
Abstract
The transcription factor oligodendrocyte transcription factor 2 (Olig2) plays a central role in specifying motor neurons and oligodendrocytes during vertebrate neural development. While transgenic reporter lines such as TgBAC(olig2:EGFP) have been instrumental in visualizing olig2 expression, they fall short in directly reporting endogenous protein levels and may not fully recapitulate native gene regulation. To address these limitations, we generated a TgKI(olig2-mNeonGreen) zebrafish line using CRISPR/Cas9-mediated knock-in at the endogenous olig2 locus. The resulting Olig2-mNeonGreen fusion protein localizes specifically to the nucleus, enabling direct live imaging and accurate quantification of Olig2-expressing cells. We confirmed that the knock-in preserves endogenous mRNA expression and protein function, and that homozygous fish develop normally. As proof of concept, modulation of Sonic Hedgehog signaling altered Olig2-mNeonGreen+ cell numbers as expected, confirming the reporter’s responsiveness to known upstream inputs. This TgKI(olig2-mNeonGreen) line offers a robust tool for studying neural progenitor dynamics in vivo.
Introduction
The oligodendrocyte transcription factor 2 (olig2) gene is a critical regulator of vertebrate neural development, especially in the specification of motor neuron progenitor (pMN) domains and the generation of both motor neurons and oligodendrocytes in the central nervous system.1,2 Numerous studies have leveraged olig2 as a marker gene for pMN cells due to its distinct spatial and temporal expression patterns during development.1,3–9 In zebrafish, several transgenic reporters, including TgBAC(olig2:EGFP), 10 TgBAC(olig2:dsRed), 11 and TgBAC(olig2:kaede) 12 have provided valuable insights into the transcriptional regulation of olig2 and its role in cell fate specification. However, these bacterial artificial chromosome (BAC)-based lines share inherent limitations. First, their cytoplasmic fluorescent signals do not clearly demarcate cell boundaries as fluorescent cells are tightly packed, complicating cell quantification and lineage tracing unless additional markers are used. Second, BAC constructs may omit distal regulatory elements or exhibit integration-dependent expression variability, which may not fully reflect endogenous regulation. Third, the fluorescent proteins expressed from these transgenes may be more stable than the endogenous Olig2 protein. Since Olig2 expression is known to be dynamic, as demonstrated in mice, 13 these reporters—while being useful for lineage tracing of cells that have previously expressed olig2—can result in persistent fluorescence even after endogenous expression ceases, leading to potential false positives. 14
To overcome these challenges, we generated a zebrafish knock-in reporter line in which mNeonGreen 15 was inserted in-frame at the C-terminus of the endogenous olig2 locus. This Olig2-mNeonGreen fusion enables live, nuclear-localized imaging of the endogenous protein, facilitating direct visualization and quantification of Olig2 dynamics in vivo. We confirmed that the knock-in does not alter olig2 mRNA expression or protein function and that homozygous fish remain viable and fertile. This knock-in line thus serves as a powerful and accurate tool for studying Olig2 expression and neural progenitor behavior during zebrafish development.
Materials and Method
Zebrafish husbandry
All zebrafish were raised, maintained, and used for experiments in accordance with guidelines approved by the Washington University and Harvard Medical School Institutional Animal Care and Use Committees. Embryos were obtained from natural matings, incubated at 28.5°C, and staged according to standard morphological criteria. 16
Knock-in design and establishment
To generate the TgKI(olig2-mNeonGreen) zebrafish line, we constructed a donor plasmid containing a 1,000 bp left homology arm and a 976 bp right homology arm flanking the olig2 genomic insertion site, along with a flexible GA linker (amino acid sequence: GAPAGGAGAA) followed by the zebrafish codon-optimized mNeonGreen coding sequence (Fig. 1A). These fragments, combined with a backbone harboring two I-SceI restriction sites to enhance genome editing efficiency, 17 were assembled via Gibson assembly.
