
Abstract
Accumulating morphological and electrophysiological evidence demonstrates that abnormal brain development is a key element in the progression of Huntington's disease (HD). Mutant huntingtin affects corticogenesis, cell migration, and differentiation. Cortical changes are reminiscent of focal cortical dysplasia, a malformation of cortical development that leads to hyperexcitability and epilepsy. Striatal development also is affected by the mutation. In animal models, recent studies provide additional evidence that neuronal morphology and intrinsic and electrophysiological properties deviate from normal development. Some changes indicate delayed development of cortical pyramidal neurons, while a subtype of striatal projection neuron displays a transient accelerated maturation. However, the brain is able to compensate for early abnormalities and, during a variable latent period, brain function appears normal. Eventually, homeostatic mechanisms begin to fail, resulting in the emergence of HD symptoms. The realization that neurodevelopment in HD is abnormal offers new insights and opens new avenues for early treatment. In this review, we present a brief summary of imaging and morphological studies from human carriers of the HD mutation followed by a more in-depth examination of recent findings in genetic animal models.
Plain Language Summary Abstract
Although Huntington's disease (HD) is considered primarily a neurodegenerative disorder, recent data has demonstrated that brain development is altered. Indeed, anatomical and functional changes occur very early in development and may set the stage for cell loss later in life. This article reviews recent evidence from clinical and basic research studies demonstrating that abnormal brain development increases the intrinsic excitability of brain cells. Although the brain tries to cope against increased excitability, compensatory mechanisms eventually fail. Early intervention is paramount to prevent brain deterioration and HD symptoms.
Keywords
Introduction
It is now beyond doubt that the presence of mutant huntingtin (mHTT) alters brain development in both humans and animal models of Huntington's disease (HD). Indeed, a paradigm shift in HD research occurred when a number of pioneering studies demonstrated that mHTT alters brain development.1–13 This was unexpected, considering that HD is predominantly a neurodegenerative disorder and that in the vast majority of cases behavioral, cognitive, and motor symptoms start during midlife, usually >35 yr of age or later. In a low percentage of cases (∼10%), symptoms can start before 20 yr of age, and represent a different pathological entity, juvenile onset HD (JOHD, also known as Westphal variant). JOHD differs not only in the number of CAG repeats but also in the severity, age of onset, and clinical symptoms. 