
Abstract
Nimadeji, Yanna Cai, and Na Gao. Genetic adaptation to Tibetan high altitudes and hepatocellular cancer risk: integrative bidirectional Mendelian randomization and single-cell RNA sequencing analysis. High Alt Med Biol. 00:00–00, 2025.
Background:
The unique genetic adaptations of Tibetans to high-altitude environments may influence disease susceptibility, including hepatocellular carcinoma (HCC).
Methods:
We employed bidirectional two-sample Mendelian randomization (MR) using genome-wide association study summary statistics: high-altitude adaptation (HAA) data from Tibetan/non-Tibetan East Asians (n = 10,295) and HCC data from BioBank Japan (1,866 cases/195,745 controls). Instrumental variables were selected via genome-wide significance (p < 5 × 10−8), with inverse variance-weighted random-effects models as primary analysis. MR-Egger regression analysis and Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) global analysis were used to evaluate the pleiotropic effect. Single-cell RNA sequencing from the Hepatocellular Carcinoma Database identified HAA-related differentially expressed genes (DEGs) in HCC tissues, followed by functional enrichment analysis.
Results:
MR results demonstrated HAA as a causal risk factor for HCC (odds ratio = 2.15 × 102, 95% confidence interval = 1.57–2.96 × 104, p = 0.032), with no reverse causality. Single-cell analysis revealed 23 HAA-associated DEGs in HCC, enriched in immune regulation, DNA metabolism, and cell growth pathways. Notably, genes such as EPAS1 and GCH1; Guanosine triphosphate, GTP, previously linked to Tibetan HAA, were found to be differentially expressed in HCC tissues.
Conclusion:
This study provides statistical and genetic evidence that HAA is a causal risk factor for HCC in Tibetans. The identification of HAA-related genes in HCC tissues offers new insights for targeted prevention and treatment strategies in Tibetan populations.
Introduction
The Qinghai-Tibet Plateau, renowned as the “Roof of the World” and the “Water Tower of Asia,” is the highest and most topographically complex plateau globally. Its climate is characterized by extreme cold, aridity, and intense solar radiation, creating a unique ecosystem with significant survival challenges (Zhang et al., 2023). The Tibetan population, indigenous to this region, has evolved distinct genetic adaptations to chronic hypoxia over millennia of natural selection, serving as a paradigm for human resilience in extreme environments (Lorenzo et al., 2014). Tibetans exhibit unique physiological traits, including elevated hemoglobin concentration, reduced arterial oxygen content, increased resting ventilation, absence of hypoxic pulmonary vasoconstriction, and enhanced oxygen utilization (Beall, 2007). While these adaptations confer survival advantages, they may also influence susceptibility to diseases such as cancer (Jiang et al., 2023).
Liver cancer ranks as the sixth most common malignancy and the third leading cause of cancer-related deaths worldwide, with over 900,000 new cases and 830,000 deaths reported in 2020 (Sung et al., 2021). Hepatocellular carcinoma (HCC), accounting for more than 80% of liver cancer cases, is among the top 3 causes of cancer mortality in 46 countries and the top 5 in 90 nations (Rumgay et al., 2022). Due to asymptomatic progression and late diagnosis, HCC has a five-year survival rate of ∼18%, exacerbating its global burden (Yuan et al., 2025). Notably, data from China’s National Mortality Surveillance System (2008–2021) reveal that Tibet has higher crude and age-standardized HCC mortality rates than other provinces (Liu et al., 2024). In addition, genetic polymorphisms in Tibetan populations, such as those in the XPD and GSTM1 genes, have been linked to HCC susceptibility (赵君慧, 李华, 迪吉, 2014). However, whether genetic adaptations to high-altitude environments causally influence HCC risk remains unclear.
Mendelian randomization (MR) is a statistical method that utilizes genetic variants as instrumental variables (IVs) to evaluate the causal relationship between exposures (such as lifestyle factors) and outcomes (such as diseases) (Smith and Ebrahim, 2003). Its fundamental concept is derived from Mendel’s laws of inheritance, whereby alleles are randomly allocated from parents to offspring during meiosis. This “natural randomization” ensures that an individual’s genetic makeup is independent of environmental confounders, such as lifestyle habits and socioeconomic status, thereby reducing the impact of confounding and reverse causation in observational studies (Bowden et al., 2015; Smith and Ebrahim, 2003). Moreover, because genetic variants are determined at birth and remain unaffected by subsequent environmental influences, they serve as reliable proxies for lifelong exposure, thus providing robust evidence for long-term causal relationships in disease development (Lawlor et al., 2008).
Therefore, we conducted a two-way MR analysis using large-scale meta-analysis data from genome-wide association studies (GWAS) on high-altitude adaptation (HAA) and HCC. We identified HAA as a causal risk factor for HCC, while no significant causal effect of HCC on HAA was observed. In addition, we explored differentially expressed genes (DEGs) in HCC tissues that may be associated with HAA using single-cell RNA sequencing (scRNA-seq) datasets. These findings provide valuable insights for liver cancer prevention and treatment strategies in Tibet.