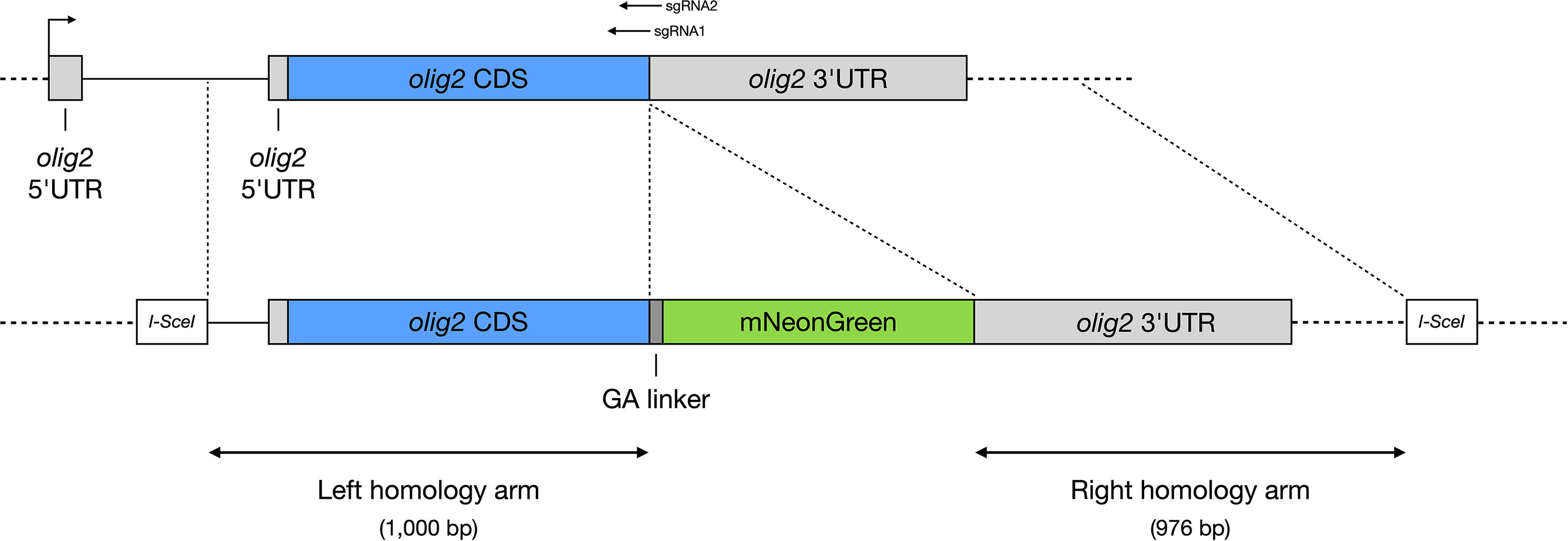
Knock-in design. Schematic representation of the knock-in strategy for generating the TgKI(olig2-mNeonGreen) reporter line. Homology-directed repair was facilitated using CRISPR/Cas9 to insert a flexible GA linker and mNeonGreen coding sequence immediately downstream of the olig2 coding sequence and upstream of its endogenous stop codon. olig2, oligodendrocyte transcription factor 2.
Guide RNAs (sgRNAs) targeting the olig2 locus near the endogenous stop codon were designed using CHOPCHOP.
18
sgRNA synthesis followed protocols outlined in Gagnon et al.
19
The sgRNA target sequences are:
sgRNA 1: GGTCAGCCGTGGCATGCTGA sgRNA 2: GTCACTTTATTTTGAGTCAC
The knock-in donor plasmid, synthesized sgRNAs, Cas9 mRNA (Trilink BioTechnologies), and I-SceI enzyme and buffer (NEB) were co-injected into zebrafish embryos at the one-cell stage. Injected embryos were screened for mNeonGreen fluorescence, raised to adulthood as founder fish, and subsequently outcrossed to establish stable knock-in lines.
Insertion validation
Genomic insertion was validated by Polymerase Chain Reaction (PCR) amplification and sequencing using the following primers:
5′ forward: CTCATTGGTTCTTGACGGGC (1492 bp upstream, external validation) 5′ reverse: ATTTGGGTTTCCAGTTCCTTGC (internal mNeonGreen sequence) 3′ forward: AGCAAGGGCGAGGAGGACAA (5′ end of mNeonGreen) 3′ reverse: TCATTTTAAAACATTTATTTAACAGTTAGGACTCA (end of 3′ untranslated region [UTR])
PCR products were purified and sequenced using Oxford Nanopore Technologies (Plasmidsaurus), confirming accurate in-frame integration and intact 3' UTR.