14 Although some aspects of JOHD are similar to those seen in adult-onset HD (AOHD), others are specific of JOHD including, among others, epilepsy, ataxia, spasticity, and pain.15,16 Disease progression of JOHD is faster compared to AOHD, particularly in the case of higher CAG repeat numbers. In this review, we will discuss evidence from human and animal studies pointing to the fact that abnormal brain development predisposes to cortical hyperexcitability in HD, particularly in cases of JOHD.
Abnormal brain development in HD: evidence from imaging and morphological studies (Table 1)
In premanifest HD patients (mean age 37 yr, > 39 CAG repeats), MRI studies provided evidence of differential changes in striatal and cortical volumes. 17 Thus, while striatal and cerebral white matter volumes were significantly decreased compared to control subjects, the volume of the cerebral cortex itself was increased, suggesting altered development in HD. Specifically, cortical morphology showed enlargement of gyral crowns and abnormally thin sulci. 18 In addition, children at risk of HD have disproportionately small heads, suggesting abnormal brain growth. 19 Surprisingly, a large study examining cortical development in children and adolescents at risk of developing AOHD (mean age 13 yr, ∼44 CAG repeats) demonstrated no significant differences in thickness and surface area across all cortical lobes, 20 suggesting that changes in cortical morphology are subtle or that compensatory mechanisms occur early in HD brain development. Another possibility is that cortical changes become more prominent with age and disease progression. Future studies in JOHD subjects with higher number of CAG repeats and presenting with seizures may reveal more detectable abnormalities in cortical morphology during early development. In contrast to the lack of gross cortical abnormalities, children carrying the mutation display biphasic changes in striatal morphology, with early striatal hypertrophy followed by rapid decline.21,22
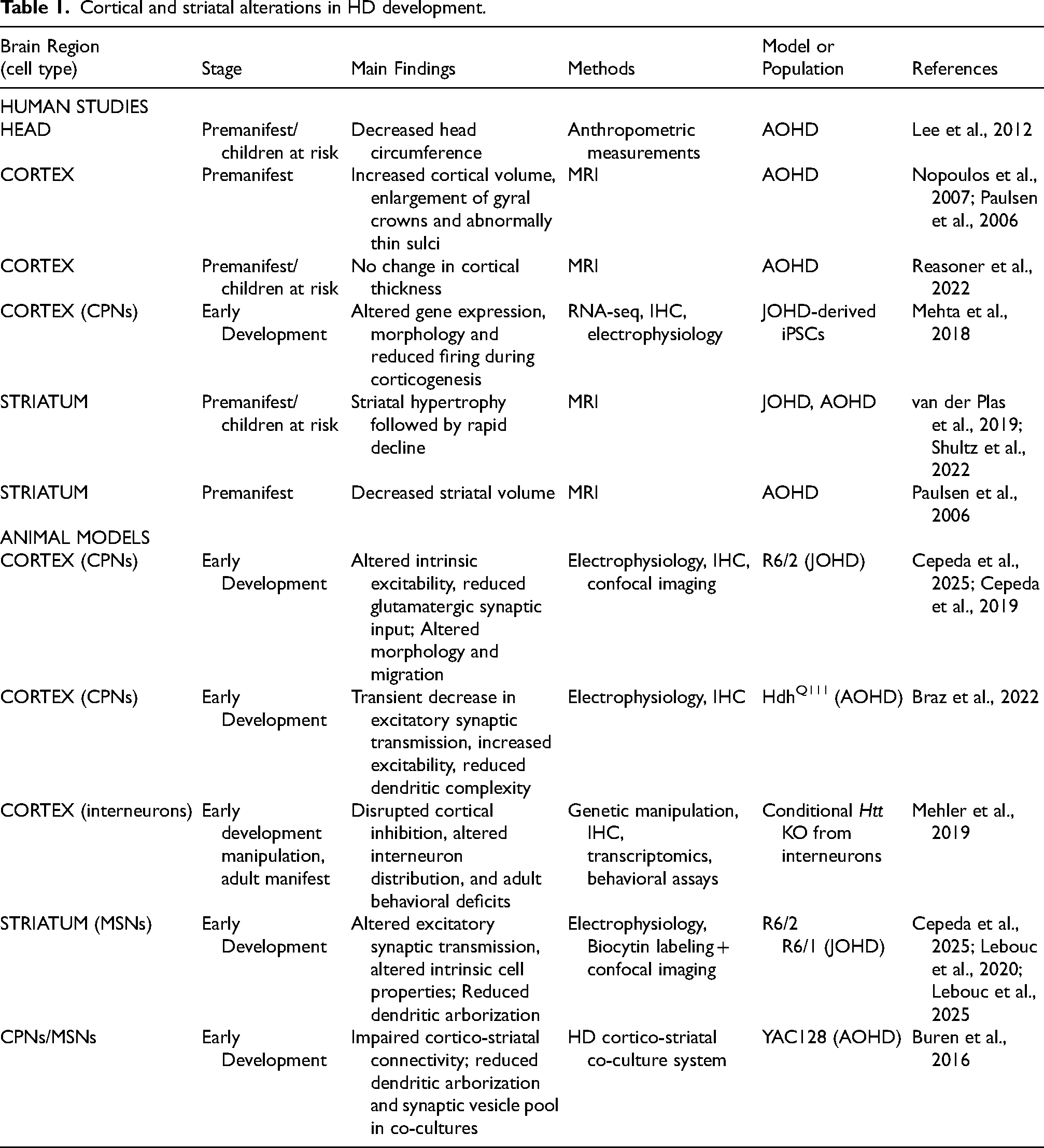
Cortical and striatal alterations in HD development.
