
Abstract
Biological materials composed of extracellular matrix (ECM) or its components have been successfully used for tissue repair and reconstruction. Preclinical studies, along with a cohort study following stage T1A esophageal adenocarcinoma (EAC) resection, have shown that ECM biomaterials can restore esophageal mucosa and submucosa without cancer recurrence. However, the molecular mechanisms underlying these effects remain largely unexplored. The present study investigates the in vitro effects of ECM degradation products from nonmalignant esophageal (eECM) and urinary bladder (ubECM) sources on EAC cell proliferation, migration, and associated signaling pathways. Both eECM and ubECM significantly inhibited OE33 cell proliferation, with eECM exhibiting a stronger effect—reducing proliferation to 25% at 24 h and 7% at 72 h compared with pepsin control (p < 0.001). A high-throughput cell surface marker screen followed by gene and protein expression analysis revealed that both ECM sources downregulated CD164 and CXCR4, reducing CXCR4 protein levels by approximately 50% (p = 0.006 for eECM, p = 0.007 for ubECM). Notably, only eECM significantly suppressed OE33 cell migration (p ≤ 0.0001) and downregulated bone morphogenetic protein 4 BMP4 expression, along with its downstream targets pSMAD1/5/8, ID2, and SNAI2, thereby reducing epithelial–mesenchymal transition. These findings support the concept that biochemical cues from nonmalignant ECM modulate neoplastic cell behavior. Given the involvement of PI3K-Akt and BMP4 signaling in EAC progression, ECM-based strategies may warrant further investigation as potential therapeutic approaches following esophageal cancer resection.
Impact Statement
The extracellular matrix (ECM) and degradation products thereof have been shown to promote a healthy homeostatic state in both normal and injured tissues. Similarly, various formulations of ECM have been shown to mitigate metaplastic and neoplastic transformation of cells in certain types of cancer, including esophageal adenocarcinoma. The molecular mechanisms responsible for these effects are largely unknown. The present study investigated the in vitro effects of ECM degradation products on esophageal cancer cell proliferation and migration and associated signaling pathways.
Keywords
Introduction
The tissue organizational field theory (TOFT) is based on the premise that cell phenotype is a product of the microenvironment. 1 A primary component of the tissue microenvironment is the extracellular matrix (ECM), including the structural and functional molecules that constitute the ECM. ECM can be isolated from tissues through the removal of cells from allogeneic or xenogeneic source tissues, such as small intestine, 2 urinary bladder, 3 dermis, 4 and esophagus; 5 i.e., the process of decellularization.6–8 Such ECM materials have been used as structural and inductive bioscaffolds for a variety of therapeutic applications: body wall repair,9–11 augmentation cystoplasty,12–14 skeletal muscle,15–18 esophageal reconstruction,19–22 myocardial repair,23–25 and dura mater replacement,26,27 among others. 8 These ECM biomaterials have been formulated as surgical meshes (sheets), powders, and hydrogels.8,28
The exposure of neoplastic cells to ECM from decellularized healthy tissues has repeatedly been shown to induce a reversion toward an epithelial cell differentiation or reduce tumor growth in vitro and in vivo.29–34 In a clinical cohort study of five patients with grade T1A esophageal adenocarcinoma (EAC), ECM sheets facilitated the restoration of a functionally normal mucosa when placed at the site of cancer resection without the recurrence of neoplasia for a period of at least 4–24 months. 20 The ECM bioscaffolds completely degraded within 2 weeks in all patients, suggesting that ECM degradation products influence the tissue remodeling outcome in the postcancer niche and postsurgical resection. ECM degradation products, specifically eECM, showed a robust decrease in neoplastic esophageal cell proliferation, PI3K-Akt, and cell cycle/DNA replication signaling and showed minimal or opposite effects on nonmalignant esophageal epithelial cells in vitro, which would be favorable for a therapy delivered to a diseased esophagus. 33 Similarly, an orally administered hydrogel form of ECM was associated with the reversion of Barrett’s esophagus (BE), a metaplastic precancerous condition, in a canine model of gastroesophageal reflux. 5 The mechanism(s) by which these ECM-mediated effects occur are largely unknown.
The present study investigated the effect of ECM degradation products derived from homologous healthy esophageal tissue (eECM) and heterologous healthy urinary bladder tissue (ubECM) on OE33 neoplastic esophageal epithelial cells. Candidate cell surface receptors were identified, and related signaling pathways were investigated for their potential role in cell proliferation and migration.
Methods
Preparation of ECM digests and cell culture
Esophageal mucosa ECM (eECM) and urinary bladder ECM (ubECM) were prepared by decellularizing porcine tissue of market weight pigs. Tissue-specific decellularization protocols were used as previously described35,36 to meet the criteria of decellularization. 7 Powdered ECM (10 mg/mL) was digested in a pepsin buffer (1 mg/mL pepsin in 0.01 M HCl) for 48 h and then neutralized to physiological pH with 0.1 M NaOH. Pepsin control (1 mg/mL) was similarly prepared without the addition of ECM and neutralized.