Materials and Methods
Study design
We performed bidirectional two-sample MR analyses to estimate causal effects between HAA and HCC. GWAS summary statistics for HAA in East Asian Tibetans and non-Tibetans were obtained from a previous study (Yang et al., 2017), while HCC data were derived from a study using the BioBank Japan (BBJ) project (http://jenger.riken.jp/en/) (Ishigaki et al., 2020). BBJ is Japan’ s largest disease-oriented biobank, established to promote personalized medicine based on individual genetic information by collecting and storing biological samples and clinical data (Tanaka et al., 2022). The authors were not involved in the BBJ project, and the GWAS summary statistics were obtained solely from publicly available sources. All of these GWAS summary statistics were generated using either linear regression (Yang et al., 2017) or logistic regression models (Ishigaki et al., 2020). Subsequently, HAA-associated DEGs in HCC tissues were identified using scRNA-seq data from the Hepatocellular Carcinoma Database (HCCDB) (http://lifeome.net:809/#/home) (Jiang et al., 2024).
GWAS summary statistics of HAA
The GWAS summary statistics for HAA were obtained from a previous study by Yang et al. (2017). The term “high altitudes” typically refers to regions at or above 2,500–3,000 meters. Subjects with strong HAA were defined as healthy individuals residing in high-altitude areas without any known medical records, chronic mountain sickness, or other high-altitude-related diseases (Valverde et al., 2015). This study performed a genome-wide analysis on 7.3 million genotyped and imputed single-nucleotide polymorphisms (SNPs) in a total of 10,295 individuals, including 3,008 Tibetans and 7,287 non-Tibetan East Asians. The Tibetan sample had a male-to-female ratio of 0.59. Data on the average age of Tibetans and the male/female ratio and the average age for non-Tibetan East Asians were not available. Supplementary Table S1 summarizes the basic characteristics of the HAA GWAS.
GWAS summary statistics of HCC
The GWAS summary statistics for HCC were obtained from a study conducted using the BBJ project (Ishigaki et al., 2020). This study performed a genome-wide analysis on 197,611 East Asian individuals from BBJ, including 1,866 cases and 195,745 controls (Nagai et al., 2017; Yang et al., 2017). The case group had a male-to-female ratio of 2.87 and a mean age of 68.0 years, while the control group had a ratio of ∼1.00 and a mean age of 61.6 years. Supplementary Table S1 summarizes the basic characteristics of the HCC GWAS.
Selection of IVs for HAA
To select the IVs for HAA, we first extracted SNPs that were significantly associated with HAA (p < 5 × 10−8). Next, using data from the 1000 Genomes Project for the East Asian population, we removed SNPs exhibiting strong linkage disequilibrium (LD) (r2 < 0.001), retaining eight SNPs (Auton et al., 2015). Third, we eliminated palindromic SNPs or those with potential allele strand mismatches between the exposure and outcome summary statistics (this step did not remove any SNPs) (Hartwig et al., 2016). Fourth, we extracted the data for these eight SNPs from the HCC GWAS summary statistics. By default, if a requested SNP was not available in the HCC GWAS, a proxy SNP in strong LD (r2 > 0.8) from the 1000 Genomes Project for the East Asian population was used. However, two SNPs and their proxies were absent from the HCC GWAS and therefore were excluded from further analysis. Fifth, using LDlink (https://ldlink.nih.gov/), we confirmed that none of the selected SNPs were associated with other risk factors for HCC. Sixth, we evaluated the strength of the IVs in the MR analysis using the F-statistic, excluding any SNP with an F value ≤ 10 (this step did not remove any SNPs) (Palmer et al., 2012; Burgess et al., 2011). For the six remaining SNPs, Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) did not detect any outliers (MR-PRESSO test p > 0.05). Finally, we selected six SNPs as the IVs for HAA. Detailed information on the IVs is provided in Supplementary Table S2.
Selection of IVs for HCC
To select the IVs for HCC, we first extracted 11 SNPs significantly associated with HCC (using a threshold of p < 5 × 10−6 to ensure that more than four SNPs were available for subsequent analyses). We then applied the same filtering steps as for the HAA IVs to obtain nine SNPs. Using LDlink (https://ldlink.nih.gov/), we found that none of these SNPs were associated with HAA. MR-PRESSO detected one potential outlier (MR-PRESSO test p = 0.09). After removing this outlier, no further outliers were detected (MR-PRESSO test p > 0.05). Finally, eight SNPs were selected as the IVs for HCC. Detailed information on the HCC IVs is provided in Supplementary Table S3.
MR analysis
MR analyses were conducted using the “TwoSampleMR” R package, applying five methods: inverse variance-weighted (IVW) with random-effects, simple mode, weighted mode, weighted median, and MR-Egger regression. Effects were reported as odds ratios (ORs) with 95% confidence intervals (CIs), with a significance threshold at p value < 0.05. The IVW random-effects model was prioritized for its conservative estimation of heterogeneity-adjusted causal effects (Zhai and Guyatt, 2024). Simple and weighted modes were conventional MR methods. The weighted median estimate was sensitive to additional or removal of genetic variants, and MR-Egger regression proved sensitive to outliers (Slob and Burgess, 2020).