Cortical architectural changes have been observed in HD patients and in animal models using other methods. In a large cohort of HD autopsy material, a significant increase in developmental malformations, in particular periventricular nodular heterotopias, were observed compared to control cases. 23 This suggested that in HD, cortical neuron genesis and migration deficits occur. A reflection of this is the observation that, in embryos of a knock-in mouse model of HD (HdhQ111/Q111), the ventricular zone was smaller than that of wildtypes (WTs, HdhQ7/Q7), whereas the cortical plate thickness was increased. 4 In agreement, the cortex was thinner in mutant embryos compared to WTs. Further, at postnatal day P21, the thickness of layer VI was reduced, whereas that of layers II-IV was increased. 4 We have proposed that some changes in cortical architecture are reminiscent of those observed in focal cortical dysplasia (FCD), a malformation of cortical development characterized by dyslamination, cortical pyramidal neuron (CPN) misorientation, and the presence of dysmorphic neurons leading to cortical hyperexcitability and epileptic seizures.7,24 MRI studies in FCD indicate abnormal gyri, focal cortical thickening or thinning, areas of focal brain atrophy, blurring of the gray-white matter junction,25,26 supporting the idea that both pathologies share some common features.
NeuN staining reveals altered cortical cytoarchitecture and disrupted alternative splicing
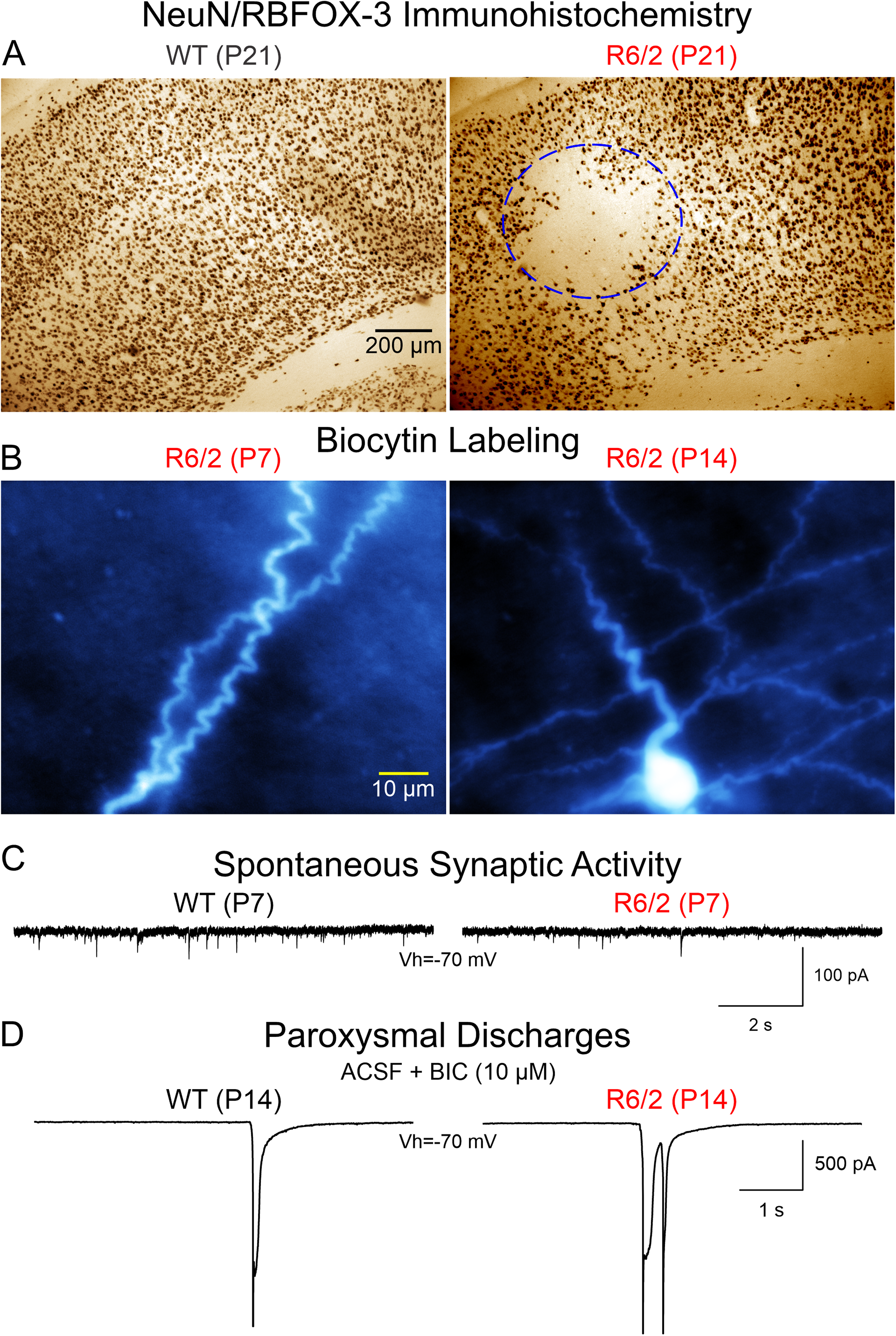
Our early morphological studies in the R6/2 transgenic model of HD using Neuronal Nuclei (NeuN) as a specific marker of postmitotic neurons, 27 identified the occurrence in symptomatic mice of morphological changes similar to those observed in FCD, including cortical dyslamination, neuronal crowding in some areas, whereas other areas are devoid of the neuronal marker. 7 Remarkably, these morphological aberrations also occur in developing R6/2 mice, starting at about P15. 