
Abstract
Several recent advances in microphysiological systems or organ-on-chip technology have demonstrated its potential for replacing traditional in vitro and animal models in the coming years. Despite the physiological relevance and cost-effectiveness of organ chips, there are several hurdles that must be overcome for widespread adoption for biological studies. Many manufacturing and scalability challenges have been overcome by a transition from polydimethylsiloxane to thermoplastics. However, challenges have arisen in these sealed, brittle systems related to end-point tissue analyses, harvest, and high-resolution imaging, which is particularly difficult for multilayer organ chips. Here, we present low-cost organ chips that are fluidically sealed but demountable, fabricated using a cut-and-assemble method without the need for cleanroom technologies. We have validated the capabilities of this method by demonstrating the culture of human aortic smooth muscle cells and induced pluripotent stem cell-derived neural cells, encapsulated in gelatin methacryloyl (GelMA) hydrogel on chip, for up to 27 days. The three-dimensional (3D) culture layer of the organ chip was removed, and high-resolution images were obtained following immunostaining. Furthermore, these organ chips facilitate rapid redesign and manufacture for alternative tissue and/or interface systems. To the best of our knowledge, this is the first innervated organ chip with multiple removable cell culture layers, as well as the first humanized nerve–artery model that includes a 3D hydrogel culture. In future work, these unique features of our platform can be utilized for investigating the crosstalk mechanisms between different cell types in coculture.
Impact Statement
We present here a new method for fabricating low-cost demountable organ-on-a-chip platforms. This method leverages our recent cut-and-assemble method for layered three-dimensional organ chips comprised of gas impermeable thermoplastics.
Keywords
Introduction
In recent years, microphysiological systems (MPSs) have gained popularity due to their increased physiological relevance and tunability over traditional two-dimensional (2D) cell culture methods and animal models.1,2 Despite extensive development at the bench, organ chips have yet to be robustly adopted. As polydimethylsiloxane (PDMS) absorbs hydrophobic molecules and is highly gas permeable,3–5 many recent designs have pivoted away from PDMS toward brittle thermoplastics.6–9 The transition to more brittle materials, however, has introduced one major challenge: the inability to directly access the inner culture chambers for imaging or biological assays after initial seeding. 10 Although complex geometries and interfacing cell culture layers provide added physiological relevance, optical, proteomic, and genetic analyses of individual cellular components become a challenge. While some analysis techniques can be carried out via extraction of cells, this fails to preserve their spatial resolution and often results in considerable loss of sample, resulting in poor yield, especially in permanently sealed systems. Tissue constructs that are three-dimensional (3D) in the z-axis possess limitations in imaging resolution, especially for multilayer organ chips or organ chips with hydrogel-encapsulated cell cultures. A potential workaround, removable components that either utilize reversibly bonded components or clamping strategies, allow access to internal components at the experimental terminus.10,11 James Hickman and Michael Shuler, along with the Hesperos Inc. team, have highlighted the utility of demountable and reconfigurable systems across an extensive library of elegant designs and utilities.12,13 However, these systems rely on soft lithography and PDMS gaskets. Shah et al. have reported the use of silicone rubber gaskets in an organ-chip model of gut epithelial monolayers and microbiome in anaerobic conditions. 14 Toward more biomimetic models, interfacing 3D tissues will be required, but disassembly of models is critical for terminal tissue harvesting, spatial genetic or proteomic analysis, and high-resolution imaging.
Here, we present a novel method for 3D MPS platforms fabricated via our well-established laser-cut and assemble method (Fig. 1).6,7,9,15,16 This process was implemented to develop a neurovascular organ chip with interfacing cell culture layers that are fluidically sealed but demountable. The organ chip uses a rapid cut-and-assemble method previously established by our lab to fabricate laser-cut polymethyl methacrylate (PMMA), an FDA-compliant (CFR 177.1010) thermoplastic, and polyethylene terephthalate (PET) layers, as well as stereolithographic 3D-printed clear resin caps.6,17 After evaluating several elastomer materials based on cost, gas permeability, ability to be laser cut, and biocompatibility, we determined that Viton, an FDA-compliant fluoroelastomer (CFR 177.2600), is most suitable for use as gaskets in our chip (Fig. 2, Table 1). This organ chip uses standard hardware to fluidically seal the various layers, which allows straightforward rearrangement of layers for different experimental configurations and demounting. Each organ chip costs ∼$8–11 and can be easily manufactured using widely available makerspace equipment. This system has been validated with complex human neurovascular cocultures to create a model of the physiological nerve–artery interface. We demonstrated the utility of this design by immunostaining for neuronal catalysts, which have transient expression in neurons and therefore often require high-resolution imaging for visualization. In addition, we demonstrate the potential of our organ chip for multichannel imaging or multiplexed imaging experiments, where we stained and imaged the 3D (aortic smooth muscle cell culture) and 2D (human aortic endothelial cell culture) layers of the organ chip independently.
