lamellipodia formation in lung’s endothelium and reactive oxygen species generation. 39 However, there is no strong evidence to support the causative effects of this gene in our patient because the variant is in heterozygous state. The variant identified in the MCPH1 gene in a patient in this study was in homozygous state. A few studies have been performed to understand the roles of the MCPH1 gene: MCPH1 has been categorized as a prominent DNA repair gene that works to prevent premature mitotic entry. 40 Its roles in HI in humans have not been previously reported. But, MCPH1 is very close to the DFNM2 locus on Chromosome 8, with both DFNM1 and DFNM2 loci being hotspots for various NSHI genes.41,42 Although the protein modeling of the variant identified MCPH1, c.2311C>G p.(Pro771Ala) did not show visible change in the protein structure (Figure 4(a)), the network interactions between the MCPH1 gene and other genes as seen in the present study (Figure 4(b)) clearly showed MCPH1 relevance in some biological activities. Furthermore, OCM (OCM1 and OCM2) gene has not been the focus in human HI. It is noteworthy to state that OCM gene alias ocm1 gene that was identified in the murine study 19 is an important paralog of OCM2 with a similar function. Some studies have described the causative effects of OCM2 gene, with a pathogenic mutation identified (c.227G>C(p.(Arg76Thr), RNA not analyzed) and associated with intellectual disability in a family-based study. 43 Also, in a genome-wide association study that found a significant variant in the OCM2 gene associated with cardiac arrest in patients with coronary artery disease. 44 Nonetheless, there is no strong evidence to support the causative effects of this gene in our patient because the variant is in heterozygous state. In this study, we identified a PLP variant in the LRIG1 gene. Unlike the previously discussed genes, genomic PLP variants in LRIG1 were recently found in an adult onset of HI in humans. 45 Interestingly, the Cameroonian patient in which we found the monoallelic LRIG1, c.1657G>A p.(Gly533Arg) was the only adult patient of the cohort (Supplementary Figure 4B), although, the significance of the identified PLP variant in this gene in terms of the age at HI onset was not investigated. The LRIG1 protein is involved in the sequential developmental events that include axon guidance and synapse formation. 46 In addition, gain and loss of function assays in mice indicate that the LRIG1 protein restricts dendrite morphology. 47
Taken together, the roles of mutations in these genes in HI will need further exploration in larger and multiple HI human populations and controls from multiple settings, to refine the disease-gene pair curation. Indeed, a high number of PLP variants in known HI-associated genes were found in the same hearing-impaired individuals, and the larger 1000 Genomes Project sample set, with unknown hearing phenotype or auditory function. 45 The population structure analysis of the WES dataset investigated in this study further highlights high genetic structure differences between the cohorts and across different African populations, a diversity associated with nearly 300,000 years of evolutionary changes. 21 This is an indication that future study on HI among Africans should recruit control individuals from the same regional and ethnolinguistic group as the patients. Indeed, in the African populations many variants may not be fully annotated because of the limited African genome data in the public genome databases. 48
The major limitation to this study is the limited sample size of individuals with HI. Therefore, our findings cannot be generalized in the Cameroonians and South African populations. Future studies should include multiplex families with available biological samples and data that will afford the possibility of performing segregation analysis. Moreover, in vitro functional analysis could have complemented our finding because gene variants may affect protein expression or mRNA expression without changing the protein structure. The present study also revealed that not all murine genes have human ortholog (4/38), suggesting that numerous mutations in mice with HI will not necessarily apply to humans.
In conclusion, among the identified rare candidate PLP variants in 3/34 human-mouse ortholog genes, only one, the homozygous variant MCPH1 p.Pro771Ala, could likely explain HI in a patient. In silico functional analysis revealed co-expression and interactions with well-established GJB2 and GJB6 HI genes suggesting that the human-mouse ortholog genes investigated could coordinate important biological functions associated with the hearing system in humans. The identification of rare potentially pathogenic coding risk variants in these of patients of African ancestry could contribute to furthering our understanding of genotype-phenotype variations in NSHI.
Supplemental Material
sj-pdf-1-ebm-10.1177_1535370220960388 - Supplemental material for Whole exome sequencing identifies rare coding variants in novel human-mouse ortholog genes in African individuals diagnosed with non-syndromic hearing impairment
Supplemental material, sj-pdf-1-ebm-10.1177_1535370220960388 for Whole exome sequencing identifies rare coding variants in novel human-mouse ortholog genes in African individuals diagnosed with non-syndromic hearing impairment by Oluwafemi G Oluwole, Kevin K Esoh, Edmond Wonkam-Tingang, Noluthando Manyisa, Jean Jacques Noubiap, Emile R Chimusa and Ambroise Wonkam in Experimental Biology and Medicine
Footnotes
AUTHORS’ CONTRIBUTIONS
OGO conceptualized the idea, performed parts of the experiments, analyses, prepared results, and drafted the manuscript. KKE and EC did parts of the analyses and results. JJN, ETW, and NM diagnosed, recruited patients, and revised the manuscript. AW is the project lead, supervised the recruitment, experiments, analyses, and revised the manuscript.
Declaration OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ETHICAL APPROVAL
The study was approved in Cameroon by the Institutional Research Ethics Committee for Human Health of the Gynaeco-Obstetric and Paediatric Hospital of Yaoundé, Cameroon (Ethics approval number No723/CIERSH/DM/2018) and in South Africa by the Human Research Ethics Committee (Ethics approval number HREC Ref: 104/2018
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by the National Institutes of Health, Bethesda, MD, USA. Grant number [U01-HG-009716] awarded to AW, and the African Academy of Science/Wellcome Trust, grant, number [H3A/18/001] awarded to AW. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
SupplementAL MATERIAL
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.