28 Discrete areas devoid of NeuN stain are more evident at P21 and P30. However, loss of NeuN does not necessarily imply neuronal demise. 29 In R6/2 mice, it has been shown that neuronal loss occurs very late in disease progression.30,31 Also, our anatomical studies using cresyl-violet did not disclose FCD-like lesions nor cell loss in R6/2 mice. In consequence, reduced expression of NeuN could have different implications. Indeed, several studies have disputed the idea that NeuN is a reliable neuronal marker, particularly during disease states. 32 NeuN was later identified as FOX-3 (also known as RBFOX3), an RNA binding protein and a member of the FOX-1 gene family that functions as a regulator of alternative splicing. 33 Importantly, alternative splicing is abnormal in HD.34–36 In particular, reduced expression of RBFOX family proteins, including RBFOX3/NeuN, which also interact with HTT, has been demonstrated in animal models and human striatal tissue. 37 Interestingly, in the context of JOHD, mutations or deletions in RBFOX3 underlie epilepsy susceptibility. 38 Thus, not only NeuN immunohistochemistry revealed abnormal cortical organization, but also a deficit of RBFOX3 in developing HD brains starting at about P15. This could have deleterious consequences in terms of alternative splicing as well as cortical excitability. Interestingly, islands devoid of NeuN were not observed in the striatum. 28 This probably means that initial mis-splicing resulting from reduced expression of FOX1 family splicing regulators starts in a maldeveloped cerebral cortex and not in the striatum.
At the cellular level, based on biocytin staining, we also observed CPN misorientation and neurons with abnormal processes including recurved or wavy dendrites and axons in R6/2 and other HD models7,39 (Fig. 1). Tortuous dendrites and axons also occur in human FCD. 40 This observation suggests that pathfinding, orientation, and final positioning of cortical progenitors is affected by mHTT. Overall, imaging and anatomical studies indicate cortical abnormalities that could set the stage for CPN hyperexcitability.