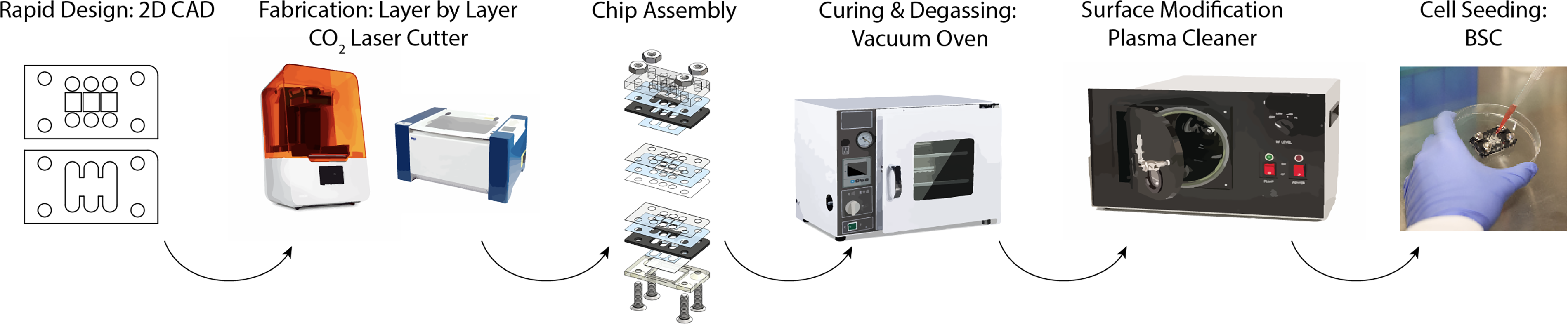
Laser-cut and assemble method for fabricating demountable 3D MPSs. After assembly, MPSs were degassed and plasma treated before seeding of cells for coculture experiments. 3D, three-dimensional; MPSs, microphysiological systems.
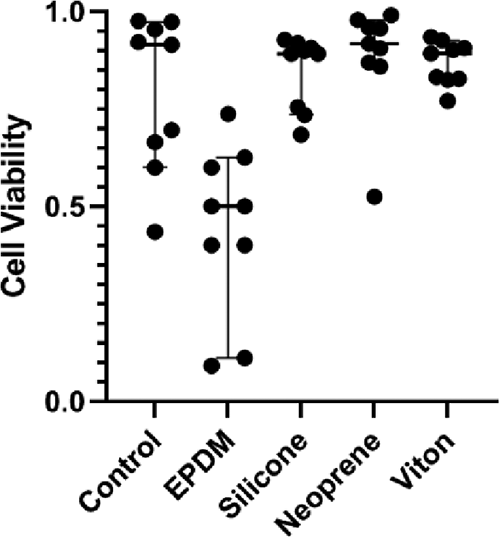
F11 cell viability cultured with elastomer samples for material selection.
Materials Selection Criteria for Elastomer Layer

EPDM, ethylene propylene diene monomer.
Bold is used to denote the material chosen for the reported microphysiological system.
Materials and Methods
Cell viability and material selection
F11 (rat dorsal root ganglia—mouse neuroblastoma hybrid) cells (Millipore Sigma #08062601-1VL) were grown in basic media composed of Advanced Dulbecco's Modified Eagle Medium (DMEM, cat: 12491023, Gibco), fetal bovine serum (10%, Atlanta Biologicals), penicillin–streptomycin (1%, cat: 15140122, Gibco), and GlutaMAX (1×, cat: 35050061, Gibco) between passage 26 and 40. Commercially available elastomers were evaluated for utility as an inert gasket layer (Table 1). Rubber samples were ordered in a sample pack (8450K11), including silicone, ethylene propylene diene monomer (EPDM), Neoprene, and Viton (1235N21), from McMaster Carr and cut into uniform square samples (10 × 10 mm). F11 cells were cultured in tissue culture-treated 24-well plates at a density of 0.5 × 106 cells/well with square samples for 7 days. Cells were then evaluated with a live/dead assay (ThermoFisher R37601), imaged, and quantified (Fig. 2).
Device fabrication
Neurovascular organ chip devices (Fig. 3) were constructed by a previously described cut-and-assemble method, with modifications (Fig. 1).6,17 Using a laser engraver system (Epilog Zing 16, Epilog Laser), double-sided adhesive tape (3M 966), 3/16″ PMMA (McMaster-Carr 8560K211), 0.0005″ PET (McMaster-Carr 8567K102), and 1/64″ Viton were cut to the shapes of the respective layers as shown in Figure 4. Each layer of the organ chip device was assembled as follows:

Demountable neurovascular MPS.