Genotyping homozygosity
Homozygosity of the knock-in allele was determined using primers flanking the wild-type olig2 stop codon (forward: GTCTCACGTTCCCGGTATGA, reverse: GGGCTAAGGAAGGTTTGCCAT). Wild-type embryos displayed a single 286 bp band, and homozygous knock-in embryos showed a single 1024 bp band.
Hybridization chain reaction RNA fluorescence in situ hybridization (HCR RNA-FISH)
HCR RNA-FISH staining was performed following protocols from Molecular Instruments. Briefly, embryos were fixed in 4% paraformaldehyde overnight at 4°C, washed, and pre-hybridized at 37°C. Hybridization occurred overnight at 37°C with 1 nM probes. Embryos were subsequently washed and incubated in hairpin amplification mixtures overnight at room temperature. Embryos were washed again before imaging.
Imaging and microscopy
Embryos were mounted in egg water on agarose molds as previously described. 20 Confocal imaging was performed using an upright Zeiss LSM980 microscope. Time-lapse imaging was conducted at room temperature (23°C), beginning at the bud stage (∼10 h post-fertilization, hpf) and continuing for 10 h, with 10-min intervals. Images were acquired with isotropic resolution (1 μm × 1 μm × 1 μm). Based on somite staging, we empirically determined that embryos developed at approximately 50% the rate of those incubated at 28.5°C, reaching the 12-somite stage after 10 h of imaging at room temperature. Nuclei were manually traced, and quantitative data were exported using the surface creation and tracing functions in Imaris (Oxford Instruments).
Dorsal–ventral expression analysis
Spinal cord and notochord regions were segmented using ilastik, 21 trained with a subset of embryos expressing Tg(sox19a:H2B-mCherry) 22 and Tg(shha:membrane-mCherry). 23 Pixel distances from the spinal cord to the notochord were calculated, and fluorescence intensities were measured at these distances. Standardized (min–max scaled to 0–1) mean intensity curves were generated per embryo, and final plots represent the mean intensity across embryos with 95% confidence intervals.
Cyclopamine treatment
Embryos were dechorionated at the dome stage and treated with 50 μM cyclopamine in 1× Danieau buffer [17.4 mM sodium chloride NaCl, 0.21 mM potassium chloride KCl, 0.12 mM magnesium sulfate MgSO4, 0.18 mM calcium nitrate Ca(NO3)2] until imaging. Control embryos were treated with 1× Danieau buffer containing 0.2% ethanol (vehicle control).
mRNA synthesis and injection
Capped mRNA was synthesized from linearized plasmid templates using the mMessage mMachine kit (Invitrogen). Zebrafish embryos at the one-cell stage were injected with approximately 1 nL of 50 ng/μL shha mRNA 24 using pulled glass needles.
Cell counting
Cell counts were performed using Imaris software’s spot creation function with default parameters (green channel, estimated diameter 5 μm, automatic background subtraction, and quality thresholding). Statistical analyses were conducted using Prism 10 (GraphPad).
Result
To precisely monitor endogenous Olig2 protein dynamics, we generated a knock-in zebrafish line TgKI(olig2-mNeonGreen) using CRISPR/Cas9-mediated genome editing. This approach inserted the fluorescent protein mNeonGreen at the C-terminus of the endogenous olig2 locus (Fig. 1). Two single-guide RNAs (sgRNAs) targeting sequences near the olig2 stop codon guided homology-directed repair using a donor DNA template with approximately 1 kb homology arms and a flexible linker fused to mNeonGreen. 17 Stable lines were derived by identifying fluorescent-positive embryos, raising them to adulthood, and outcrossing founder fish. The genomic insertion was confirmed by sequencing upstream of the left homology arm and through the entire olig2 3' UTR, verifying correct in-frame fusion directly downstream of olig2 coding sequences, immediately before the native stop codon.