The human EAC cell line, OE33 (Sigma-Aldrich), was cultured according to the manufacturer’s specifications in Roswell Park Memorial Institute 1640 medium containing 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 1% penicillin/streptomycin. Cells were cultured in a two-dimensional (2D) monolayer at 373,737°C in 5% CO2.
OE33 cell proliferation
Cell proliferation was measured using a 5-bromo-2-deoxyuridine (BrdU) colorimetric immunoassay (Roche), following the manufacturer’s instructions. Briefly, OE33 cells were seeded in a 96-well plate at 104 cells/well. After 18 h, the cells were treated with media, supplemented by ubECM (250 µg/mL), eECM (250 µg/mL), or pepsin control (25 µg/mL). BrdU was added 2 h before the endpoint. BrdU incorporation during DNA synthesis was measured by absorbance at 370/492 nm using a spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Proliferation was normalized to the medium group, with assays performed in quadruplicates, each performed in three biological replicates (n = 3).
OE33 cell migration
Cell migration assays used a 48-well chemotaxis chamber with 8 μm polycarbonate filters (Neuro Probe) coated with 0.05 mg/mL Collagen I. The bottom wells contained ubECM (250, 400 μg/mL), eECM (250, 400 μg/mL), or pepsin control (25, 40 ug/mL), while the top wells had 5 × 104 OE33 cells. After 4 h, the nonmigratory cells were scraped from the top, and the migratory cells were fixed in 95% methanol for 5 min, stained with DiffQuik Stain, and imaged with a brightfield microscope. The migrated cells were quantified manually. Experiments were performed with four technical and three biological replicates (n = 3).
Flow cytometry analysis
OE33 cells (1.5 × 106 cells/flask) were seeded in a T175 flask overnight. ubECM (250 µg/mL), eECM (250 µg/mL), or pepsin control (25 µg/mL) was added for 24 h. The cells were collected with AccutaseTM, washed twice with ice-cold PBS, and cell viability was assessed by Trypan blue staining using a hemocytometer. Cell surface profiling was performed as previously described.37,38 Nonspecific binding sites and Fc receptors were blocked with mouse serum for 20 min, and the cells were divided in equal parts for fluorescence-minus-outcomes and DAPI only controls. The cells were divided into a 96-well plate containing FITC, PE, and APC-conjugated antibodies specific to 228 cell proteins (BP80394, BDT FACSCAP Lyoplate, BD Biosciences). Plates were incubated in the dark for 30 min, washed with PBS, and centrifuged at 500 × g for 7 min. Cells were fixed with 2% methanol-free formaldehyde for 20 min, permeabilized with 0.05% saponin in indirect fluorescence antibody (IFA) buffer, and washed with IFA buffer. Finally, DAPI was added to the cells, and flow cytometric data were collected on a Fortessa SORP flow cytometer (BD Biosciences), as described previously. 39 The instrument was set up using CS and T beads (BD Biosciences), and PMT voltages were adjusted to predetermined target channels using the seventh peak of an eight-peak Rainbow Calibration Particles (Spherotech, Lake Forest, IL, RCP-30-5A) as a reference point. FITC, APC, and PE Calibrite beads (BD Biosciences) and single-stained antibody capture beads (BD Biosciences) were used as spectral compensation standards, along with cells stained with DAPI alone. Listmode data were analyzed with Venturi I software (Applied Cytometry, Dinnington, UK). The complete expression profile results can be found in Supplementary Data.
CD164 and EGFR single stain were performed for confirmation. Titration experiments were performed for CD164 and EGFR to optimize the concentration of the antibodies (Supplementary Fig. S2). The cells were collected with AccutaseTM and washed three times with PBS. The cells were first stained with Zombie NIR™ Fixable Viability Kit, washed, blocked with mouse serum for 20 min at 4°C, divided into equal volume (about 3 × 105/tube), and stained with 10 μL CD164 (BD Biosciences, clone N6B6) or 10 μL CD164 EGF Receptor (BD Biosciences, clone EGFR.1) and unstained as a negative control at 4°C for 20 min. Cells were washed and then fixed with 0.5% paraformaldehyde before flow cytometric on the Fortessa flow cytometer. The instrument was set up using Quantum™ MESF beads (Bangs Laboratories, Inc.), and PMT voltages were adjusted to predetermined target channels using the seventh peak of an eight-peak Rainbow Calibration Particles as a reference point. Data were analyzed using VenturiOne software (Applied Cytometry Systems, Dinington, UK), as described previously. 40
qPCR analysis
OE33 cells (3 × 105 cells/well) were seeded in a 6-well plate overnight and treated with ubECM (250 µg/mL), eECM (250 µg/mL), or pepsin control (25 µg/mL) for 24 h. RNA was extracted using TRIzolTM and reverse transcribed with the high-capacity RT kit (ABI). qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) for target and housekeeping gene β-glucuronidase (BGUS) on a QuantStudio 6 Flex. Primer details are in Supplementary Table S1. Results were analyzed by the ΔΔCt and normalized to the pepsin control. Experiments were performed in technical duplicates with 3 biological replicates (n = 3).