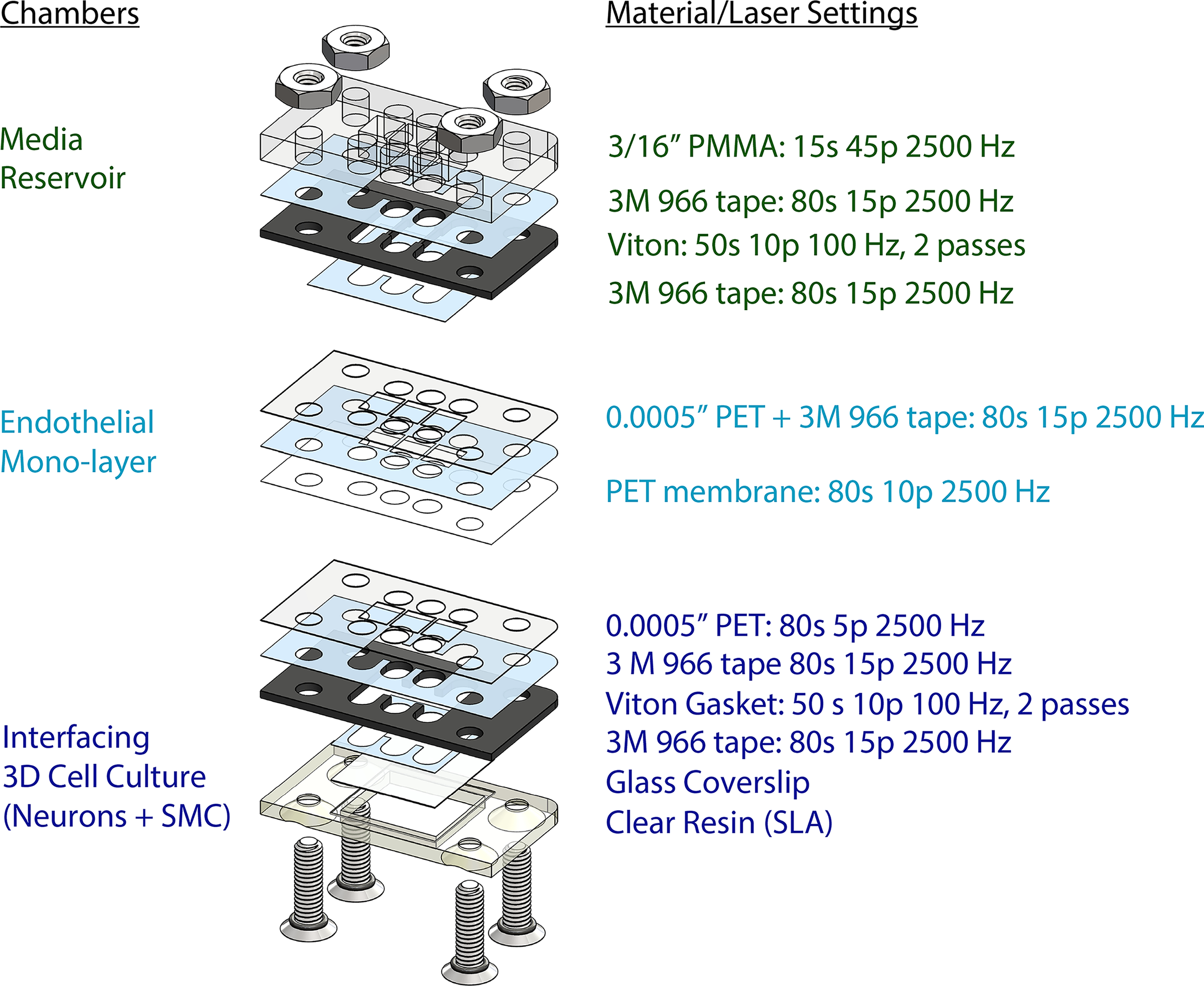
Layer-by-layer design of neurovascular chip and laser settings for cutting the different polymer materials throughout the chip assembly.
To establish a media reservoir, a Viton gasket piece was attached to a 3/16″ PMMA piece with a piece of double-sided tape. This assembly was heat-pressed at 53°C for ∼15 s. Then, a partial piece of double-sided tape was attached to the exposed side of the Viton gasket.
To establish a 2D cell culture layer, a double-sided tape sheet was attached to a sheet of 0.0005″ PET on one side. The assembly was carefully attached to a PET membrane with 1 µm pore size (it4ip, 2000M12/620M103). Then, the media ports of the assembly were laser cut (Fig. 4). The assembly was then heat-pressed at 53°C for ∼15 s. The partial piece of double-sided tape (which was previously attached to the media reservoir) was used to attach the 2D cell culture layer. Then, the media reservoir + 2D cell culture layer assembly was heat-pressed again at 53°C for ∼15 s.
To establish a 3D cell culture layer, a 0.0005″ PET piece (Fig. 4) was laser cut. A Viton gasket piece was attached to this PET piece with a double-sided tape, cut to the same shape as the Viton gasket. The assembly was then heat-pressed at 53°C. A partial double-sided tape piece, which includes an 18 mm × 18 mm portion and a border to be detached, was attached to a #1 thickness 18 mm × 18 mm coverslip. For attachment of the glass coverslip (Fisherfinest 12548A), double sided adhesive tape was cut to an 18 mm × 18 mm square with borders. The coverslip was attached to the center of the 18 mm × 18 mm tape, and the borders of the tape were then removed and discarded. The coverslip holder piece was fabricated by a stereolithographic 3D printer (Form 3B+, FormLabs) using Clear Resin (FormLabs). The coverslip with tape was then attached to the gasket on the assembly, and the assembly was placed on the stereolithographic (SLA) 3D-printed clear resin coverslip holder. The assembly was heat-pressed at 53°C for ∼15 s.
The three layers of the organ chip were assembled with 5/8″ stainless steel screws with o-rings (McMaster Carr 98070A162) and nuts (McMaster Carr 91841A009). Chips were tightened to 0.12 Nm with an Anpuds digital Torque Wrench (3 Nm) to ensure proper sealing without overtightening. Assembled organ chips were vacuum incubated at 53°C for at least 96 h. Then, organ chips were UV-sterilized and plasma-treated (Harrick Plasma PDC-001) for 180 s.