The resulting TgKI(olig2-mNeonGreen) embryos displayed robust mNeonGreen fluorescence specifically localized to known olig2 expression domains in the brain and spinal cord throughout development (Fig. 2).1,3,4 Confocal microscopy at 1 day postfertilization (dpf) confirmed clear nuclear localization of the mNeonGreen fluorescence, consistent with Olig2’s role as a nuclear transcription factor. Signal enrichment was observed specifically in ventral spinal cord cells within the pMN domain, excluding medial and lateral floor plate regions (Fig. 3A).1,3,4,6,10,25,26 In the brain, expression matched previously documented olig2 patterns, highlighting regions in the diencephalon and rhombomeres 5 and 6 (r5, r6) (Fig. 3B).1,12,27,28
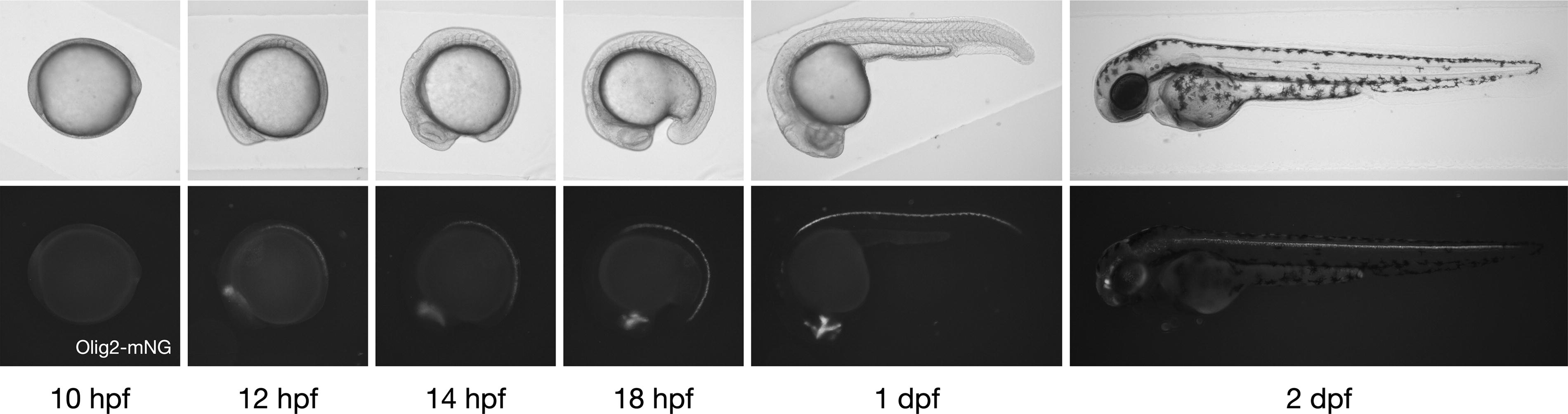
mNeonGreen signal in TgKI(olig2-mNeonGreen) embryos throughout development. Time-course imaging of brightfield (upper panel) and Olig2-mNeonGreen fluorescence (lower panel) in homozygous TgKI(olig2-mNeonGreen) embryos from 10 hpf to 2 dpf. Embryos are positioned in left lateral view, with anterior to the left and dorsal at the top. dpf, day postfertilization; hpf, h postfertilization.
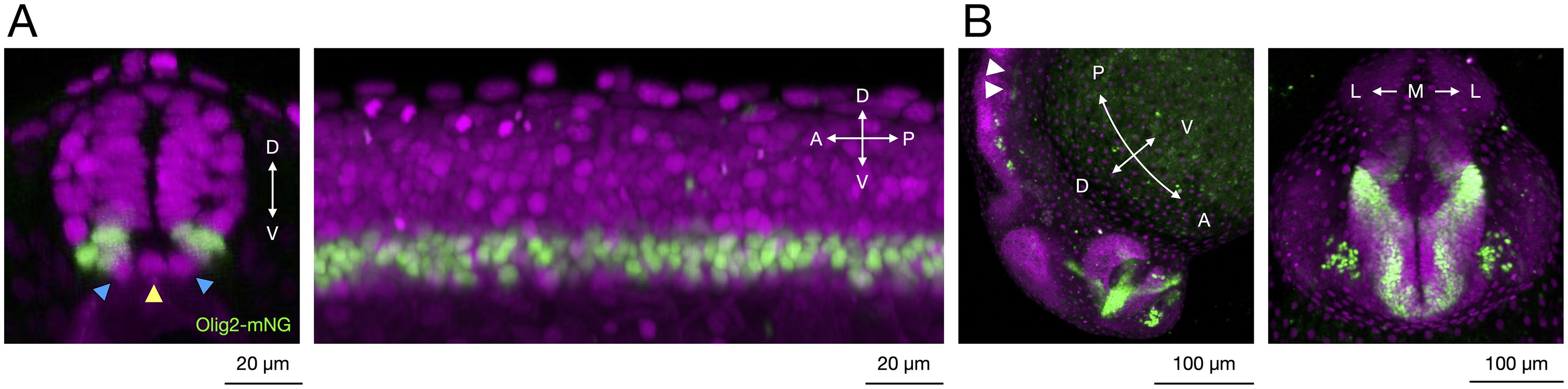
Confocal microscopy of TgKI(olig2-mNeonGreen) at 1 dpf.