Western blot
OE33 cells (106 cells/well) were treated with ubECM (250 µg/mL), eECM (250 µg/mL), or pepsin control (25 µg/mL) for 24 h. Protein was extracted with RIPA buffer and loaded onto a 4%–20% Mini-PROTEAN® TGX™ Gel (Bio-Rad). After transferring to PVDF, blots were blocked with 5% nonfat milk in TBS/0.05% Tween-20 at room temperature for 1 h and then incubated overnight at 4°C with primary antibody (Supplementary Table S2). After washing, blots were incubated with HRP-labeled secondary antibody (1:5000, Dako) for 1 h at room temperature. A signal was detected with Clarity™ ECL Substrate (Bio-Rad) using the Chemidoc Tough imaging system (Bio-Rad Laboratories, Inc.) and analyzed with ImageJ software (National Institutes of Health). Bands were normalized to β-actin or tubulin. Western blots were performed in triplicate (n = 3).
Statistical analysis
Data were analyzed with a one-way analysis of variance and post hoc Tukey test using GraphPad Prism 7.0 statistical software (GraphPad Software, Inc., San Diego, CA), with p < 0.05 considered significant. Data are reported as mean ± standard error of the mean unless otherwise stated.
Results
ECM influences cell behavior in part via cell surface-receptor-initiated events. 41 The effect of ECM degradation products on the proliferation, migration, and upstream cell surface receptor expression of an EAC cell line OE33 was investigated.
Both ubECM and eECM treatments reduced the proliferation of OE33 cells compared with pepsin control at 24, 48, and 72 h (p < 0.05) (Fig. 1A). At 24 h, the proliferation rate of OE33 cells decreased to 59 ± 12% (p = 0.002) with ubECM treatment and to 25 ± 2% (p < 0.001) with eECM treatment. At 48 h, the proliferation rate decreased to 42 ± 4% with ubECM treatment (p < 0.001) and to 9 ± 0.4% with eECM treatment (p < 0.001). At 72 h, the proliferation rate decreased to 40 ± 13% (p < 0.001) with ubECM treatment and to 7 ± 1% (p < 0.001) with eECM treatment. Pepsin and medium control were not different at the three timepoints. There was no noticeable difference in cell morphology with any treatment, but the cell density reduced with ubECM and eECM treatments compared with pepsin control (Supplementary Fig. S1).
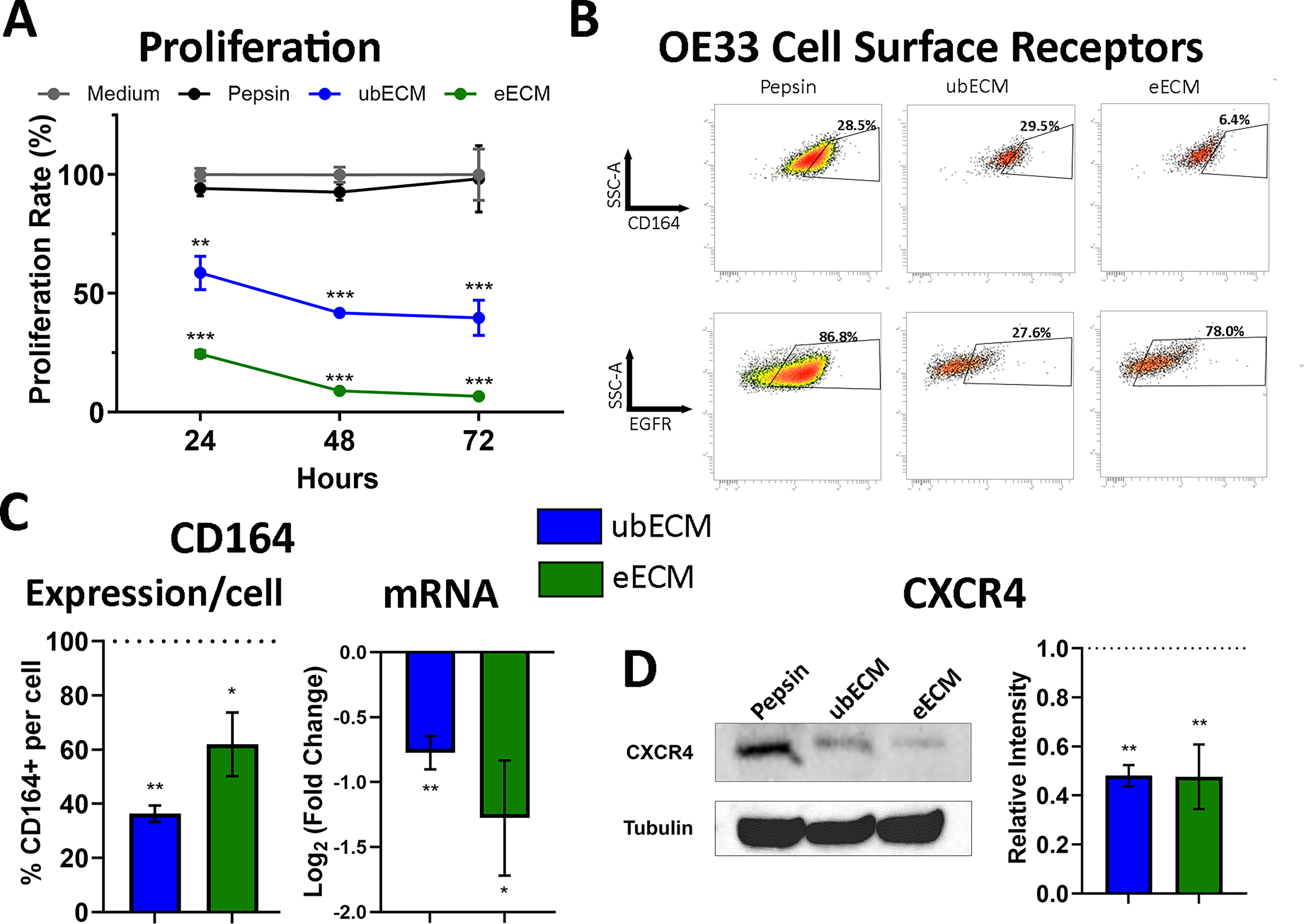
ECM degradation products inhibited the proliferation of the OE33 cancer cell and reduced CD164/CXCR4 expression. OE33 cells were treated with ubECM (250 mg/mL), eECM (250 mg/mL), or pepsin control (25 mg/mL).
A screening panel of 242 cell surface markers was performed on OE33 cells treated with pepsin, ubECM, and eECM to identify candidate cell surface receptors that changed with ECM treatment. The expression of EGFR and CD164 was reduced on OE33 cells after treatment with ECM degradation products (Fig. 1B) compared with pepsin control. eECM reduced the expression of CD164 (from 28.5% to 6.4%) and EGFR (from 86.8% to 78.0%) expression. ubECM reduced the expression of EGFR (from 86.8% to 27.6%) and had no effect on CD164 expression (from 28.5% to 29.5%).