Heterogeneity test
We assessed heterogeneity among the individual genetic variant estimates using Cochran’s Q test, as implemented in both MR-Egger regression and the IVW method. A p < 0.05 from either method indicates significant heterogeneity in the MR analysis.
Horizontal pleiotropy test
MR analysis relies on three main assumptions: (1) the genetic variants are associated with the exposure; (2) the genetic variants are independent of confounding factors; and (3) the genetic variants influence the outcome solely through the exposure, with no alternative pathways (Davies et al., 2018). Associations that violate assumptions (2) and (3) are typically indicative of horizontal pleiotropy. We assessed horizontal pleiotropy using MR-Egger regression. In addition, we performed a global test using MR-PRESSO (MR Pleiotropy RESidual Sum and Outlier test) to identify and remove outlier SNPs contributing to heterogeneity in the causal effect estimates (Verbanck et al., 2018). A p > 0.05 indicates that the independent HAA genetic IVs do not exhibit significant pleiotropy in the HCC GWAS, and vice versa.
Leave-one-out analysis
A leave-one-out analysis was conducted by sequentially removing each SNP and re-running the MR analysis on the remaining SNPs to assess the influence of each individual SNP on the overall causal estimate (Hemani et al., 2018).
ScRNA-seq datasets collection and data processing
We accessed the HCCDB (http://lifeome.net:809/#/home) and downloaded scRNA-seq datasets comprising 20 normal liver samples, 3 tumor-adjacent liver tissue samples, and 32 HCC tissue samples (Jiang et al., 2024). These data include two publicly available gene expression datasets from the Gene Expression Omnibus [GSE192742 (Ma et al., 2021) and GSE151530 (Guilliams et al., 2022)] as well as sequencing data generated by HCCDB. Batch effects among the datasets were removed using canonical correlation analysis (Jiang et al., 2024). During quality control, HCCDB excluded cells with elevated mitochondrial mRNA levels (mt > 20%) or with exceptionally high or low feature counts (fc < 200 or fc > 10,000) (Jiang et al., 2024). Principal component analysis was performed to project cells into a lower-dimensional space defined by the first 30 principal components, and clustering was visualized using Uniform Manifold Approximation and Projection. In total, 7 major lineages and 18 minor lineages were identified (Jiang et al., 2024).
Screening for HAA-related DEGs in HCC tissues
We used the Seurat R package (version 5.1.0) and its FindMarkers function to identify DEGs between HCC tissues and other tissue types (Benjamini–Hochberg adjusted p value < 0.05 and |log2fold change| > 0.5). We then intersected these DEGs with Tibetan HAA-related genes extracted from a whole-genome sequencing study of 1,001 indigenous Tibetans (Zheng et al., 2023) to obtain HAA-related DEGs in HCC tissues. These DEGs were visualized using volcano and dot plots to display their expression across different tissues and cell types.
Functional enrichment analysis of HAA-related DEGs
We performed functional enrichment analysis on the HAA-related DEGs using Metascape (https://metascape.org/gp/index.html#/main/step1). Metascape is a robust online tool designed for gene and protein list annotation, enrichment analysis, and functional analysis (Zhou et al., 2019). Gene Ontology (GO) analysis was conducted to reveal the enriched biological processes (BP), cellular components (CC), and molecular functions (MF) associated with these genes, using a significance threshold of p < 0.01 to identify key GO pathways (Ashburner et al., 2000; Yu et al., 2012).
Results
Characteristics of selected IVs
To assess bidirectional associations between HAA and HCC, we first selected IVs for HAA and HCC separately. GWAS summary statistics for HAA were derived from a previous GWAS study of Yang et al. (2017), while HCC data were obtained from the GWAS study of Ishigaki et al. (2020) (Supplementary Table S1). After screening, we retained six SNPs as IVs for HAA (Supplementary Table S2) and eight SNPs for HCC (Supplementary Table S3). The mean F-statistics for these SNPs ranged from 22.205 to 65.454 (Supplementary Tables S2 and S3), indicating a low likelihood of weak instrument bias.
Bidirectional MR estimates between HAA and HCC
To evaluate the causal effect of HAA on HCC, we employed five MR methods: IVW, simple mode, weighted mode, weighted median, and MR-Egger regression. Forest and scatter plots of causal estimates are shown in Figure 1 and Supplementary Figure S1. IVW analysis (primary method) revealed a significant causal effect of HAA on HCC risk (OR = 2.15 × 102, 95% CI = 1.57–2.96 × 104, p = 0.032; Table 1). The IVW method typically offers the highest statistical power. Although the four supplementary MR methods did not reach statistical significance (p > 0.05), all methods consistently showed positive effect estimates (OR > 1), with effect directions aligned with the IVW finding. This consistent directional trend reinforced the IVW finding, suggesting that genetic predisposition to HAA is associated with an increased risk of HCC.