To demonstrate the utility of the TgKI(olig2-mNeonGreen) line for live imaging, we performed time-lapse imaging during neural plate convergence (10–15 hpf), capturing dynamic changes in Olig2 localization during neural progenitor cell division (Fig. 4A). The Olig2-mNeonGreen signal transiently dispersed from the nucleus into the cytoplasm during mitosis and subsequently re-accumulated in the daughter nuclei following karyokinesis. Quantification of nine cell division events based on nuclear fluorescence intensity revealed a decrease beginning ∼10 min before karyokinesis, followed by recovery to pre-division levels within 20 min after division (Fig. 4B).
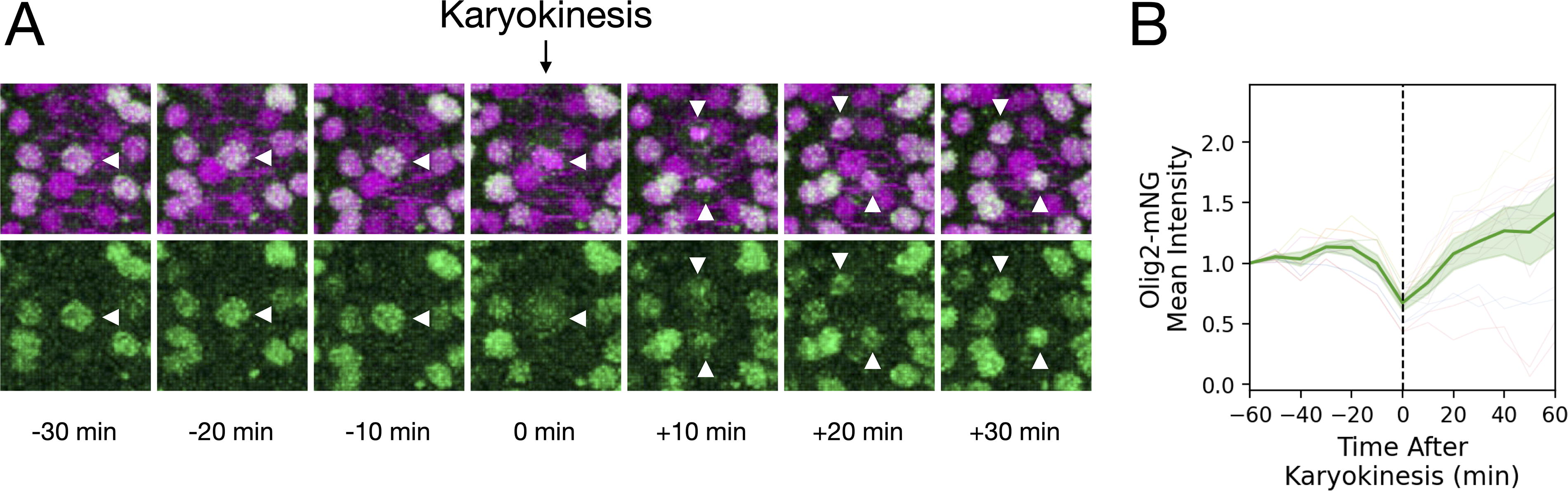
Visualization of Olig2-mNeonGreen dynamics during neural progenitor cell division.