The cell surface marker screening panel results were validated by two methods: single-stain flow cytometry and qPCR. The Quantum™ MESF kit (Bangs Laboratories, Inc., Fishers, IN) was used to quantify the number of surface protein receptors per OE33 cell. eECM reduced the expression of CD164 on the individual cell surface to 62 ± 12% (p = 0.01) and ubECM reduced to 36 ± 3% (p = 0.002) (Fig. 1C) compared with pepsin control. The results were corroborated by qPCR wherein eECM and ubECM reduced the expression of CD164 on OE33 cells (Fig. 1C). Gene expression was reduced to 45 ± 16% (p = 0.01) with eECM treatment and 59 ± 7% (p = 0.002) with ubECM treatment, each compared with pepsin control. There was no difference between the ubECM and eECM treatments on CD164 expression (p = 0.17 for flow cytometry, p = 0.32 for qPCR). There was no difference in EGFR expression of OE33 cells treated with ubECM or eECM degradation products compared with pepsin according to flow cytometry and qPCR results (Supplementary Fig. S1), and this receptor was no longer evaluated.
It has been shown that CD164 interacts with CXCR442,43 and that CXCR4 is an upstream regulator of the PI3K/Akt pathway. We have previously shown that ubECM and eECM degradation products have an inhibitory effect on the PI3K/Akt pathway. 33 After a 24 h treatment, western blot was performed on the OE33 lysates for CXCR4 (Fig. 1D) and showed that ubECM and eECM reduced the expression of CXCR4. The level of CXCR4 in OE33 cells treated with ECM degradation products was approximately half the level in cells treated with pepsin control (p = 0.006 for eECM, p = 0.007 for ubECM) (Fig. 1D). There was no difference between the ubECM and eECM treatment groups (p = 0.98).
The Boyden chamber assay was performed to determine the effect of ECM treatment on OE33 cell migration. There was a decrease in cell migration with eECM treatment compared with pepsin control at 250 μg/mL (p ≤ 0.0001) and 400 μg/mL (p ≤ 0.003) (Fig. 2A, B), but there was no difference between ubECM (250 μg/mL) treatment and pepsin control (p = 0.08). Only the highest concentration of ubECM (400 μg/mL) had an inhibitory effect on the migration of OE33 cells (p ≤ 0.03). However, the cells at this high ECM concentration also showed low viability, and inhibition of migration could not be separated from cell death. This high concentration was not tested in the following experiments. In all, eECM was more effective at inhibiting the migration of OE33 cells compared with ubECM.