Structural alterations in the HD cerebral cortex predispose to neuronal hyperexcitability
Although no significant changes in gross anatomy of the cerebral cortex have been demonstrated, at least in AOHD, subtle abnormalities in cortical architecture and CPN morphology, similar to those observed in FCD seem to occur. These changes can affect intrinsic membrane properties as well as intracortical and cortico-striatal connectivity and excitability in HD. Indeed, transcranial magnetic stimulation (TMS) studies have demonstrated increased intracortical facilitation and reduced short interval intracortical inhibition in premanifest and early manifest HD patients41–43 indicating altered excitatory/inhibitory balance. Further, using paired-pulse TMS paradigms, it has been suggested that GABA receptor-mediated cortical inhibition is deficient in both pre-symptomatic and symptomatic patients. 44 Notably, inhibitory TMS (1 Hz) of the supplementary motor cortex significantly reduced choreic movements in HD patients. 45 Reduced cortical inhibition and hyperexcitability have also been demonstrated in animal models.46,47 For example, in the BACHD mouse model, decreased layer II/III parvalbumin (PV)-interneuron excitation and decreased CPN inhibition at 6 months was reported. 47 Interestingly, R6/2 mice have fewer perisomatic PV-positive terminals on CPNs than WT CPNs, an observation consistent with findings in HD autopsy brains. Importantly, this reduced inhibition was reflected by increased cortical activity measured via in vivo calcium imaging. 48 These observations raise some important questions; what causes cortical hyperexcitability? When does it start? How does it affect striatal development and function?
Intrinsic and synaptic alterations of CPNs and MSNs during development in HD models
There is a general consensus that HD is, above all, a synaptopathy.52–55 Our group was the first to demonstrate significant reductions in glutamate receptor-mediated synaptic activity along the cortico-striatal pathway in early and late symptomatic R6/2 mice. 56 Alterations in the thalamo-striatal and thalamo-cortical pathways also have been reported.57–60 HTT is expressed very early in developing brains 61 and in R6/2 and YAC128 models, mHTT aggregates have been observed in developing axonal tracts. 62 This suggests that synaptic transmission in CPNs and along the cortico-striatal pathway could be affected very early during brain development. In agreement, Braz et al. showed that during the first postnatal week excitatory synaptic activity was decreased compared to that of WT CPNs, due to reduced expression of glutamate AMPA receptors. 11 This synaptic deficit also recovered by the second postnatal week. Notably, early pharmacological intervention by systemic injections of an Ampakine (Cx516) during the neonatal period rescued these deficits and preserved sensorimotor function, cognition, and spine density in adult mice. 11
Our studies on synaptic function in developing CPNs confirmed deficits in glutamatergic transmission at P7 but it recovered by P14, again suggesting that HD brains are capable of compensating for early synaptic deficits (Fig. 2). In addition, we also demonstrated that GABAergic synaptic transmission is reduced at P7 and GABAA receptor-mediated giant depolarizing potentials (GDPs), commonly observed in CPNs at this age, are more prevalent in R6/2 mice, indicating that the GABA developmental switch, from depolarizing to hyperpolarizing, which normally occurs between the first and second