We validated that the insertion did not disrupt endogenous olig2 transcription by performing HCR RNA-FISH at 14 hpf. Wild-type and homozygous knock-in embryos exhibited similar dorsal-ventral olig2 mRNA distribution patterns, and mNeonGreen mRNA in knock-in embryos precisely mirrored wild-type olig2 mRNA expression, confirming proper transcriptional regulation at the modified locus (Fig. 5).

Validation of olig2 and mNeonGreen mRNA expression profiles in the spinal cord.
To assess functional integrity, homozygous knock-in embryos were raised to adulthood and evaluated for viability, fertility, and gross morphology. Homozygous TgKI(olig2-mNeonGreen) adults were inbred for two generations without any observable abnormalities. Additionally, HCR RNA-FISH staining for isl2a (at 1 dpf) and sox10 (at 2 dpf)—markers known to be disrupted upon olig2 loss 1 — revealed normal motor neuron and oligodendrocyte formation, indicating the preserved functionality of the Olig2-mNeonGreen fusion protein (Fig. 6). To confirm homozygosity, we used a primer pair flanking the wild-type olig2 stop codon, which yields a 286 bp amplicon in unmodified alleles. In both the brightest 25% of embryos from heterozygous incrosses and in adult fish producing exclusively Olig2-mNeonGreen-positive offspring, only the expected 1024 bp fusion amplicon was detected, indicating true homozygosity and ruling out additional untagged olig2 copies.
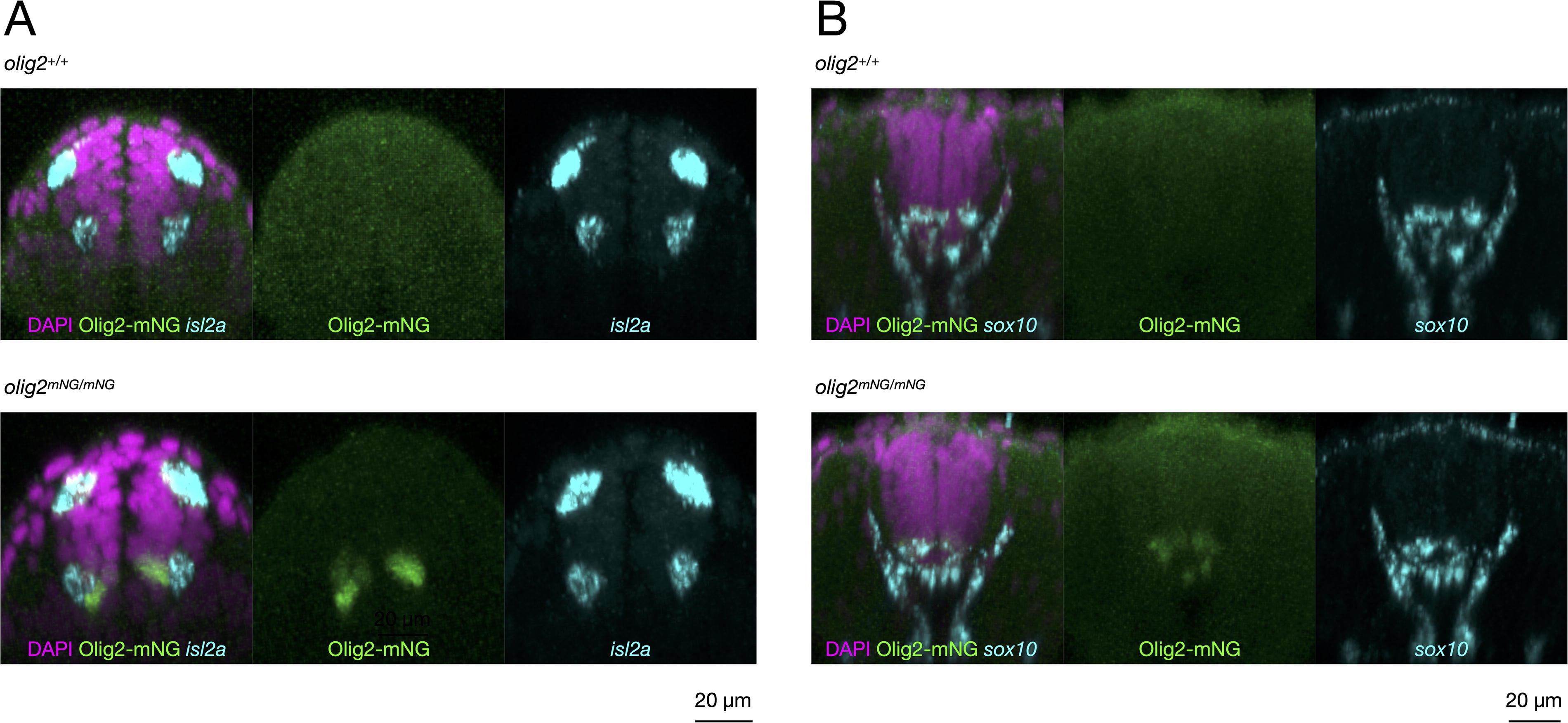
Comparable motor neuron and oligodendrocyte specification and differentiation in homozygous knock-in embryos.