eECM inhibited OE33 cell migration via the BMP4 pathway. The direct effect of ubECM or eECM degradation products on the migration of OE33 cells was evaluated in a Boyden chamber assay for 4 h of treatment (n = 2, with three technical replicates and three images per sample).
Bone morphogenetic protein 4 (BMP4) is highly expressed in EAC 44 and is an upstream signaling molecule in epithelial–mesenchymal transition (EMT), an important regulator of EAC cell migration. 45 eECM (250 μg/mL) reduced the expression of BMP4 (fourfold, p < 0.001) in OE33 cells (Fig. 2C). ID2, a downstream target of BMP4, was inhibited in OE33 cells treated with eECM (1.3-fold, p ≤ 0.004) (Fig. 2C). Phosphorylated SMAD (pSMAD) 1/5/8 is another downstream target of BMP4, and eECM degradation products inhibited pSMAD 1/5/8 activation by western blot analysis (1.7-fold, p ≤ 0.02) (Fig. 2D).
Snail2 is a transcriptional repressor that directly binds to the E2-Homeobox promotor region of E-cadherin, leading to E-cadherin downregulation. Snail2 activates the expression of vimentin by an indirect mechanism. 46 eECM downregulated SNAI2 (Fig. 2C) (1.4-fold, p = 0.04), which is consistent with downregulating EMT. The gene expression of hallmark EMT markers was evaluated including the epithelial marker E-cadherin (CDH1) and mesenchymal marker Vimentin (VIM) after ubECM and eECM treatments. eECM showed a small increase in CDH1 (1.4-fold, p = 0.048) and decreased VIM (2.1-fold, p = 0.01) compared with pepsin control (Fig. 2C). There was no significant difference in CDH1 (p = 0.91) and VIM expression (p = 0.99) in the ubECM treatment group compared with pepsin control (Fig. 2C). These results are consistent with the results of the migration assay.
In conclusion, eECM and ubECM degradation products inhibited the proliferation of the OE33 cancer cell and reduced the CD164/CXCR4 complex upstream of the PI3K-AKT pathway. Furthermore, only eECM showed an enhanced effect on OE33 cells by also reducing the migration of OE33 cells, with correlated evidence to support the downregulation of the BMP4 pathway. The proposed mechanisms of ECM on OE33 cancer cell phenotype are summarized in Figure 3.
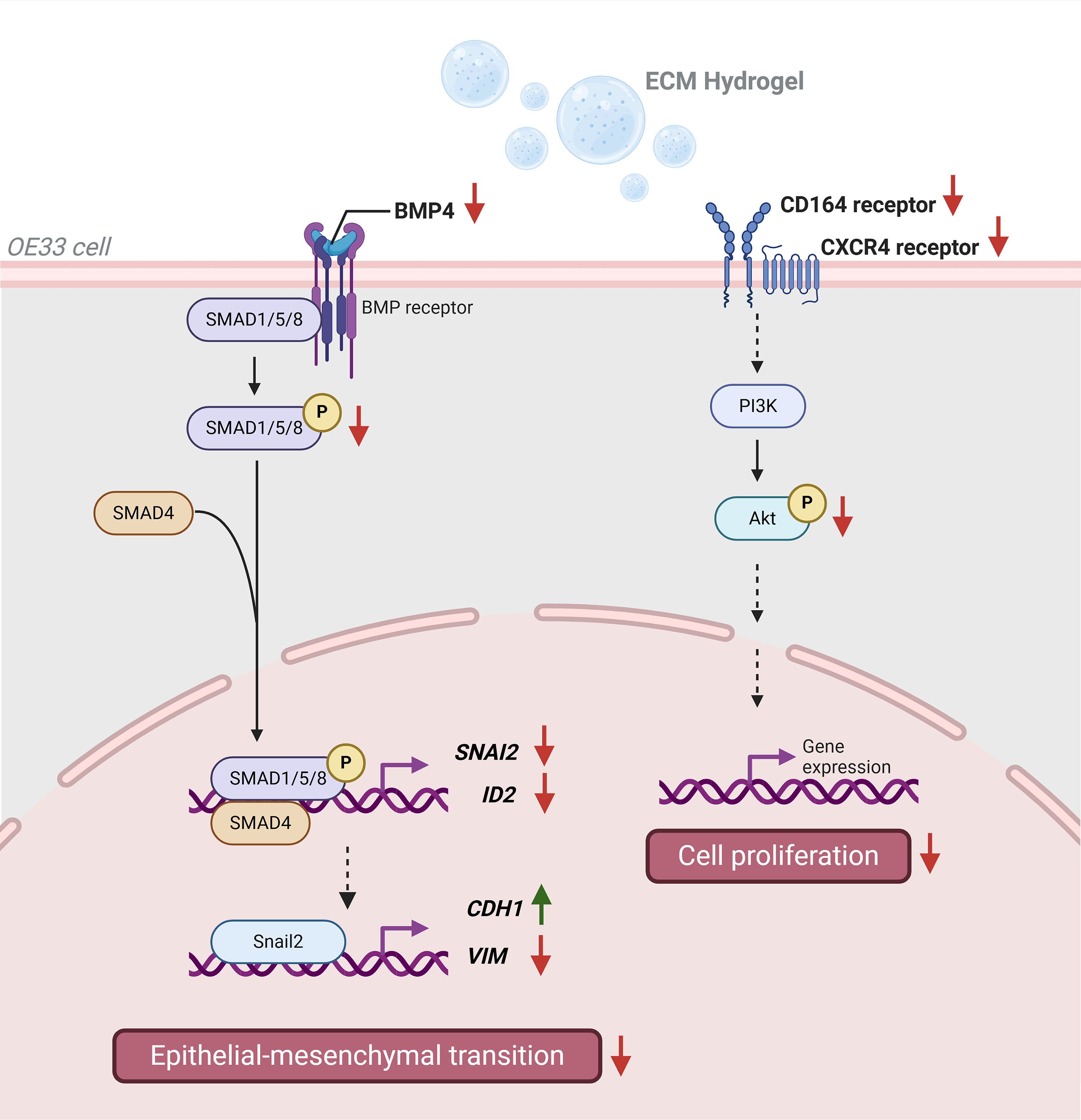
Proposed mechanism of ECM hydrogel-mediated downregulation of BMP4 and CD164/CXCR4 pathways. ubECM and eECM inhibited OE33 cell proliferation via the CD164-CXCR4 receptor complex upstream of the PI3K-Akt pathway. Only eECM inhibited OE33 cell migration via the BMP4 signaling pathway, which was associated with the suppression of epithelial–mesenchymal transition. Created with BioRender.com/book.
Discussion