postnatal week, takes longer to transition in R6/2 mice. In addition, tonic GABA currents also are reduced at P7 and P14. This developmental delay could have deleterious consequences as GABA plays a critical role not only as a neurotransmitter but also as a trophic factor during early development, supporting cell migration, proliferation, differentiation, and synaptogenesis.63,64 If, as we showed, the GABA developmental switch is abnormal in HD brains, this also could contribute to the generation of FCD-like areas in the cortex.
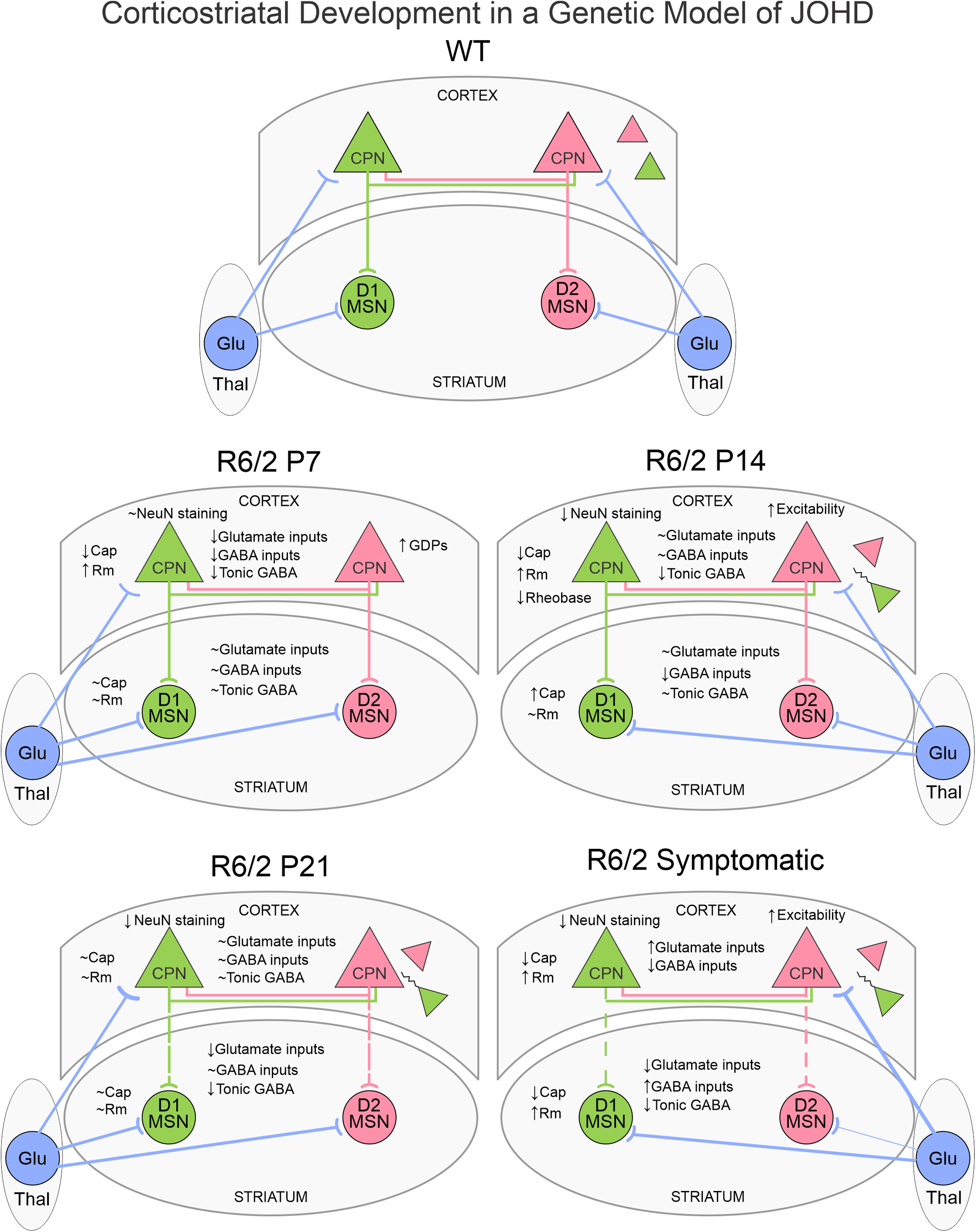
A recent study on very early striatal development of MSNs in R6/1 mice (from P0 to P10), demonstrated that D2-MSNs undergo abnormal development in R6/1 and CAG140 mouse models, with abnormal dendritic growth and functional maturation during early postnatal periods. 50 A significant reduction in D2-MSN density was detected in the matrix, but not in the striosomes, at P8 in R6/1 mice. Changes in intrinsic excitability also occurred. At P0–2, but not later, D2-MSNs in the matrix compartment of R6/1 mice exhibited early signs of maturation, manifested by more robust AP firing, more hyperpolarized resting membrane potentials, larger cell capacitance, and increased complexity of dendritic fields compared to WT MSNs, supporting the idea that this specific striatal circuit matures more rapidly in HD mice. This finding is in contrast with the general observation that CPN development seems delayed and emphasizes differential changes in cortex and striatum in terms of the rate of maturation. It is worth noting that a proof of developmental delay in striatal neurons was found in a cell culture model in which YAC128 (a full-length HD model) and WT striatal neurons were co-cultured with cortical neurons. 71 This study demonstrated that cortico-striatal connectivity in WTs develops rapidly and continuously from 7 to 21 days in vitro (DIV), whereas YAC128 connectivity showed no significant growth from 14 DIV onwards. This effect implicated a reduction in post-synaptic dendritic arborization and the size and replenishment rate of the presynaptic readily releasable pool of excitatory vesicles. 71
With regard to glutamatergic synaptic activity, a reduction in the amplitude, but not the frequency, of glutamatergic cortico-striatal inputs was observed in R6/1 D2-MSNs at the beginning of the second postnatal week, 50 suggesting a postsynaptic reduction in AMPA receptors, as also demonstrated in the HdhQ111 mouse model. 11 These studies indicate that deficits in glutamatergic inputs occur early in HD models and become more prominent with disease progression, as shown in early and late symptomatic mice. 56 Interestingly, in spite of a number of developmental alterations, no behavioral symptoms could be demonstrated in R6/1 mice, at least during early development. 50
Interneuron development in HD
Cortical and striatal interneurons are primarily derived from the medial and caudal ganglionic eminences (MGE and CGE) during embryogenesis. The MGE predominantly gives rise to PV- and somatostatin (SST)-expressing interneurons, whereas the CGE generates interneurons expressing other markers such as vasoactive intestinal peptide (VIP) and calretinin.72,73 These interneurons undergo tangential migration into the cortex or radial migration into the striatum between embryonic day E11 and E18 in mice.74–76 PV and SST interneurons are the two most abundant classes of cortical inhibitory neurons, comprising approximately 40% and 30% of GABAergic interneurons, respectively, in mouse cortex. 