Human iPSC cultures and differentiation toward autonomic neuron populations
Human induced pluripotent stem cells (hiPSCs, iPS(IMR90)-4) were sourced from WiCell. Cells were maintained at standard cell culture conditions (5% CO2, 37°C) in mTeSR Plus medium (STEMCELL Technologies 100-0276) on 6-well plates (Corning 07-200-83) coated with 1:100 growth factor reduced Matrigel (Corning CB-40230) in DMEM-F12 (Gibco 11-320-033). Full media exchanges were performed daily. Accutase (Corning 25058CI) was used to disassociate cells when they approached confluency. ROCK inhibitor (Sigma Y0503) was added to cell culture media (10 μM) for ∼24 h after thawing, before passaging, and after passaging.
hiPSCs were differentiated following an adapted version of a protocol previously published by Takayama et al. 18 (Fig. 5). Briefly, iPSC at P18 + 42 were plated (Day −1) into a 6-well plate coated with 1:100 growth factor reduced Matrigel in DMEM-F12 at 2 × 105 cells/well and maintained at standard culture conditions (5% CO2, 37°C) in mTeSR Plus medium and 10 μM ROCK inhibitor. The following day (Day 0), the ROCK inhibitor media was removed. hiPSCs were then differentiated with three separate base media compositions: a knockout serum replacement (KOSR)-based medium, an N2 supplement-based medium (N2), and a B27 supplement-based medium (B27). The KOSR-based medium was composed of DMEM-F12, 20% KOSR (Gibco 10828010), 1% nonessential amino acids (Gibco 11140050), 0.5 mM monothioglycerol (Spectrum Chemical M11771), and 1% penicillin-streptomycin (Gibco 15140122). The N2-based medium was composed of DMEM-F12, 1% N2 supplement (Gibco 17502048), 1% nonessential amino acids, and 1% penicillin-streptomycin. Lastly, the B27 medium was composed of 50× B27 Plus Supplement (Gibco A3582801) diluted to 1× in Neurobasal-A (Gibco 10888022). The differentiation protocol was initiated on Day 0, and full media exchanges were completed every other day until Day 13, after which the frequency was every third day. On Day 16, medium was replaced for both sympathetic-like and parasympathetic-like cultures with B27 medium, and both cell types were maintained in B27 medium until culture termination.
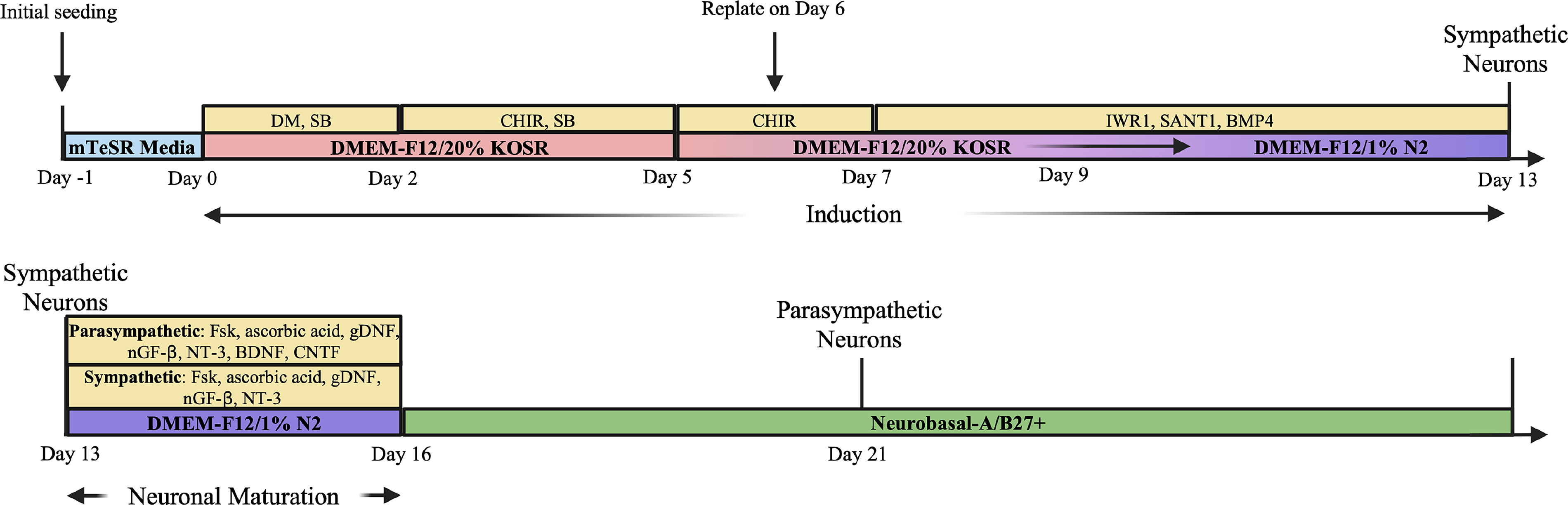
Differentiation protocol for sympathetic and parasympathetic motor neuron populations.
HAEC and HASMC cultures
Human primary aortic endothelial cells (HAEC) obtained from American Type Culture Collection (ATCC, PCS-100-011, Lot: 80923231) were maintained in Bovine Brain Extract (BBE) Endothelial Cell medium (Vascular Cell Basal Medium, ATCC, PCS-100-030 with BBE Endothelial Cell Growth Supplement, ATCC, PCS-100-040) at standard cell culture conditions (5% CO2, 37°C) in cell culture flasks.
Human Aortic Smooth Muscle Cells (HASMC) were obtained from ATCC (PCS-100-012, Lot: 70008916) and maintained in ATCC Vascular Cell Basal Medium with Vascular Smooth Muscle Cell Growth Kit (ATCC, PCS-100-042).
When cultures reached confluency, cells were dissociated with 0.25% v/v trypsin-EDTA (Gibco 25200056) and seeded at densities between 2500 and 7500 cells/cm2.