The inflammation-driven transformation of normal esophageal mucosa to metaplastic mucosa and/or neoplasia is accompanied by marked changes in ECM structure and composition.47–49 Conversely, supplying nonmalignant (i.e., healthy tissue derived) ECM to a neoplastic microenvironment mitigates cancer cell behavior and phenotype, in both human esophageal cancer and a preclinical animal model.5,20,33 The intracellular pathways and specific signaling molecules that are involved in the ECM-induced effects on cancer cell phenotype have been largely unexplored.
The present study showed that degradation products of ECM, harvested from healthy urinary bladder and healthy esophageal mucosa tissues, both inhibited the proliferation of the OE33 cancer cell line, potentially via the CD164/CXCR4 receptor complex upstream of the PI3K-Akt pathway. However, only esophageal mucosa ECM inhibited the motility of these cancer cells and mitigated BMP4 signaling and the associated EMT events. Specifically, esophageal mucosa ECM downregulated the expression of BMP4, and its downstream targets pSMAD 1/5/8, ID2, and SNAI2. Snail2 is a critical transcription factor of EMT, further supporting the protective effects of degradation products from normal esophageal ECM. Hallmark events of EMT downregulation were shown with an increase in CDH1 and decrease in VIM gene expression. In addition, an enhanced tissue-specific effect was shown with solubilized normal eECM downregulating two tumorigenic signaling pathways and associated cell functions when compared with the effects of solubilized ubECM.
The continuous cross talk between ECM and resident cells is well established and has been referred to as “dynamic reciprocity.”41,50 TOFT extends the dynamic reciprocity concept and stipulates that normal ECM normalizes cancer cell phenotype. 1 The effects of ECM on cell phenotype have been attributed to ECM structure, 51 mechanical properties, 52 topographical features, 53 and specific biochemical components of the ECM such as laminin, fibronectin, and Lysyl oxidase.52,54 These variables, and the relative effect of each on cell phenotype, are not mutually exclusive. ECM degradation products were added to the media in the present study, suggesting that the observed effects were predominantly biochemical. However, it is important to consider that the physical properties of the media—particularly viscosity—may also contribute to cellular responses. Bera et al. demonstrated that elevated extracellular fluid viscosity enhances cell migration and promotes the dissemination of cancer cells, in both 2D and three-dimensional environments. 55 These findings suggest that ECM degradation products, which may retain sufficiently high molecular weight fragments, could alter media viscosity and thereby influence cell behavior through physical cues in addition to biochemical signaling. This consideration further supports the concept that ECM remodeling impacts the cellular microenvironment via both chemical and mechanical pathways.