The nuclear-localized mNeonGreen fluorescence facilitated automated quantification of Olig2-positive cells. Given the established direct regulation of olig2 by Sonic Hedgehog (Shh) signaling,3,6,24 we tested the responsiveness of the reporter line to Shh pathway perturbations. Treatment with the Shh inhibitor cyclopamine from the dome stage significantly reduced Olig2-positive cell numbers, whereas activation via shha mRNA injection expanded the Olig2-positive domain, demonstrating the line’s robust utility for quantitative analyses of neural progenitor dynamics in response to signaling perturbations (Fig. 7).
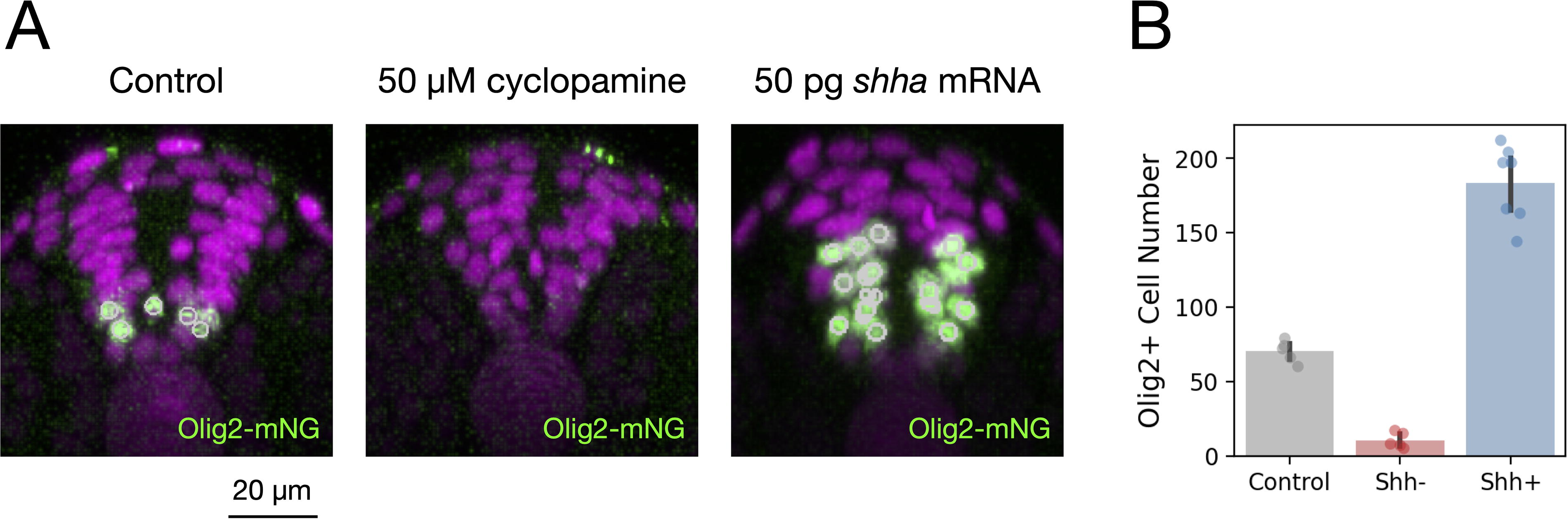
TgKI(olig2-mNeonGreen) as a quantitative tool to study pMN specification.