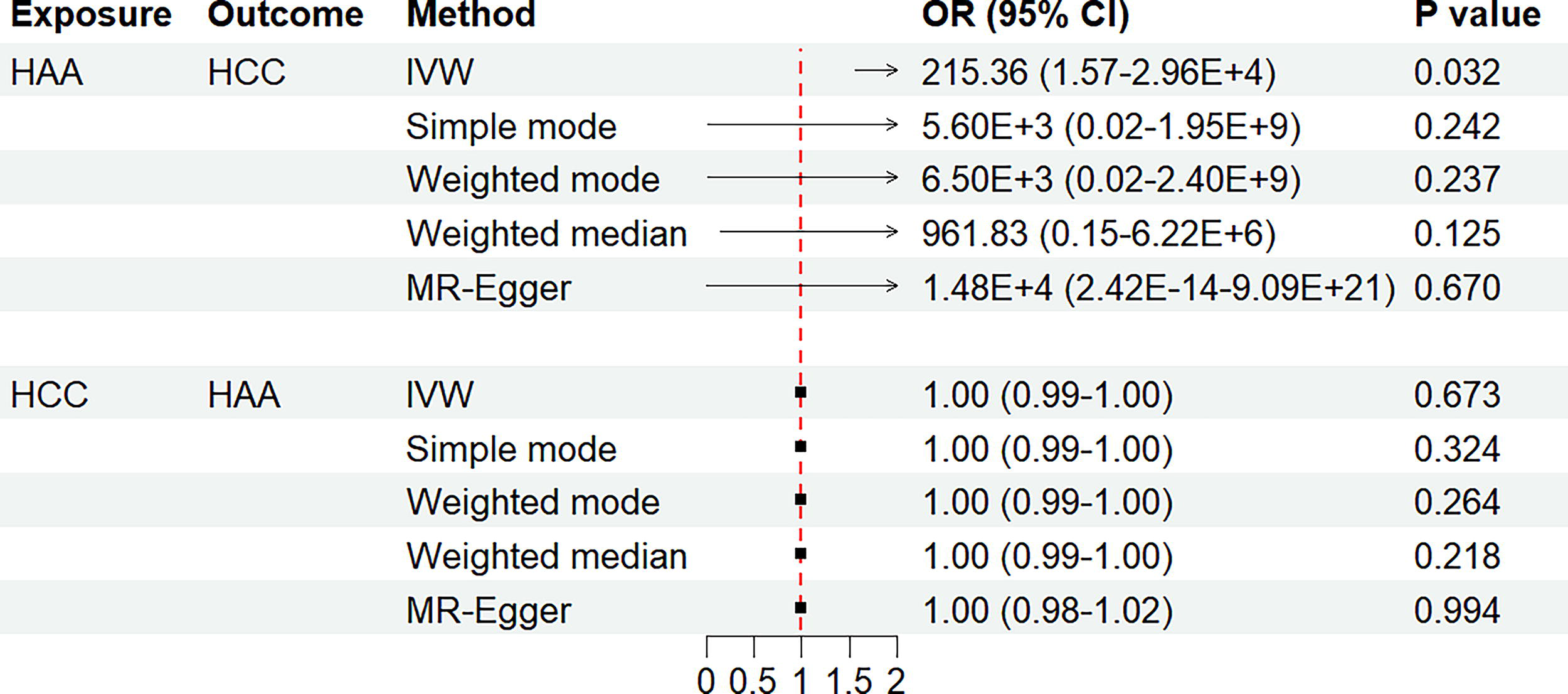
Bidirectional Mendelian randomization estimates for the association of high-altitude adaptation with hepatocellular carcinoma. The summarized Mendelian randomization (MR) effect sizes of the causal relationships between the high-altitude adaptation (HAA) and the hepatocellular carcinoma (HCC). We used five methods, including the inverse variance-weighted (IVW), simple mode, weighted mode, weighted median, and MR-Egger regression methods, for MR analyses. IVW method with a random-effect model was used as the main MR analysis. A two-sided p value of less than 0.05 was considered to be statistically significant. CI, confidence interval; OR, odds ratio.