ubECM and eECM showed similar yet distinctive effects on OE33 function and signaling pathways, which may be explained by the similarities and differences of the ECM protein profiles and biochemical components.5,34,56 These differences support the concept that ECM provides biological signals tailored to the tissue’s microenvironment that influence cell attachment, growth, and differentiation to maintain tissue homeostasis. 57
Consistent with this tissue-specific concept, ECM hydrogels demonstrate distinct rheologic properties and biological functions depending on their tissue of origin. 28 These facts reflect the distinctive cellular and extracellular environments of each tissue, as well as the processing methods required for hydrogel formation. 28 Moreover, factors such as the age and species of the tissue source can affect biological outcomes. For instance, degradation products from human fetal skin ECM showed greater chemoattractant activity for keratinocyte progenitor cells than those derived from adult human skin. Similarly, degradation products from porcine adult skin ECM showed greater chemoattractant potential than their human adult counterparts. Notably, human fetal skin ECM degradation products showed the strongest chemoattractant activity overall, supporting the concept that ECM scaffold remodeling involves the recruitment of lineage-directed progenitor cells by bioactive degradation products and both the developmental stage and species origin of the ECM source modulate this effect. 58
Adding another layer of complexity, matrix-bound nanovesicles (MBVs), which are ubiquitous components of the ECM, carry proteins and microRNAs that vary across tissues. A study examining six tissue sources—urinary bladder, small intestine, dermis, esophagus, cardiac, and liver—describes these differences. 59 Moreover, while MBV from various tissues share a baseline of cytokines, each MBV subset expresses a distinct set of cytokines. These findings suggest a regulatory role shaped by the tissue-specific cellular composition and function.
This work further reinforces the notion that ECM’s influence on cell behavior depends not only on its structural and mechanical properties but also on its tissue-specific context that drives distinct cellular responses. Indeed, ECM derived from central nervous system (CNS) tissue also shows distinct chemotactic and differentiation effects on neural cells compared with ECM from non-CNS sources, reinforcing the concept of site-specific bioactivity. 60 These observations support the broader principle that ECM bioactivity is highly context-dependent, governed by the unique combination of structural and signaling molecules retained from the tissue of origin. In the context of the present study, the inability of ubECM to modulate BMP4 signaling may reflect a lower abundance or absence of specific inductive cues that are preserved in eECM, likely due to its anatomical origin and functional role in esophageal tissue homeostasis.
The results of the present study also show that the effects mediated by ECM bioscaffolds act at least partially at the level of cell surface receptors, the point at which cells receive various cues from the microenvironment. The results identified the cell surface receptor complex CD164 and CXCR4 as being downregulated at the gene and protein levels in OE33 cancer cells following exposure to the degradation products of ECM. CD164 is a glycoprotein and a type I integral transmembrane sialomucin. 61 Relatedly, it is well established that mucin synthesis and composition change as an injury response to gastroesophageal reflux disease (GERD) 62 and sialomucins (nonsulfated acidic mucins) are selectively expressed in Barrett’s epithelium and absent in non-neoplastic epithelium. 63 CD164 is upregulated in several types of cancer, including colon and ovarian cancer, and is associated with increased ovarian epithelial cell proliferation, colony formation, and decreased apoptosis. However, less is known about the role of CD164 in EAC. CD164 forms a complex with CXCR4 upstream of PI3K-Akt signaling, 61 and it has previously been reported that ECM degradation products inhibit PI3K-Akt signaling in OE33 cells but not in non-neoplastic esophageal epithelial cells (Het-1A). 33 PI3K-Akt regulates cell proliferation, metabolism, and survival and is the most commonly dysregulated pathway in human cancers 64 including EAC. 65 Since PI3K-Akt is also activated in the wound healing response of nonmalignant cells, 66 ECM signaling molecules derived from healthy ECM may represent a therapeutic option for diseased esophagus.
A new mechanism by which eECM treatment can downregulate neoplastic esophageal cells was identified in the present study with downregulation of BMP4 signaling mediated EMT. BMP4 is a protein belonging to the transforming growth factor-β family and is a potent inducer of EMT. 67 BMP4 is increased during the progression of the GERD-BE-EAC sequence45,68 and regulates the early transdifferentiation of squamous esophageal epithelium to a columnar cell phenotype in BE. 69 Upon binding its transmembrane receptor, BMP4 signals by phosphorylating the cytoplasmic SMAD 1/5/8 protein, which translocates to the nucleus and increases target gene ID2 and the transcriptional repressor Snail2. Snail2 binds to the E-cadherin gene (CDH1) by the E2 homeobox promoter, thereby directly decreasing epithelial E-cadherin and indirectly regulating mesenchymal vimentin by an unknown mechanism. 45 The net effect of these intracellular mediators results in EMT, a process by which epithelial cells lose their cell–cell and cell–ECM connections, undergo multiple biochemical changes (e.g., reorganization of their cytoskeleton, reprogramming gene expression, etc.), and transdifferentiate to a mesenchymal phenotype that exhibits enhanced invasiveness.