Discussion
The transcription factor Olig2 plays a pivotal role in vertebrate neural development, particularly in specifying pMN domains and promoting differentiation into motor neurons and oligodendrocytes.1,2 Our TgKI(olig2-mNeonGreen) zebrafish line provides a faithful readout of endogenous Olig2 protein dynamics, with the nuclear-localized Olig2-mNeonGreen fusion enabling precise visualization and quantification of Olig2-expressing cells in live embryos without additional nuclear markers.
Olig2 expression is directly regulated by Shh signaling during neural development.3,6,24 Shh secreted from the notochord and floor plate establishes a ventral-to-dorsal concentration gradient essential for defining neural progenitor domains, including the pMN domain where olig2 is expressed. Previous studies have demonstrated that increasing or decreasing Shh signaling levels can respectively expand or shrink the olig2-expressing domain, making olig2 a reliable readout of Shh pathway activity.3,6,24 Our data using cyclopamine inhibition and shha mRNA overexpression confirm that our TgKI(olig2-mNeonGreen) reporter faithfully responds to these established regulatory inputs.
Despite significant advantages, the TgKI(olig2-mNeonGreen) reporter has certain limitations. Compared to traditional BAC transgenic reporters, the knock-in line generally exhibits lower fluorescence intensity due to the rapid turnover rate of the endogenous Olig2 protein, potentially necessitating more sensitive imaging systems for detailed long-term live imaging or lineage-tracing experiments.
Our study focused primarily on a single knock-in allele. Given the endogenous nature of this knock-in, allele-specific effects are likely minimal; however, we cannot completely rule out the presence of closely linked genetic variations. Nonetheless, this line has been extensively outcrossed (>3 generations) in two separate zebrafish facilities without observed silencing or loss of expression across generations.
Notably, detailed records of targeting efficiency and germline transmission were not retained. Researchers aiming to generate similar lines should consider employing current genome-editing techniques, which likely offer higher efficiency and reproducibility.
Moving forward, the TgKI(olig2-mNeonGreen) zebrafish line opens avenues for advanced research, including investigations into dynamic neural progenitor behaviors, real-time responses to signaling pathways like Hedgehog, and broader studies on neural differentiation, regeneration, and disease modeling. This knock-in reporter provides a valuable and robust tool, significantly enhancing the zebrafish and neuroscience communities’ capabilities to quantitatively study neural development.
Conclusion
In summary, the TgKI(olig2-mNeonGreen) zebrafish line provides an accurate, direct method for visualizing and quantifying endogenous Olig2 protein expression in vivo. By overcoming key limitations of previous transgenic lines, this reporter line facilitates robust quantitative studies of motor neuron and oligodendrocyte development and neural progenitor dynamics during vertebrate embryogenesis.
Footnotes
Authors’ Contributions
C.T.C.: Experimental design and conceptualization. T.K.: Characterization of the knock-in transgenic fish. S.N.: Design of knock-in strategy. S.G.M. and T.Y.-C.T.: Generation of knock-in allele. C.T.C. and T.Y.-C.T.: Overseeing the entire project, literature review. C.T.C. and T.Y.-C.T.: Data acquisition/interpretation/curation/statistical analysis. C.T.C. and T.Y.-C.T.: Writing of original manuscript. C.T.C., T.K., S.N., S.G.M., and T.Y.-C.T.: Reviewing and editing of article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by the National Institute of General Medical Sciences (R35GM150759 to C.-T.C. and T.Y.-C.T., R01GM154339 to S.M., K99GM151398 to S.N.), the Taiwanese Ministry of Education Fellowship (C.-T.C.), the Pew Biomedical Scholar Award from the Pew Charitable Trusts (T.Y.-C.T.), and the Edward Mallinckrodt Jr. Foundation New Investigator Grant (T.Y.-C.T.), S.N. was the Philip O'Bryan Montgomery, Jr., MD, Fellow of the Damon Runyon Cancer Research Foundation (DRG-2339-18).