The Results for the Mendelian Randomization Analyses of Associations Between the High-Altitude Adaptation and Hepatocellular Carcinoma
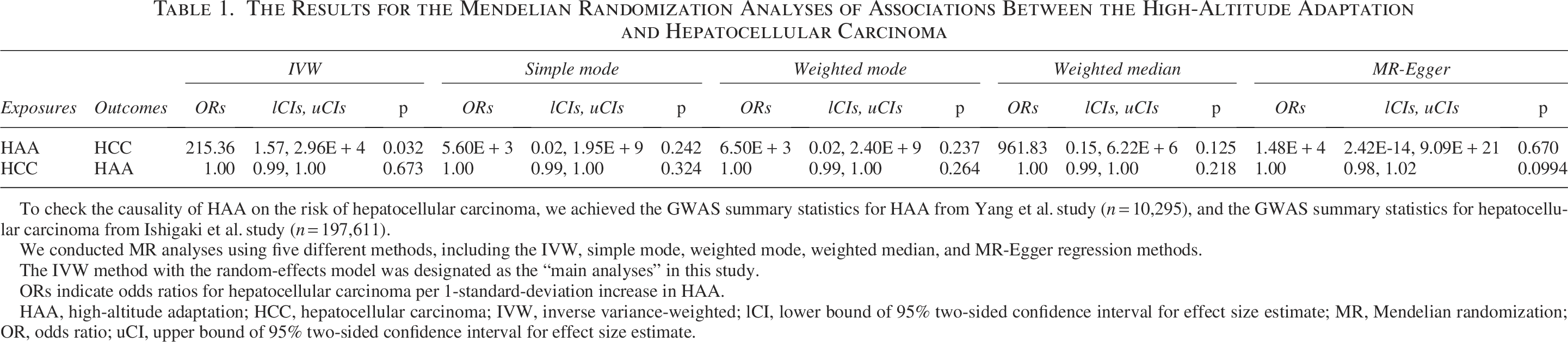
To check the causality of HAA on the risk of hepatocellular carcinoma, we achieved the GWAS summary statistics for HAA from Yang et al. study (n = 10,295), and the GWAS summary statistics for hepatocellular carcinoma from Ishigaki et al. study (n = 197,611).
We conducted MR analyses using five different methods, including the IVW, simple mode, weighted mode, weighted median, and MR-Egger regression methods.
The IVW method with the random-effects model was designated as the “main analyses” in this study.
ORs indicate odds ratios for hepatocellular carcinoma per 1-standard-deviation increase in HAA.
HAA, high-altitude adaptation; HCC, hepatocellular carcinoma; IVW, inverse variance-weighted; lCI, lower bound of 95% two-sided confidence interval for effect size estimate; MR, Mendelian randomization; OR, odds ratio; uCI, upper bound of 95% two-sided confidence interval for effect size estimate.
Subsequently, we investigated the causal effect of HCC on HAA. No causal effect of HCC on HAA was detected (p of all MR methods > 0.05; Table 1), as shown in the forest and scatter plots in Figure 1 and Supplementary Figure S1.
Sensitivity analyses
To assess the robustness of the causal effect estimates between HAA and HCC, we conducted several sensitivity analyses, including tests for horizontal pleiotropy, heterogeneity, and leave-one-out analysis. Both the MR-Egger intercept test and MR-PRESSO indicated no evidence of horizontal pleiotropy (all p > 0.05; Table 2). In addition, Cochran’s Q test revealed no significant heterogeneity (all p > 0.05; Table 2). To mitigate any potential impact of heterogeneity, the fixed-effects IVW model was employed as the primary MR method. The leave-one-out analysis demonstrated that no single SNP substantially influenced the overall causal estimate (Supplementary Fig. S2). Collectively, these results suggest that our MR findings are robust.
The Results for the Pleiotropy and Heterogeneity Tests

To evaluate the robustness of the causal effect estimates of HAA on hepatocellular carcinoma, and vice versa, we performed several sensitivity analyses, including the horizontal pleiotropy tests and heterogeneity tests.
Horizontal pleiotropy analyses were conducted by MR-Egger regression and MR-PRESSO methods, and the results showed that there is no evidence of horizontal pleiotropy in IVs of either HAA or hepatocellular carcinoma (all p > 0.05).
In the heterogeneity tests, the Cochran’s Q statistics showed that there is no evidence of significant heterogeneities (all p > 0.05).
HAA, high-altitudes adaptation; HCC, hepatocellular carcinoma; IVW, inverse variance-weighted; MR, Mendelian randomization; MR-PRESSO, MR-Pleiotropy RESidual Sum and Outlier; SE, standard error—not available.
Identification of HAA-related DEGs in HCC tissues
After quality control, the HCCDB annotated the single-cell data into 7 major cell lineages and 18 minor lineages (Supplementary Figs. S3 and Figs. S4) (Jiang et al., 2024). We analyzed HCC tissues samples and control samples (normal liver tissue and liver tissue adjacent to the tumor) and identified a total of 3,013 DEGs, with 1,347 genes upregulated and 1,666 downregulated in HCC tissues (Fig. 2). Subsequently, we intersected these DEGs with 192 HAA-related genes extracted from a whole-genome sequencing study of 1,001 indigenous Tibetans (Zheng et al., 2023) and identified 23 HAA-related DEGs in HCC tissues (Fig. 2; Supplementary Fig. S4). Four genes (AK3, EPAS1, DDRGK1, and MRPL23) were upregulated, while 19 genes (SP110, ARAP2, LNPEP, SSH2, APBA2, FOXN3, RILPL2, FCGR2A, ITCH, JADE2, TRABD, GCH1, NEK7, FOXP1, STXBP3, GOPC, SBF2, C1orf21, and TSC22D2) were downregulated. Figure 3 displays the expression patterns of these genes across different cell types.