Neurovascular MPS seeding
Gelatin methacryloyl (GelMA) was synthesized as previously published, using fish gelatin. 19 GelMA was diluted to 5% w/v in a Day 5–7 medium solution with 0.5% w/v LAP. Day 6 NPC derived from hiPSC and P6-P7 HAMSC were each resuspended in 5% GelMA precursor solution, each at 5 × 106 cells per mL. Twenty-seven microliters of HASMC/GelMA precursor solution were seeded into the middle chamber of the organ chip and crosslinked for 45 s with blue light (405 nm, 10 W), with the glass side of the chip facing the lamp. Twenty-seven microliters of NPC/GelMA precursor solution were seeded into each outer chamber of the organ chip and crosslinked for 130 s with blue light, with the glass side of the chip facing the lamp. Medium was exchanged at ∼20 min after seeding to remove any uncrosslinked GelMA precursor solution. On Day 5 of culture, P5 HAEC were seeded at 5 × 105 cells/cm2 onto the middle 2D chamber.
Disassembly
For disassembly, first, the screws and nuts were removed from each MPS. The layers were then loosened by gently swiping a screwdriver along the edges of the assembly. The media reservoir + 2D layer was separated from the 3D layer by twisting the pieces away from each other and pulling them apart. The 2D layer was then gently pulled apart from the media reservoir (Fig. 6).

Demountable chambers support disassembly at culture endpoints.
Immunostaining
Following the disassembly of the organ chip, 3D layers of the organ chip were placed in a 6-well plate. Samples were rinsed with 1x HBSS in deionized water (Gibco 14065-056). Samples were then fixed in 4% paraformaldehyde (Thermo Scientific, 043368.9M) in HBSS for 40 min and permeabilized in 0.1% Triton X-100 (Thermo Scientific A16046.AP) in HBSS for 30 min at room temperature, each followed by three 10-min washes in HBSS. The samples were then blocked by 2.5% goat serum (Sigma-Aldrich G9023) in HBSS (blocking solution) overnight (∼16 h), followed by an overnight (∼16 h) incubation with primary antibodies diluted in the blocking solution, both at 4°C on a rocker. Following four 1-hour HBSS washes on a rocker to remove excess primary antibody solution, the samples were incubated with secondary antibodies diluted in blocking solution overnight (∼16 h) at 4°C on a rocker. Following four 1-hour HBSS washes on a rocker to remove excess secondary antibody solution, samples were incubated with DAPI (Invitrogen D1306) diluted 1:1000 in blocking solution for 10 min. Finally, three 10-min HBSS washes were performed on a rocker to remove excess DAPI solution.
The 2D layer was separated from the assembly (Fig. 6). The edges of the layer were cut, and samples were placed into a 12-well plate. Following a rinse with HBSS, samples were fixed in 4% paraformaldehyde in HBSS for 10 min and permeabilized in 0.1% Triton X-100 in HBSS for 10 min at room temperature, each followed by three 10-min washes in HBSS. The samples were then blocked by 2.5% goat serum in HBSS overnight (∼16 h), followed by a 2-hour incubation with primary antibodies diluted in the blocking solution at room temperature. Following three 5-min HBSS washes on a rocker to remove excess primary antibody solution, the samples were incubated with secondary antibodies diluted in blocking solution for 1 hour at room temperature. Samples were washed three times for 5 min each with HBSS to remove excess secondary antibody solution. Samples were mounted with Prolong DAPI (Invitrogen P36931) and a #1 thickness 18 × 18 mm coverslip. Samples were then sealed using nail polish.
All samples were imaged using an inverted fluorescence microscope (Zeiss Axio Observer). Images were processed using ImageJ/FIJI software.
Computational fluid simulations
The laminar flow of cell culture medium through the organ chip flow component was modeled in COMSOL. First, SolidWorks was used to create the 3D flow channel, which was then exported as an STL file and imported into COMSOL as the geometry. Fluid properties of water were used, with the viscosity manually changed to 9.35 × 10−4 Pa·s, averaging previously reported values, 20 to more closely model the viscosity of DMEM cell culture medium. Using the laminar flow module, no-slip walls and fully developed flow were selected, and a volumetric flowrate of 8 × 10−8 m3/s was set at the inlet (equivalent to 4.8 mL/min, a flowrate compatible with most peristaltic pumps, which are frequently used in vascular studies). Backflow was not suppressed at the outlet to model any possible recirculating fluid. A stationary study was run with meshing set to “normal,” and then, the shear stress was modeled by the input “spf.sr * spf.mu” to multiply the shear rate and dynamic viscosity.
Experiment
Elastomer selection and impact on viability
A materials selection matrix weighted for stability, gas permeability, cost, machineability (laser cutting), and optical transmission yielded four commercially available elastomers. Small samples were added directly to F11 2D cultures on well plates and evaluated for toxicity. Silicone, neoprene, and Viton did not impact viability compared to controls (Fig. 2), while EPDM negatively impacted F11 cell health. Viton was ultimately selected for its inertness, low gas permeability, and ease of manufacturing (Table 1).
MPS with removable layers
In our design, a combination of cut-and-assemble fabrication and SLA 3D-printed components were successfully utilized to construct a 3D neurovascular MPS, allowing us to construct precise geometries with tunability in the x-, y-, and z-axes. SLA was also used to fabricate the example flow component (Figs. 3 and 9), reducing the need for multiple tape layers, which therefore also reduces the possibility of leaks from loosening of the assembly. In addition, our design avoids any contact of cells directly with the 3D-printed components, thus reducing the importance of resin biocompatibility, as the cells do not directly contact the 3D-printed coverslip holder or flow components.