77 While PV interneurons have been traditionally associated with critical period plasticity and exhibit rapid postnatal development of fast-spiking properties, emerging evidence suggests that SST interneurons may integrate into cortical circuits earlier, receiving strong thalamic input and expressing key synaptic regulators during the second postnatal week. In the striatum, fast-spiking PV interneurons and low-threshold spiking SST interneurons regulate MSN activity and are critical for shaping output from the basal ganglia.78,79 Studies on the early developmental trajectories of cortical and striatal interneurons in HD are still scarce. However, our studies in R6/2 mice demonstrating early deficits in GABAergic synaptic inputs onto cortical and striatal projection neurons anticipate abnormalities in local circuit interneurons. In a landmark study, 80 the authors demonstrated that conditional ablation of HTT from cells expressing subpallial markers (Gsx2-Cre for MSNs, SST, and calretinin interneurons or Nkx2.1-Cre for forebrain PV, SST and cholinergic interneurons) displayed patterns of basal ganglia degeneration similar to that seen in human HD. Early deficits of SST and Reelin interneurons as well as age-dependent loss of PV-positive neurons in Nkx2.1-Cre mice also were reported. Not surprisingly, interneuron loss led to frequent occurrence of epileptic seizures in conditional mice, which also are typical in JOHD and in the R6/2 model.
Based on this and other studies, we could postulate that GABAergic interneurons play a key role in cortical hyperexcitability. Indeed, although diverse classes of interneurons are spared in HD, they undergo altered intrinsic and synaptic properties.81–84 Notably, histological analyses in R6/2 mice and human HD autopsy cases reveal a reduction in peri-somatic inhibitory synaptic contacts on layer II/III CPNs. 48 Loss of PV interneurons also occurs in the striatum. 85 Further, based on altered GABAergic synaptic activity in both cortex and striatum, it could be speculated that disrupted interneuron development may contribute to early circuit imbalance in cortico-striatal networks. Disrupted excitatory/inhibitory balance has been demonstrated in cortical circuits, with very early developmental deficits, followed by upregulated GABA inhibition, ending in a progressive reduction of GABAergic inputs.28,46,47 In striatum, early deficits are followed by progressive upregulation during the symptomatic stage.28,56,86
The existence of biphasic changes in synaptic activity and dynamic changes in excitatory-inhibitory balance leads us to revisit the question of what causes epileptic seizures in JOHD and in some genetic animal models? During early development, initial deficits in GABA synaptic activity quickly recover and revert to upregulation. 87 At this stage, only application of GABAA receptor antagonists (e.g., picrotoxin or bicuculline) can reveal cortical hyperexcitability. Thus, what keeps hyperexcitability in check is an early upregulation of cortical inhibition. However, with disease progression there is a loss of inhibitory contacts on CPNs and this failure of inhibition leads to epileptic activity and eventual neuronal loss. The striatum also mounts countermeasures to reduce cortico-striatal overexcitation but eventually it also fails. However, these changes appear to be rooted in abnormal cortical development, i.e., FCD-like architecture, as well as reduced expression of FOX1 family alternative splicing regulators.