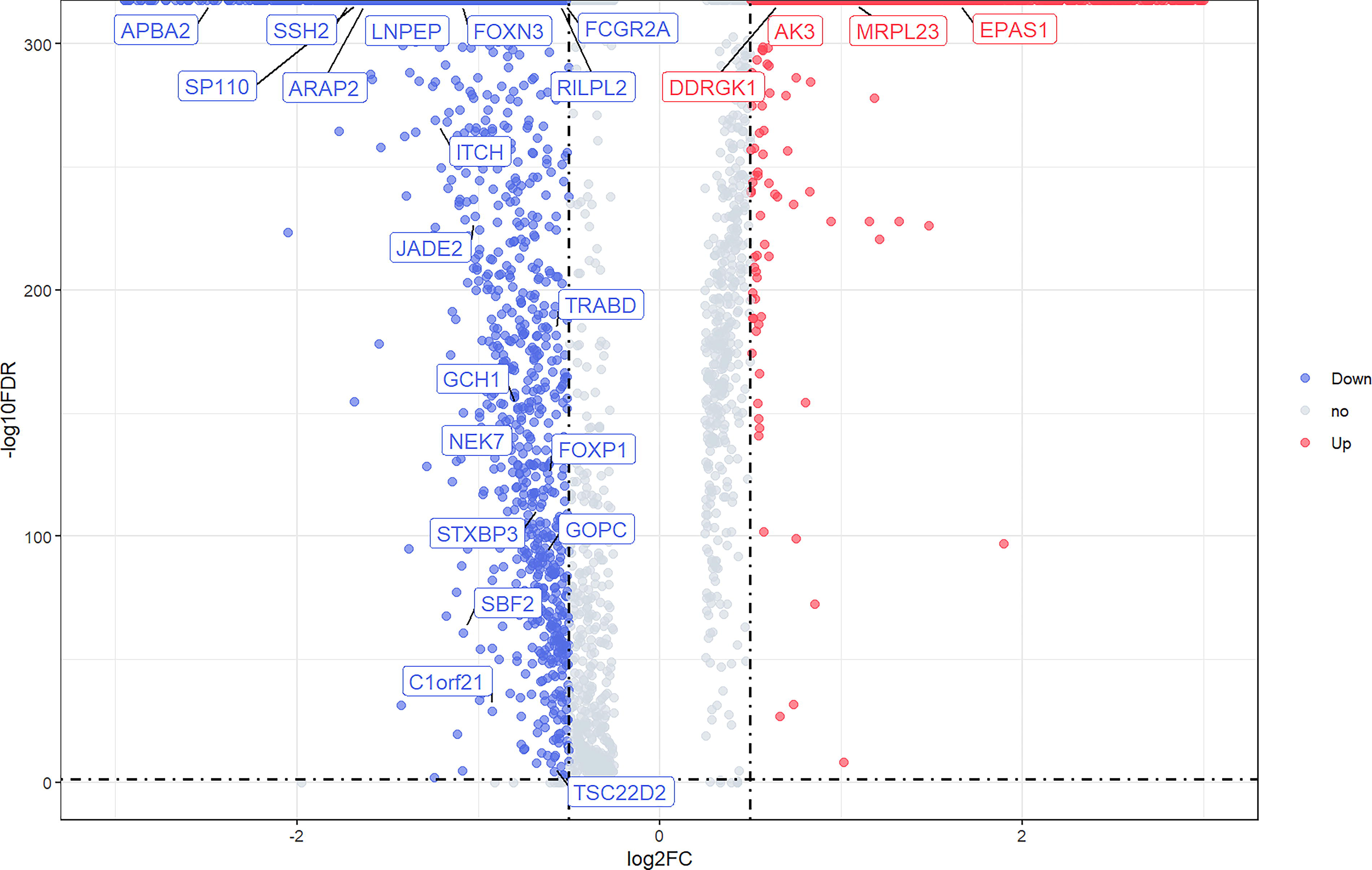
Volcano plot of genes differentially expressed between hepatocellular carcinoma tissue and controls (normal liver tissue and liver tissue adjacent to the tumor) in the HCCDB single-cell sequencing dataset. Blue nodes represent downregulation in hepatocellular carcinoma tissue; red nodes represent upregulation; and gray nodes represent no significant difference from controls. Differentially expressed genes related to high-altitude adaptation are labeled in the figure. HCCDB, Hepatocellular Carcinoma Database.

Dot plot of high-altitude adaptation-related differentially expressed genes in different cell types. The percentage of cells expressing the gene (pct.exp) is represented by the size of the point.
Functional enrichment analysis of HAA-related DEGs
Using Metascape, we performed GO analysis on the 23 HAA-related DEGs with a significance threshold of p < 0.01, identifying enriched BP, CC, and MF (Fig. 4; Supplementary Table S4). The GO analysis revealed that these DEGs are primarily enriched in BP related to the regulation of DNA biosynthetic process, protein modification by small protein conjugation, positive regulation of immune response, protein modification by small protein conjugation or removal, regulation of cell growth, immune effector processes, regulation of DNA metabolic process, skeletal system development, regulation of response to biotic stimulus, regulation of system process, and protein ubiquitination. In terms of CC, the genes were enriched in the vacuolar membrane, while in MF, they were associated with RNA polymerase II-specific DNA-binding transcription factor binding, DNA-binding transcription factor binding, and transcription factor binding.