The neurovascular MPS includes three internal structures: a 3D cell culture layer, a 2D cell culture layer, and a media layer (Figs. 3 and 4). The 3D cell culture layer allows the contact of adjacent hydrogel culture chambers through GelPins, 17 forming a contiguous culture, and contains a glass bottom to facilitate high-resolution microscopy. In the 2D culture layer, cells are seeded on a semipermeable membrane, allowing contact with the hydrogel-encapsulated cells below. Fabricated components are assembled layer-by-layer, employing 3M 966 double-sided adhesive tape, which has been well-established as biocompatible and suitable for cell culture through our previous work.6,7,9,16,17,21 This is evidenced by robust cell morphology at culture endpoints in this study, and furthermore, no delamination was observed over the 27-day culture period. The three layers are stacked with a 3D-printed piece at the bottom of the assembly and held together using stainless steel screws and nuts. Sealing hardware was tightened to 0.12 Nm to ensure alignment of all assembled layers and avoid overcompression or warping of materials. Components were checked regularly and retightened as needed. This ensured no observable leaks occurred across all chips (n = 11) over the 27-day culture period. The three layers of the organ chip were easily removed from each other by disassembling the hardware and gently pulling the layers apart (Fig. 6), allowing direct access to each individual cell type (Fig. 7).

Demountable platform facilitates endothelial, smooth muscle, and neuron cultures for up to 27 days on chip.
Immunostaining and visualization of neural outgrowth in 3D culture
Organ chips with sympathetic or parasympathetic neurons were disassembled on Day 19 or Day 27 of neuron differentiation, and the 3D culture layer was set aside for imaging (Fig. 7B). The 3D culture layers were then fixed, permeabilized, and immunostained. Samples were imaged using an inverted fluorescence microscope (10× objective) to visualize expression of β-III tubulin (B3T), a neuronal marker, and cell nuclei (counterstained with DAPI). Z-stack images were taken, and maximum intensity projections of slices 1–95 were generated of parasympathetic neurons after 27 days in culture (Fig. 7). As expected, expression of B3T was present, and the cells were distributed throughout the height of the hydrogel, as observed in the orthogonal views, demonstrating innervation of the chamber. Therefore, using our nerve–artery organ chip platform, we were able to demonstrate the visualization of expression and distribution of neuronal markers in differentiating neural cells on chip.
After immunostaining, organ chips were imaged using a 63× oil immersion objective (Fig. 8). For immunostaining, organ chips with sympathetic or parasympathetic neurons were disassembled on Day 19 or Day 27 of neuron differentiation, and the 3D culture layer was set aside for imaging. The 3D culture layer was then fixed, permeabilized, and immunostained for B3T (neuronal marker) and tyrosine hydroxylase (TH—sympathetic neuron marker; Fig. 8A) or choline acetyltransferase (ChAT—parasympathetic neuron marker; Fig. 8B) for organ chips with sympathetic or parasympathetic neurons, respectively, and cell nuclei were counterstained with DAPI. As the 3D culture layer of the chip can be easily maneuvered after removal from the overall chip assembly, we were able to image the chip easily with a 63× oil immersion lens. Qualitatively, there is a presence of TH in the sympathetic differentiation group on Days 19 and 27, while there is an increased presence of ChAT on Day 27 of the parasympathetic differentiation group compared to Day 19.
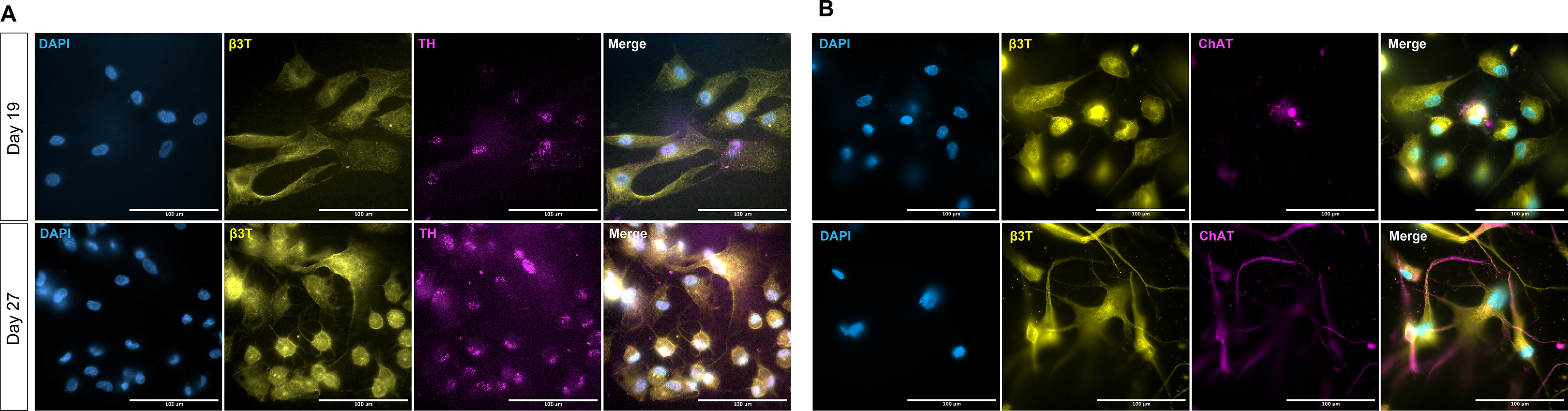
Demountable chambers support high-resolution imaging of 3D tissue chambers, 63× oil immersion, on a standard inverted fluorescent microscope, as demonstrated by staining of neural and
Computational fluid simulations demonstrate high and low shear stress arterial flow on chip
Computational fluid simulations were run in COMSOL software to demonstrate flow on chip (Figs. 3C and 9). Flow channels were designed in SolidWorks and exported to COMSOL for flow simulations. Use of CAD allowed rapid redesigning of the flow channels, and this was utilized to achieve a range of physiologically relevant flow rates and shear stresses by varying the height of the flow channel while preserving the rest of the geometry. A channel height of 0.25 mm was used for the high shear stress flow simulation, while a channel height of 0.5 mm was used for the low shear stress flow simulation. In the EC region (roughly outlined in black), a shear stress of ∼1.1 and 0.3 Pa was achieved over the endothelial region to model high and low shear stress in arteries. To demonstrate potential for application in benchtop experiments, an example flow attachment was 3D-printed by SLA and attached to the chip, and distilled water with blue food coloring was flown through by a syringe pump for visualization (Fig. 3C). Direct comparison of the high versus low shear stress shows relatively uniform shear across the cell seeding area (Fig. 9).