In addition, given the central role of PV and SST interneurons in regulating oscillatory activity and maintaining excitatory/inhibitory balance, their disruption in HD likely contributes to early dysfunction of cortico-striatal circuitry.88–90 In mouse models, aberrant gamma and beta oscillations have been recorded in both the cortex and striatum prior to overt neurodegeneration, indicating early network imbalance.91,92 Reduced synchrony and impaired phase coupling between cortical and striatal regions suggest compromised information flow that parallels early behavioral impairments such as motor inflexibility and anxiety.93,94
Therapeutic insights and future perspectives
Recent insights into abnormal neurodevelopment in HD have opened a new perspective on how to approach underlying mechanisms of disease and unveil timely therapeutic strategies. A perfect example is the use, soon after birth, of an Ampakine to rescue reduced glutamate AMPA receptor-mediated synaptic transmission. 11 Another possible approach is targeting abnormal alternative splicing. It has been suggested that decreased expression of RBFOX family of splicing factors might be one of the earliest alterations underlying the mis-splicing signature in HD. Accordingly, correcting the expression of Rbfox genes might have a higher impact in normalizing alternative splicing than that of other splicing factors. 37 Indeed, moderate overexpression of RBFOX1, but not other splicing factors such as U2AF2, corrects multiple HD-associated mis-splicing events and alleviates HD mice neuropathology and motor symptoms. 95 It will be important to elucidate whether overexpressing RBFOX3 has similar therapeutic effects, considering that it is a neuron-specific splicing factor, plays a critical role in neuronal differentiation, and is reduced during early development in HD models. 28
Another potential target is the GABA interneurons, in particular the PV-expressing fast-spiking interneurons. The developmental trajectory of interneurons is increasingly recognized as a potential contributor to circuit dysfunction, particularly in JOHD. Disruption of interneuron specification, migration, and maturation may precede and even shape the subsequent degeneration of projection neurons. Investigating the early-life trajectory of GABAergic interneurons, and their functional integration into cortico-striatal circuits, may yield valuable insights into the pathophysiology of HD and identify new therapeutic windows for early intervention.
While early intervention in affected individuals has always been emphasized by the research community, one surprising finding is that the developing HD brain has a unique ability to counter alterations that could potentially harm the organism, similar to the concept of anticipatory homeostasis. Regardless of the animal model used, or the timing of morphological and functional alterations, by the end of brain development the morphology and electrophysiology of cortical and striatal neurons appear indistinct from WT animals. This suggests the existence of compensatory mechanisms that prevent phenotypic manifestations for long periods of time, in the case of AOHD,96,97 but could be less effective in the case of JOHD. We could hypothesize that HD brains can sense that aberrant development may lead to deleterious consequences later in life. In response, an anticipatory compensation occurs in the cerebral cortex by means of increased GABAergic inhibition to diminish cortical hyperexcitability. In the case of human AOHD, it takes many years before overt symptoms become apparent. However, when cortical inhibition falters, symptoms emerge, starting with cognitive and psychiatric disturbances followed by overt motor symptoms.
The observation of biphasic changes in gross striatal morphology, i.e., early hypertrophy followed by rapid decline in children carrying the HD mutation is very revealing21,22 and may be a prime example of homeostatic anticipation. Other biphasic changes also occur including increased glutamate release followed by significant reductions, 98 early deficits in GABA transmission followed by upregulation,28,86 exuberance of dendritic fields followed by reductions in D2-MSNs. 50 Discovering and promoting intrinsic compensatory mechanisms, in conjunction with early intervention, will aid in finding the best therapeutic approaches for HD.
Footnotes
Acknowledgments
We would like to acknowledge Pr. Michael S. Levine for his continuous support, critical comments, and encouragement.
Funding
This work was supported by USPHS grant NS111316 (CC).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.