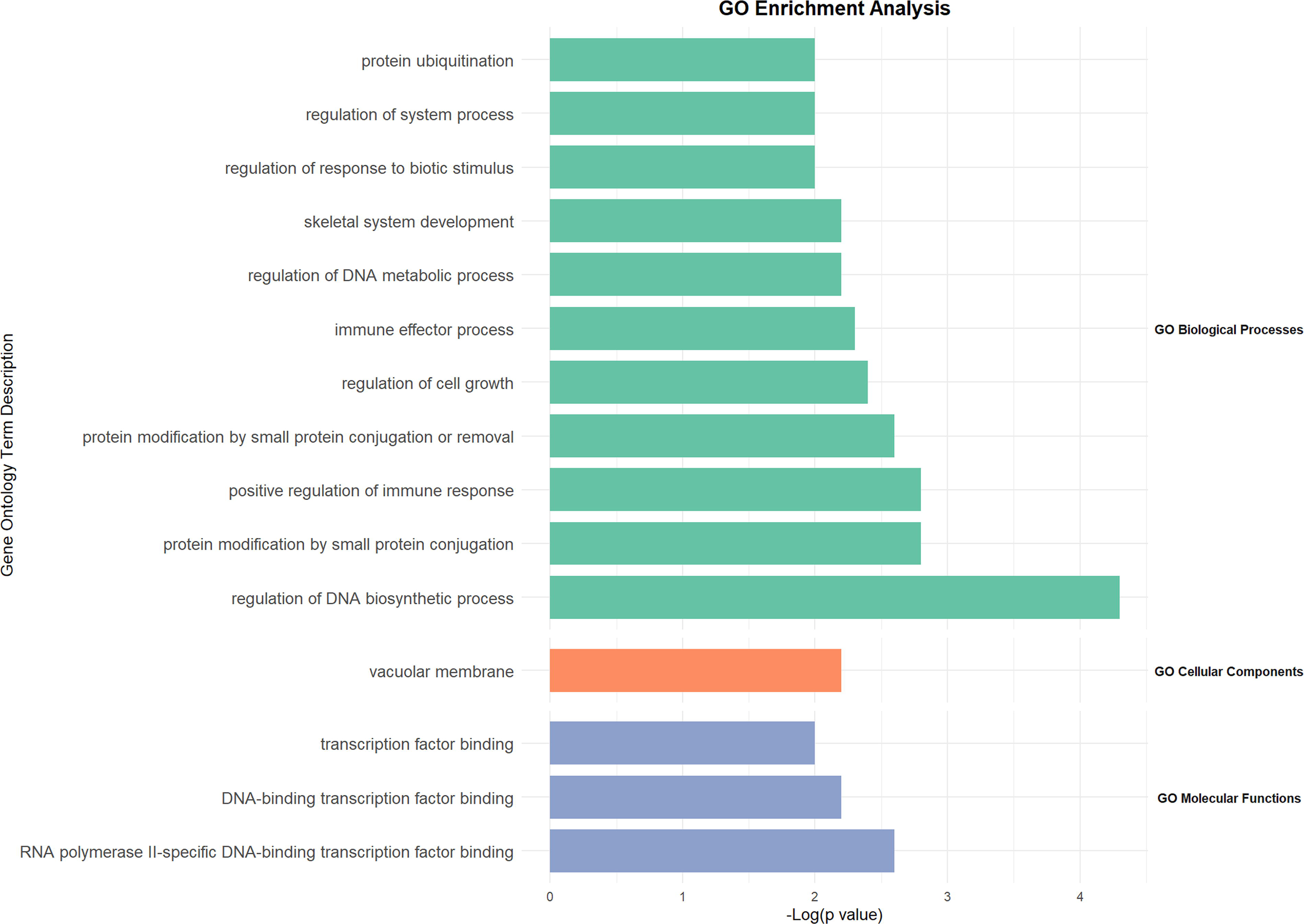
Bar graph of the top six enriched terms across differentially expressed genes related to high-altitude adaptation, colored by p values.
Discussion
Our MR study leveraging GWAS summary data identified HAA as a causal risk factor for HCC with no reverse causality. ScRNA-seq analysis revealed 23 HAA-associated genes in HCC tissues, enriched in pathways related to immune regulation, cell growth, DNA metabolism, and transcriptional regulation, and responses to biological stimuli, suggesting multi-mechanistic biological links between Tibetan genetic adaptation and HCC pathogenesis.
HCC mortality rates in Tibet are higher than in other provinces of China, and age-specific mortality increases with advancing age (Liu et al., 2024). However, because of limitations such as insufficient medical resources and challenges in data collection, few studies have examined the prevalence and pathogenesis of HCC in Tibetan populations, with etiological research often confined to non-genetic factors such as viral hepatitis and alcohol consumption. Our study is the first to employ MR to analyze the potential causal relationship between HAA and HCC. Our findings suggest that long-term HAA may lead to genetic and immunological differences in Tibetans compared with other populations, rendering them more susceptible to HCC, although further research is needed to confirm this finding.