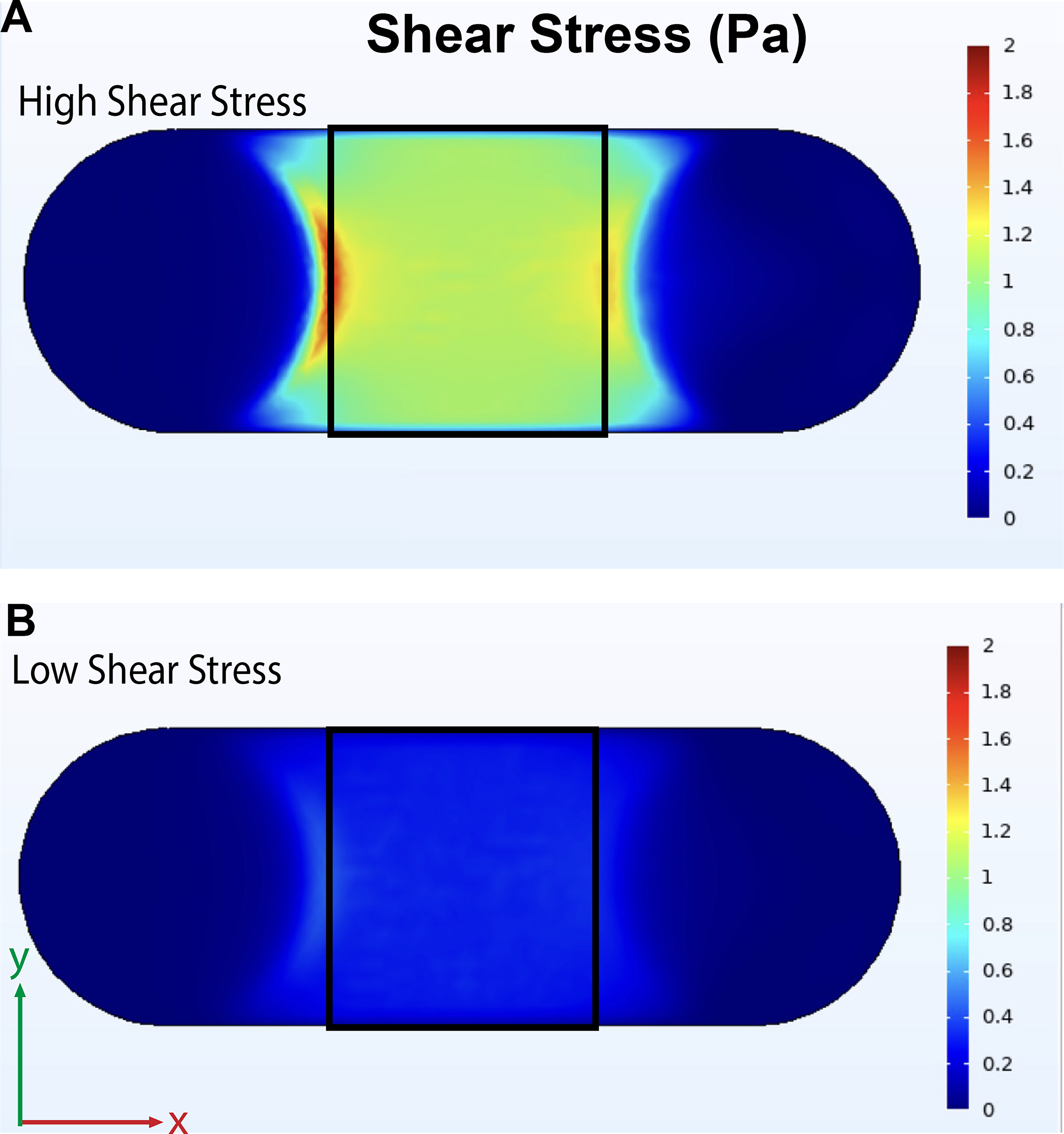
High and low shear stress arterial flow on chip. The view from the cellular region is shown, with the inlet on the left side.
Discussion
Future translation of MPS technologies relies on approaching the complexity and heterogeneity of in vivo tissue yet retaining ease of use, including capabilities for traditional microscopy techniques without the need for expensive specialized equipment.21,22 Our layer-by-layer approach to MPS assembly allows limitless fabrication of elements or chambers in the z-direction. While beneficial for increasing the complexity and biorelevance of these platforms, this engineering advantage is at the detriment of traditional optical microscopy techniques in irreversibly sealed devices. Therefore, demountable platforms allow high-resolution imaging with standard inverted fluorescent microscopes equipped with standard working distance objectives.