The results of the present study show that eECM treatment markedly downregulates BMP4 and the associated intracellular mediators of the pathway including pSMAD 1/5/8, with smaller decreases in ID2 and SNAI2. Furthermore, eECM treatment showed a small increase in E-cadherin gene expression and decreased vimentin expression, consistent with a net downregulation of BMP4-mediated EMT. Interestingly, ubECM did not have the same effect on this pathway as eECM. It remains unknown whether a more complete EMT downregulation could be seen at later timepoints with eECM treatment but provides evidence that eECM can downregulate BMP4-mediated EMT as early as 24 h after treatment.
The present findings are in agreement with and expand upon a study showing the use of eECM to inhibit the PI3K-Akt pathway and the first to propose BMP4 as a possible signaling pathway affected by eECM treatment. 33 Understanding the molecular mechanism for ECM treatment of EAC may identify avenues for tailored treatments such as (1) selecting a patient population showing increased CD164/CXCR4 activation or increased BMP4 and EMT signaling and (2) selecting the route for administration. For example, targeting the diseased epithelium could be accomplished via an oral delivery of mucoadhesive ECM hydrogel that would maximize contact of ECM signaling molecules. The eECM hydrogel used in the present study was delivered orally in a canine model of BE, a precursor to EAC, and reverted the metaplastic epithelium in four of six animals within 30 d, suggesting proof of concept of this modality as a therapeutic strategy. 5
Finally, the results of the present study showed that 2 EAC signaling pathways were downregulated following eECM exposure to neoplastic esophageal epithelial cells, that is, a multifactorial treatment to normalize dysregulated cancer signaling pathways. The ECM has evolved since early metazoan evolution to regulate the stoichiometric balance of proteins, thereby mitigating the frequency of cancer formation beyond what might be otherwise anticipated. As Bissell and Hines argue in their review, 70 with more than 10 trillion cells in the body exposed to tumorigenic pressures, “it is a feat of evolutionary biology [that we do not get more cancer]” and attribute normal tissue microenvironments for this protective effect. In a similar vein, the concept of cancer immunosurveillance, proposed by Sir Frank Macfarlane Burnet in 1957, further contributes to this protective effect.71,72 Current biological agents to treat cancer are predominantly single protein antibodies or inhibitors and can develop cancer resistance by escaping to alternative signaling pathways. However, just as the biochemical complexity of native tissue can be an advantage, it also serves as a challenge for clinical translation that requires an understanding and ability to reproducibly manufacture ECM products from a biological source. 73 Having reproducible and validated read-out systems, such as changes in the cell signaling pathways identified in the present study, can become an important quality control metric in manufacturing.
There were limitations to the present study. First, only one cell type was evaluated, OE33 EAC cells. This cell type was selected based upon its previous use in the literature45,74 and upon a previous study in which OE33 cells showed the most pronounced phenotypic change with ECM treatment. 33 Second, the analysis of ECM-derived features, such as surface topography, stiffness, and porosity, in relation to cell phenotype is constrained by technical limitations inherent to standard culture formats, where the incorporation of intact ECM often results in inconsistent and nonquantifiable exposure and contact between cells and the matrix. Third, the changes in signaling molecules (CD164/CXCR4 and BMP4-mediated EMT) and cell function (proliferation and migration, respectively) were correlated, but causation was not shown. Future work would elucidate the definitive ECM-induced molecular mechanism of these signaling pathways in vitro and the translational relevance of these signaling pathways in vivo using preclinical models of EAC and/or patient specimens.
It is clear, however, from the results of the present study that both ECM types downregulate OE33 proliferation and CD164/CXCR4 expression upstream of PI3K-Akt signaling and a tissue-specific, enhanced effect was shown with eECM downregulating OE33 migration and BMP4-mediated EMT signaling. Understanding the mechanisms by which non-neoplastic ECM downregulates neoplastic cell phenotype is important to advance the therapeutic use of ECM-based materials for neoplasia.
Authors’ Contributions
X.L., D.J.R., and L.T.S. contributed to study design, data collection, data analysis, article drafting, and review/editing. L.Z. and L.M.Q. contributed to data collection and analysis. V.S.D. contributed to study design, data collection, and article review. S.F.B. supervised the study and contributed to study design, data analysis, article drafting, and review/editing.
Footnotes
Acknowledgment
The authors thank Prof. Dr. B.L.A.M. (Bas) Weusten for his helpful review of the article.
Author Disclosure Statement
L.T.S. is VP at ECM Therapeutics. S.F.B. is cofounder and CSO at ECM Therapeutics. X.L., D.J.R., L.Z., L.M.Q., and V.S.D. declare no interests.
Funding Information
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number F31CA210694 (L.S.) and the China Scholarship Council No. 201706940004 (X.L.).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.