After establishing the causal link between HAA and HCC, we further identified DEGs in HCC tissues potentially associated with HAA. Notably, the hypoxia pathway gene EPAS1 has been identified as one of the most positively selected genes in Tibetans and has been shown to be associated with differences in hemoglobin concentration at high altitudes (Huerta-Sánchez et al., 2014). A prior study reported that EPAS1 is expressed in 69.5% of HCC cases, 55.6% of adjacent non-tumor tissues, and 0% of normal liver tissues, and that its expression is significantly correlated with tumor grading, venous invasion, intrahepatic metastasis, necrosis, and capsule infiltration, suggesting an important role in HCC progression and prognosis (Bangoura et al., 2007). Moreover, GCH1, which is involved in maintaining nitric oxide synthase function and normal blood pressure, carries numerous adaptive variants in Tibetans and is significantly associated with various physiological characteristics, including blood nitric oxide levels, oxygen saturation, and hemoglobin concentration (Guo et al., 2017). GCH1 encodes Guanosine triphosphate (GTP) cyclohydrolase I, the rate-limiting enzyme in the de novo biosynthesis of tetrahydrobiopterin (BH4), and has been identified as a novel metabolic regulator in HCC (Guo et al., 2017). Epigenetic silencing of GCH1 can promote HCC growth by inhibiting de novo BH4 synthesis and subsequently activating superoxide-mediated Apoptosis signal-regulating kinase 1 (ASK1)/p38 signaling (Zhong et al., 2021). These findings provide biological plausibility for the causal impact of high-altitude hypoxic adaptation on HCC.
It is noteworthy that environmental factors may act synergistically with HAA in the development of HCC in the Tibetan population, and this interaction can be indirectly explored using existing data from this study. On the one hand, the core environmental characteristics of the Tibetan region include chronic hypobaric hypoxia and intense ultraviolet radiation. On the other hand, hypoxia itself has been established as a significant biological feature in HCC metabolism (He et al., 2024). Our findings suggest a potential pathogenic pathway in which chronic environmental hypoxia may contribute to HCC progression through the mechanisms of HAA, mediated by adaptively expressed genes such as EPAS1 and GCH1.
Our findings have important clinical implications for improving HCC prevention and treatment strategies in Tibetan regions. The potential causal relationship between HAA and HCC suggests that health education among Tibetan residents should be enhanced and that efforts should be made to control synergistic risk factors such as hepatitis virus infection, alcohol consumption, and aflatoxin exposure. In addition, early screening strategies for HCC could be developed based on HAA-related DEGs (e.g., EPAS1 and GCH1), or personalized therapies targeting HAA-related pathways could be explored.
Our study has several strengths. First, the bidirectional two-sample MR approach uses genetic variants as IVs, which reduces confounding from environmental factors and reverse causation, thus providing a more accurate estimation of the causal relationship between exposures and disease. Second, the use of high-quality scRNA-seq data to identify HAA-related DEGs in HCC helps overcome the limitations posed by scarce biological data in Tibetan populations and lays the groundwork for subsequent functional studies.
However, our study has some limitations. First, the GWAS summary data used are predominantly from East Asian populations, and the MR analysis results may not be directly generalizable to high-altitude populations from other regions. Second, the sample size of the single-cell data is relatively small. Further studies with larger cohorts and additional functional experiments are needed to validate these findings.
Conclusion
By demonstrating statistical and genetic evidences, our study supports that Tibetan HAA is a potential causal risk factor for HCC. Furthermore, by pinpointing 23 HAA-associated DEGs in HCC tissues, we propose a mechanistic pathway: local environmental factors like chronic hypoxia may promote HCC progression by engaging HAA mechanisms, mediated by adaptively expressed genes such as EPAS1 and GCH1. This finding underscores the role of genetic susceptibility in the Tibetan population and offers a mechanistic basis for novel intervention strategies.
Authors’ Contributions
N.: Conceptualization, formal analysis, validation, writing—review and editing; Y.C.: Data curation, methodology, visualization, writing—original draft; N.G.: Supervision, project administration, resources, funding acquisition (if applicable), writing—review and editing.
Footnotes
Acknowledgments
The authors are grateful to the researchers of HAA- and HCC-related studies for making their GWAS summary statistics publicly available, which were essential for the analyses. The authors thank the HCCDB for providing publicly available single-cell RNA sequencing data. Special thanks to the participants and investigators of the original GWAS studies, including Tibetan and non-Tibetan East Asian populations in the high-altitudes adaptation study and the individuals in the BBJ cohort.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research received no specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data Availability Statement
This study utilized publicly available data. GWAS summary statistics for HAA were obtained from cnsgenomics.com/data/yang_et_al_2017_pnas.html. GWAS summary statistics for HCC were downloaded from https://gwas.mrcieu.ac.uk/datasets/bbj-a-158/. Single-cell sequencing data for HCC were accessed from ![]() .
.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.