The removable interfacing of 2D and 3D cell cultures on our organ chip provide several advantages, from high-resolution endpoint analyses to inclusion of flow components. In addition to the direct advantages of higher imaging resolution and accessibility for endpoint assays, separation of 2D and 3D layers allows for specifying incubation times for specific live and/or fixed assays on each layer. Compared to a sealed 3D system, the demountable chambers also enable tissue-specific immunocytochemical staining for each chamber to allow for each filter cube/laser setting/secondary antibody to be utilized for each MPS section, providing expanded resolution of standard fluorescent microscopy. Several studies have employed multiplexed imaging for investigations in oncology research.23–25 However, only a few studies have explored multiparametric imaging on organ chips.21,26 Overall, the removable, interfacing chambers of our organ chip can be easily redesigned, expanded, and employed for understanding developmental mechanisms and disease pathophysiology, especially by employing multiple differentiating human cell types derived from stem cells.
The use of widely available CAD and CFD software, as well as common makerspace equipment, reduces time and cost of organ chip fabrication and redesigns. Because of these advantages, the use of 3D-printed parts for organ chips has recently been explored. Biocompatibility is a primary concern with using 3D-printed resins for cell culture, and there has been some investigation on material composition, resolution, and postprint processing for use in cell culture devices.27–30 Particularly, there have been several reports of the use of FormLabs Clear Resin, a methacrylate-based resin which we used here. Hart et al. have reported HL-1 (immortalized rat cardiomyocyte) viability of greater than 96% with FormLabs Clear Resin following a 70% isopropanol wash and a UV postcure. 31 However, others report lower viability and/or cell attachment to FormLabs clear resin after isopropanol washes and a UV postcure,32,33 including no significant growth of human umbilical vein endothelial cells after 24 h. 34 As our organ chip design avoids direct contact of cells with the FormLabs clear resin, possible biocompatibility concerns are mitigated. 3D printing may contribute to bridging the gap to widespread adoption of organ-on-chip, though further standardized studies on biocompatibility and other designs avoiding contact of 3D-printed parts with cellular components must be explored.
Manually resealing the chip may cause slight changes in alignment and/or nonuniform load, and hardware components are prone to mechanical wear and tear, as previously discussed. 10 Affordable torque screwdrivers ($50–100) are valuable tools for dialing the proper torque to eliminate the risk of leaking, while not overtightening and compressing the assembly. A compressive force of 0.7 MPa was calculated (Supplementary Data) as a function of fastening torque and nut factor, well below measurable compressive strain for Viton.35,36 For our assembly, a torque setting of 0.12 Nm met this need and was readily checked over the duration of the experiment. Due to the thermal cycles of normal cell culture from incubation to room temperature feeding, changes in fastening torque were observed and required correction.
Cell culture inserts offer one solution for demountable devices,14,37–41 some of which include commercially available Transwell® cell culture inserts.42–52 Transwell inserts are a standard approach for membrane-separated systems, and their inclusion in organ chip devices enhances reproducibility and also allows the addition of flow. 42 Several cell culture insert-based organ chips have components that can be autoclaved and/or reused,44–46,49 and several allow increased throughput,40,45,46 which may both improve reproducibility and reduce the time needed for repeated device fabrication. However, these systems are limited in their ability to recapitulate 3D tissues and impart controllable flow regimes to apply physiologically relevant shear stress. Further development of organ chip systems with rapidly produced or commercially available components, especially those that incorporate 3D cell cultures, may improve accessibility and utility of organ chip platforms for mechanistic studies.
As suggested by Teixeira Carvalho et al., our organ chip provided a physiologically relevant environment for stem cell differentiation by controlling the spatial orientation of cell types. 10 In addition, all cell culture chambers can be directly accessed for endpoint assays, which is often not possible in permanently sealed systems. This feature can be further utilized to extract the cells from the organ chip, especially by using hydrogels compatible for such assays. For example, RNA extraction methods have been reported from several clamped organ chips. 10 Some methods of protein extraction have also been reported for hydrogel encapsulated cells but are comparatively limited.37,53 Application and further development of these methods for the organ chip could be very useful for mechanistic investigations of multiple cell types in coculture.
Though we demonstrated a neurovascular culture in this work, the applicability of the organ chip presented here is not limited to the nerve–artery interface. Various cell types can be cultured to model other vascularized or innervated systems, or other systems altogether. In addition, a membrane with larger pores could be employed to study cell migration in various environments, such as cancer cell migration, angiogenesis, or immune cell migration, in multitissue systems. Overall, the unique capabilities of this platform may be useful for mechanistic studies of complex multitissue systems.
Authors’ Contributions
S.B., A.K., G.D., and R.K. conceived the project. S.B., R.B., and L.A.-A. performed all experimental work and wrote the article. S.B. and R.B. designed and fabricated organ chips, and B.S. designed and optimized the fabrication of stereolithography-printed components. S.B. and D.M. conducted CFD studies. S.B. and D.M. performed image analysis, with support from A.K. and R.K. A.K., R.K., and G.D. provided intellectual feedback and support. All authors edited and provided feedback on the article. R.K. supervised the work.
Footnotes
Acknowledgments
The authors would like to thank Dr. Max Winkelman for preliminary work that led to the development of this work, and Nolan Burson for assistance with cell culture. Also, the authors would like to thank Dr. Richard West’s Lab at Northeastern for COMSOL support.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the